

Abstract
CRM1 (Chromosomal Maintenance 1, also known as Exportin 1) is the major mammalian export protein that facilitates the transport of large macromolecules including RNA and protein across the nuclear membrane to the cytoplasm. The gene encoding CRM1 was originally identified in yeast as required to maintain higher order chromosome structure. In mammalian cells, CRM1 was found to bind several nuclear pore proteins hence its role in nuclear-cytosolic transport was discovered. In addition to nuclear-cytosolic transport, CRM1 also plays a role in centrosome duplication and spindle assembly, especially in response to DNA damage. The crystal structure of CRM1 suggests a complex protein that binds the Ran protein bound to GTP, allowing for a conformational change that facilitates binding to different cargo proteins through a nuclear export signal (NES). Included in the cadre of cargo are multiple tumor suppressor and oncoproteins as p53, BRCA1, Survivin, NPM, and APC, which function in the nucleus to regulate transcription or aid in chromosomal assembly and movement. An imbalance in the cytosolic level of these proteins has been observed in cancer cells, resulting in either inactivation (tumor suppressor) or an excess of anti-apoptotic activity (oncoprotein). Thus, the concept of inhibiting CRM1 has been explored as a potential therapeutic intervention. Indeed, inhibition of CRM1 by a variety of small molecules that interfere with cargo-NES binding results in cancer cell death. Whether all of these proteins together are responsible for this phenotype or whether specific proteins are required for this effect is unclear at this time.
Keywords: CRM1, nuclear pore complex, leptomycin B, p53, Survivin, APC, p27, NPM, BRCA1
The nuclear-pore complex of proteins
In the eukaryotic cell, diverse molecular functions such as DNA synthesis, RNA transcription and translation and protein processing, occur within distinct intracellular compartments. As a result, macromolecules that participate in these processes must be exchanged between these compartments. While small molecules (20-40 kD) can passively diffuse across compartments, larger molecules, including most proteins and RNAs, are transported by signal- and energy-dependent mechanisms [1].
The site of active transport between the nucleus and the cytosol occurs at a multi-protein complex called the nuclear pore complex (NPC, Figure 1) [1-3]. The NPC is composed of approximately 30 individual nucleoporins (Nups), with an approximate molecular mass of 125 MDa [4-6]. The core structure is symmetrical, consisting of a spoke-ring component surrounding the central-transporter. Short fiber-like structures extend from the NPC into the cytoplasm while basket-like structures extend into the nucleus [5,7]. Four transmembrane Nups that anchor the NPC in the double lipid bilayer of the nuclear envelope have been described in vertebrate cells [8-12]. Approximately 15 different structural Nups form subcomplexes that participate in the formation of the NPC building blocks, including the nuclear basket and nuclear/cytoplasmic rings. Ten additional Nups are rich in phenylalanine-glycine repeats that mediate the interaction of the NPC with soluble nuclear transport receptors [13-15]. Each NPC rapidly transports hundreds of macromolecules bidirectionally [1]. A large number of proteins and mature snRNP particles are actively imported into the nucleus [1,16], while export cargoes include proteins, mRNA, rRNA, tRNA, and snRNAs [17].
Figure 1.
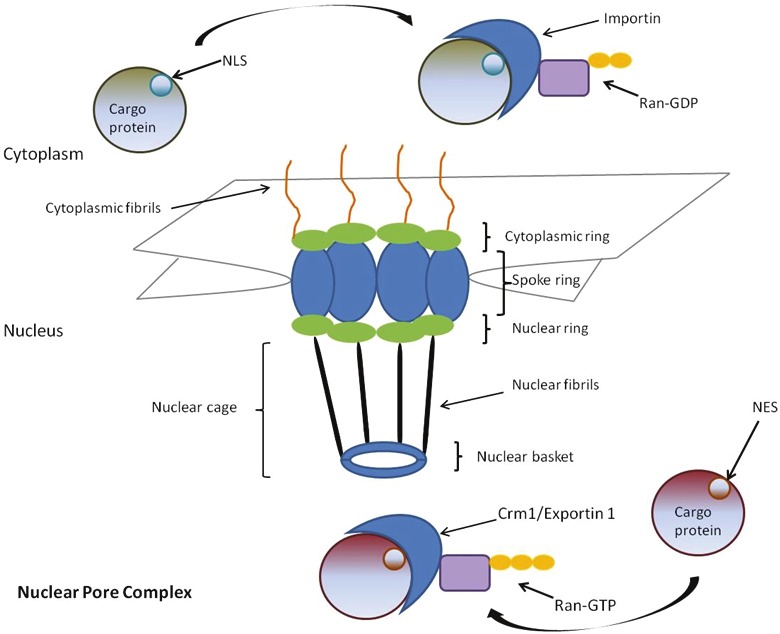
Model of the mammalian nuclear pore complex (NPC). The NPC is composed of nuclear pore proteins (Nups) surrounding a channel with a central transporter. The cytoplasmic fibrils and the nuclear basket assist in NES/NLS recognition and protein anchoring. Nuclear import is mediated by an importin bound to Ran-GDP. Nuclear export is mediated by the CRM1/Exportin 1 receptor bound to Ran-GTP.
In most cases, energy-dependent, signal-mediated nuclear import and export involves nucleocytoplasmic shuttling receptors, which bind to their cargoes either directly or via adaptor molecules [1,16,18]. They are called importins or exportins, depending on the major direction of cargo transport, or collectively karyopherins [19]. After cargo molecules interact with their cognate receptors in the originating compartment, the transport complexes undergo stepwise translocation through the NPC to the destination compartment, whereupon the cargo dissociates from the receptor and the latter is recycled. Several different classes of transport signals have been described for nuclear import (nuclear localization signals, or NLS) and export (nuclear export signals, or NES). A “classical” NLS is composed of a short amino acid sequence enriched in basic amino acids that interacts with an import receptor consisting of an importin β and importin α heterodimer. A common type of NES is a short peptide segment enriched in leucine residues [20], which interacts directly with the nuclear export receptor CRM1/Exportin 1 [21-23].
The small GTPase Ran, which shuttles between the nucleus and the cytoplasm, plays a key role in determining the directionality of nuclear transport [24,25]. Many importin β nuclear transport receptors are RanGTP (Ran complexed with GTP) binding proteins [26]. The binding of RanGTP has distinct effects on import and export receptors. RanGTP dissociates cargo from nuclear import receptors [27,28], while it promotes the association of cargo with nuclear export receptors [29-31]. Since the GTPase-activating protein for Ran (RanGAP) [32,33] is sequestered in the cytoplasm and its guanine nucleotide exchange factor (RCC1) is restricted to the nucleus [34], most nuclear Ran is in the GTP-bound form, while most cytoplasmic Ran is in the GDP-bound form. Thus, the nucleocytoplasmic compartmentalization of Ran effectors in cells and the resulting asymmetric distribution of RanGTP are important for the loading and unloading of nuclear transport receptors in the nucleus.
The CRM1 mammalian export protein
The crm1 (chromosome region maintenance) gene was originally cloned in yeast after it was identified in a screen for genes essential for maintaining higher-order chromosome structure [23,35]. Cold-sensitive (cs) crm1+ mutants of the fission yeast Schizosaccharomyces pombe resulted in deformed nuclear domains (abnormal chromosomal structures) at the restrictive temperature [35]. The crm1+ gene encodes a 115-kD protein that preferentially localizes to the nucleus and the nucleocytoplasmic junction and was originally found to interact with the AP-1 like transcription factor, pap1 [35]. Mutations in the fission yeast crm1 gene led to upregulation of pap1 [36] and functionally to a multidrug resistance phenotype, including resistance to caffeine and the anti-fungal agent leptomycin B [37-39]. A mechanism for these effects became apparent when the nuclear export function of CRM1 was discovered [40].
Human CRM1/Exportin 1 was originally cloned as a 112 kDa protein that interacted with at least two proteins associated with the human nuclear pore complex, the nucleoporins CAN/Nup214 and Nup88 [21,41,42]. Yeast two hybrid assays demonstrated interactions between CRM1 and several nucleoporins, as well as Rev and Ran proteins [43,44]. Human CRM1 was subsequently identified as the human homolog of S. cerevisiae crm1 (47% identity 67% similarity) and to the S. pombe homologue (52% identity, 69% similarity). Further analysis revealed that the N-terminus of hCRM1 shared significant homology to that of importin β [42]. Cross-species comparison of the CRM1 protein sequence and structure indicates significant homology from yeast to humans (Figure 2).
Figure 2.

The central conserved domain (CCR) of CRM1 from six disparate species was aligned by ClustalW [149] and the results presented by PRALINE [150]. The part of the alignment containing the human Cys-528 residue that covalently binds to leptomycin B is marked by a box at 562 on the alignment number scale. Note that species that are leptomycin B resistant (here represented by Saccharomyces cerevisiae and Neurospora crassa) lack the cysteine at this position.
The initial understanding of CRM1-mediated nuclear export mechanisms was greatly influenced by studies of the HIV-1 protein Rev [45]. The role of this protein in the viral life-cycle is to recognize and promote the export of unspliced and partially spliced viral RNAs. Rev and the protein kinase inhibitor (PKI) of cAMP-dependent protein kinase (cAPK) are the first proteins in which a NES was identified [46,47]. Studies of the Rev NES led to independent identification of CRM1/Exportin 1. CRM1 was subsequently found to export a very broad range of substrates (4, 5, 7-11).
CRM1 crystal structure
The mouse and human CRM1 crystal structures were independently solved by two groups of investigators in a complex with the cargo protein snurportin 1 (SPN1). SPN1 is an import adapter protein that assists in transporting mature m3G-capped spliceosomal small nuclear ribonucleoproteins (snRNPs) into nuclei during their biogenesis [48]. To mediate import cycles, SPN1 is returned to the cytoplasm by CRM1 [49]. The crystal structure of the CRM1 complex confirmed prior protein sequence analyses and showed 20 HEAT repeats, each of which included two anti-parallel helices (A and B) lining the convex and concave sides of the protein (Figure 3) [50,51]. In the complex, the CRM1 protein is bent to a distorted toroid structure, with HEAT 21 touching helices 2B and 5A, as well as the loop between HEATs 4 and 5. RanGTP is enclosed inside the toroid and stabilizes the ring closure through its multiple contacts. In contrast to other structural interactions with importin-β [52-54], the cargo protein SPN1 is not enveloped by CRM1 but rests outside of the CRM1 toroid. Further structural studies showed that CRM1 uses the same hydrophobic binding pocket for its interaction with different cargos, forcing NES peptide sequences to adapt their conformation to the rigid binding-site on CRM1 [55]. Depending on the spacing of key hydrophobic residues, the affinities of NES-containing cargos for their cognate receptor CRM1 can vary dramatically, with low-affinity cargos prevailing [56,57]. Weak affinities seem to be important for efficient disassembly of export complexes on the cytoplasmic side of the NPC [58]. Furthermore, they prevent cargos from binding to CRM1 in the cytoplasm in the absence of RanGTP [56]. Another level of regulation of cargo binding to CRM1 appears to be the available concentration of CRM1 which is rate-limiting for export [59].
Figure 3.
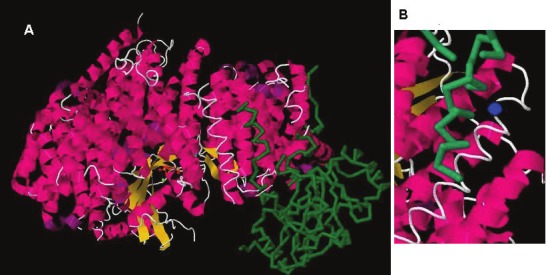
A. Crystal structure of CRM1, snurportin1 and RanGTP. The heat repeat anti-parallel alpha helices are in red, RanGTP is in yellow, and snurportin is in green. B. A portion of the crystal structure image of CRM1, snurportin1 and RanGTP highlighting the alpha helix containing Cys-528 and the hydrophobic cleft that binds a cargo protein’s NES domain. The blue circle marks the position of the Cys-528 residue. The green backbone represents the amino terminal end of SNP1 which contains the NES domain of the protein. Image adapted from the RCSB PDB (www.pdb.org) of PDB ID 3GJX [50].
Expression and function of CRM1 in normal development
Regulation of the gastrula-neurula transition (GNT)
Developmental expression and function for CRM1 in higher eukaryotes has been explored genetically. In Xenopus, the CRM1-encoding mRNA is maternally expressed and is present throughout early development, which corresponds with its constitutive protein expression [60]. XCRM1 localized within the nucleus, both before and during the gastrula stage. A specific nuclear membrane associated localization (‘adult-type’) CRM1 was observed beginning at the time of the neurula stage and CRM1 was unable to recognize the NES until this time period. In functional studies, XCRM1 RNA microinjections into stage 1 embryos showed arrested development before, during or just after the neurula stage, followed by death of the developing embryo, supporting a conclusion that unprogrammed activity of CRM1 greatly impairs normal Xenopus development and that the GNT is a critical period for its activity. Treatment of the embryos with the anti-fungal CRM1 inhibitor leptomycin B blocked development at the neurula stage, again suggesting that CRM1 is essential for procession through this stage and that inhibition of CRM1 activity does not affect normal development before this time [60]. Therefore, the intranuclear localization of CRM1 is under a developmentally controlled process and the GNT appears as an important period in the regulation of CRM1 activity during early Xenopus development.
Regulation of larval progression
In Drosophila, a screen for expressed sequences in chromosomal region 29C led to the isolation of a class of cDNAs encoding a polypeptide [61] with strong homology to the S. pombe crm1 protein [35]. Although originally named crm1 [62], the drosophila homolog was renamed embargoed (emb) to reflect the nuclear export defects observed in the emb mutant flies [61]. The emb transcript was ubiquitously expressed at all stages of embryonic development. Specific tissues in which expression was relatively high were brain, hind gut, and posterior spiracles shortly before dorsal closure and the ventral nerve cord, midgut, and somatic musculature shortly after dorsal closure [61]. In each case, emb expression levels increased when the tissue was mature, suggesting that it is required for the maintenance of these tissues rather than for their formation. Hemizygous emb flies showed an arrest in larval development at the transition from the second to third instar stage [61].
Regulation of centrosome duplication and spindle assembly
The centrosome is the principal microtubule-organizing centre of mammalian cells, and functions to direct the assembly of a bipolar spindle during mitosis [63,64]. Centrosome duplication is initiated at the G1/S boundary and is completed at S phase of the cell cycle, which coincides with DNA replication. Over the last decade several proteins that require CRM1 for their activity have been implicated in the regulation of centrosome duplication. These studies provide evidence of functions for CRM1 beyond nuclear-cytoplasmic shuttling.
Nucleophosmin
Nucleophosmin (NPM) is a centrosome-associated protein that is dependent on CRM1 for its nuclear-cytoplasmic shuttling during the cell cycle [65]. Mutation within the NES in NPM or disruption of CRM1 function by addition of leptomycin B, results in the dissociation of NPM from centrosomes and the initiation of premature centrosome duplication in addition to its effects on nuclear-cytoplasmic transport [65].
NPM is also a tumor suppressor protein in mice that likely serves the same functions in humans. This hypothesis is supported by the existance of NPM-associated chromosomal translocations as well as aberrant cytoplasmic protein expression in several hematologic malignancies [66-69]. The mechanism of NPM tumor suppressor function is likely to be controlled by its association with CRM1.
BRCA1
The breast and ovarian cancer susceptibility protein 1 (BRCA1) is a tumor suppressor protein encoded by a gene that when mutated is a risk factor for the development of breast and ovarian cancer [70-72]. BRCA1 regulates the DNA damage response and functions in the nucleus to stimulate DNA repair and at centrosomes to inhibit centrosome duplication after DNA damage [73-75]. At the centrosome, BRCA1 binds and ubiquitinates γ-tubulin which controls centrosome amplification and microtubule nucleation [76]. Inactivation of CRM1 impairs BRCA1 centrosome localization and mutation of the BRCA1 NES blocks BRCA1 regulation of centrosome amplification [77], suggesting that its interaction with CRM1 is essential for its function as a centrosome DNA damage checkpoint protein.
CRM1 can only bind to the undimerized form of BRCA1, as the NES is a core part of the binding domain used in heterodimerization of BRCA1 with its partner protein BARD1. Both BRCA1 and BARD1 NES become masked upon heterodimer formation [78]. A hypothesis based on these findings suggests that the ability of CRM1 to drive BRCA1, BARD1, and several BRCA1-BARD1 substrates, to the centrosome increases the proteins’ proximity to one another, driving BRCA1-BARD1 dimer formation to catalyze ubiquitination of downstream substrates that are required to regulate centrosome amplification during the DNA damage response [77].
CRM1-mediated export of cancer proteins
An increasingly large number of cancer-associated proteins that shuttle into and out of the cell nucleus, as tumor suppressor and oncogenic proteins, require CRM1 for their nuclear exit (recently reviewed in [79]). Interestingly, mutations and/or dysregulation of such proteins in cancer cells, including BRCA1, p53, p27, and APC can lead to an aberrant high level of expression within the cytosol which disables them from performing their normal functions within the nucleus. We review here four proteins with distinct roles in cancer cells that are dependent on CRM1-mediated nuclear export.
Survivin
Survivin is a bifunctional protein involved with regulating cell division when expressed in the nucleus and controlling apoptotic pathways when expressed in the cytoplasm [80,81]. The smallest member (16.5kDa) of the inhibitor-of-apoptosis protein family (IAP), survivin is believed to exist predominantly as a homodimer [82,83]. Survivin export from the nucleus to the cytoplasm is mediated by the CRM1/Ran-GTP axis [84-86]. Survivin contains two amino acid sequences involved with its nuclear export: the first is the leucine-rich NES located in the linker region between the N-terminus and BIR domains of the survivin homodimer and the second is a non-classical sequence in the C-terminus [85,87]. The central NES is partially masked by the homodimer interface and is therefore primarily active when survivin is a monomer. The monomeric configuration of survivin can be induced by HDAC6-mediated deacetylation at lysine 129 [88,89]. Upon interruption of the CRM1/Ran-GTP axis via disruption of the Ran-GTP gradient or by leptomycin B, survivin localizes to the nucleus, a process that thus far is thought to occur by passive diffusion as no classical NLS exists within the protein.
In the nucleus, survivin is primarily associated with the chromosome passenger complex (CPC), a core complex of proteins including Borealin, Aurora B kinase, INCENP and survivin, involved in ensuring the correct attachment between the centromere and the mitotic spindle, among other related checkpoint regulation pathways of mitosis [90,91]. CRM1 is a necessary effector in co-localizing the CPC with the centromere during G2/M phase of the cell cycle by interacting with survivin [84,85]. While not essential in CPC function or anchoring, CRM1 is required as a transient transporter of the CPC to the centromere [84,92]. Aurora B kinase also accesses the CRM1-dependent nuclear export pathway for nucleocytoplasmic localization in a manner very similar to the survivin-CRM1 interaction - by interacting with the non-catalytic N-terminal domain of Aurora B [93,94]. Mutational studies demonstrated that CRM1 interacts with the CPC through the NES of survivin [84]. The nuclear survivin-CRM1 interaction, independently of the CPC, may also negatively regulate transcription factors, such as oncogenic STAT3 [88].
In the cytoplasm, survivin exercises its cytoprotective function by actively inhibiting apoptotic pathways, to prolong cell life [95,96]. Many studies have shown that survivin is both upregulated and localized in the cytoplasm of cancer cells [97,98]; its nuclear export required for its anti-apoptotic and tumor-promoting function [99,100]. Experiments disrupting the survivin NES sequence or CRM1 function in cancer cells result in nuclear localization of survivin, which increases the susceptibility of these cells to conventional chemotherapy and radiation treatment. The withdrawal of survivin by the collapse of the CRM1/Ran GTP axis followed by ubiquitin-proteasome degradation indicates an active regulatory mechanism that promotes cell death progression [100].
A variety of human splice variants for survivin exist, though not all isoforms are uniformly present or unambiguous [101,102]. The NES is present in the canonical survivin, survivin2B, and survivin3B, but not in survivinDEx3 or survivin2α [103]. Though currently still ill-defined in vivo, expression of these isoforms has been suggested to correlate with certain disease models and clinical outcomes for cancer patients.
p27KIP1
The Cdk inhibitor p27 is an important regulator of G1 progression in normal cells. It is highly expressed in G0, where it binds tightly and inhibits cyclin E-Cdk 2 [104-106]. In mid-G1, p27 also plays a role in the assembly and nuclear import of D-type cyclin-Cdk complexes [107]. p27 levels are regulated by translational controls and by proteolysis, and decrease as cells progress from G1 to S phase [108,109]. Detectable p27 is exclusively nuclear in G0 and early G1, with a transient appearance in both the nucleus and cytoplasm as cells progress through G1, before its disappearance in late S phase [110]. The dramatic increase in p27-CRM1 binding during G1 progression and the transient appearance of cytosolic p27 at the G1/S transition suggested a link between nuclear export of p27 and its degradation [110]. The timing of the cellular p27-CRM1 interaction and the observation that p27 is exported more rapidly from G1 nuclei than from G0 nuclei suggested that p27, the CRM1-Ran-GTP export machinery, or both may undergo periodic post-translational changes to facilitate p27 export in early G1. The phosphorylation status of serine 10 (S10) critically regulates p27-CRM1 binding and export [111-113].
p27 was demonstrated to be a tumor suppressor protein in mice after its genetic deletion led to multi-organ hyperplasia, increased body size and susceptibility to carcinogen-induced tumors [114,115]. In contrast to other tumor suppressors however, mutation or deletion of p27 is rare in human cancers. Instead, deregulated receptor tyrosine kinases are believed to activate Src/BCR-ABL and Ras/MEK/MAPK, or PI3K/AKT signaling which induce p27 loss or subcellular mislocalization, respectively [116,117]. Interestingly, when p27 localizes in the nucleus it inhibits proliferation however when expressed in the cytosol it promotes cytoskeleton remodeling, suggesting a potential mechanism for tumor promotion if the balance between its expression in the nucleus and cytosol was tipped toward increased cytosolic levels. Indeed, downregulation of p27 within the nucleus or its mislocalization to the cytosol consistently correlate with poor prognosis of several different cancers [117]. In one example in breast tumors, progressive p27 loss within cell nuclei has been observed during the histopathological progression of neoplasia from benign to in situ and invasive cancers and reduced nuclear p27 levels has been shown to be an independent prognostic indicator of disease relapse or death [118-120].
p53
A master regulator of the cell cycle and of genomic integrity, p53 is a tumor suppressor protein required for homeostasis of mammalian cells [121,122]. When activated, p53 initiates several signaling pathways to arrest cell cycle progression, to repair DNA damage, and if necessary, to induce apoptosis. Approximately 50% of malignant tumors express mutant forms of p53 or have a genetic deletion of the p53 gene [123]. p53 activity is highly dependent on its subcellular localization to the nucleus, which is regulated primarily by the CRM1 nuclear export pathway [124,125]. Interestingly, p53 and CRM1 are involved in a reciprocal regulatory loop. While nuclear p53 can repress CRM1 transcription [126], increased levels of CRM1 can promote p53 mislocalization and dysfunction, as occurs in some cancer cells [127].
In normal cells, p53 is detectable at low-levels, as it is tightly regulated by proteasome-mediated degradation and sequestration in the cytosol [125]. Both processes are mediated by one of two NES in p53: one located in the N-terminal region and one in the C-terminal region [128,129]. A number of accessory proteins also use these latter regions to regulate p53 activity and its subcellular localization, including MDM2, PARP-1, and HPV-18 E6 [130,131].
APC
The tumor suppressor adenomatous polyposis coli (APC) is a large protein with minimal sequence homology to other known proteins [132]. Truncating mutations in the Apc gene represents an early step in the progression of the majority of colorectal cancers, including inherited and sporadic cases [133-135]. While the mechanisms of these mutations in tumorigenesis are not fully understood, the best documented function of APC is to oppose a Wnt signal by targeting β-catenin for proteasome-mediated degradation in the cytoplasm.
A major component of the Wnt signaling pathway, β-catenin is a transcription cofactor that functions in the nucleus [136]. In normal cells, APC regulates the levels of β-catenin by promoting its nuclear export through binding to CRM1 [137,138]. Once exported, APC is degraded in the cytoplasm through the proteolysis pathway. This process is mediated by a complex of proteins, including APC, β-catenin and the scaffolding protein Axin. Together, the complex promotes GSK-3β phosphorylation of β-catenin, targeting it for degradation. APC has been viewed as a “chaperone” protein for β-catenin, essential for its nuclear export; however, recent studies also suggest that β-catenin can engage in nuclear-cytoplasmic shuttling independent of APC and CRM1[139].
The APC protein has five different nuclear export signals, two N-terminal Rev-type NESs (NES1 and NES2) and three non-functional central signals that are deleted in the APC mutant proteins [137,138,140]. Of these, NES1 has the strongest signal and is the only sequence that is not truncated in the APC mutant forms. Truncated forms of human APC do accumulate in the nucleus following leptomycin B-treatment of colorectal cancer cell lines, suggesting that the nuclear-cytoplasmic shuttling ability of APC is not lost in some colorectal cancer cells. While the dynamic nuclear-cytoplasmic shuttling of APC has been suggested to be directly involved in mediating intracellular cell signaling through unknown means, APC tumor suppressor function is primarily recognized as a regulator of β-catenin [141].
Immunohistochemical studies using human colon tissue support a role for nuclear APC expression in human tumor suppression [135,142]. While the mutant, truncated APC protein remained strongly nuclear in colon polyps [143], the frequency of APC expression within the cytoplasm increased with progression towards malignant tumors, with 60% of colon carcinomas showing cytoplasmic APC expression compared to only 4% of normal colon tissues [143]. This finding suggests that without the ability to localize to the nucleus, APC is unable to maintain its regulatory tumor suppressor role.
Inhibiting CRM1 function
As many tumor suppressors and oncoproteins use CRM1 for their nuclear export and these proteins lose their normal function once they have exited the nucleus, leaving the cancer or pre-cancer cell vulnerable to constitutive growth factor and pro-survival signals, many efforts have been expended towards the development of compounds that inhibit CRM1 activity. Several of these drugs were recently discussed in an excellent review [79] therefore we will briefly mention two classes of these agents here.
Leptomycin B (LMB) was originally isolated as an antifungal antibiotic from a Streptomyces strain [144]. In mammalian cells, LMB resulted in cell cycle arrest at both G1 and G2 phases of the cell cycle. Upon removal of the drug, cell cycle analysis showed cells that had bypassed mitosis and become tetraploid [145]. The LMB resistance gene was identified to be a mutant of the CRM1 gene. Analyses of the mutant strongly suggested that the CRM1 protein was the molecular target of LMB [37].
LMB covalently binds to a single cysteine residue (Cys-528 in human) to inactivate CRM1 by a Michael-type addition [146]. Cys-528 is located in a central conserved region (CCR) and is conserved in LMB-sensitive organisms [146] (Figure 2). Crystal structure studies of the hCRM1 protein confirm that Cys-528 is located within a hydrophobic cleft, which explains why LMB-modified CRM1 cannot bind export cargoes that rely on this cleft [50,51] (Figure 3). LMB is believed to compete with the NES for RanGTP-dependent formation of a stable CRM1-NES complex. LMB may also prevent the physical movement required for the required conformational change in CRM1by disrupting a hydrogen bond caused by selective alkylation at the cysteine residue, which is important for the CCR function.
Although LMB was a potent inhibitor of CRM1 and an effective cell death agent in multiple cancer cell types in vitro, it failed clinical trials in patients due to its toxicity. Therefore, several additional agents are currently in the process of development. One such class of agents is the Selective Inhibitors of Nuclear Export (SINE, KPT compounds) developed by Karyopharm Therapeutics, Inc. These are oral small molecule inhibitors which use a similar mechanism as LMB of irreversible binding to CRM1 through Cys-528. Thus far, these agents are well-tolerated in several small and large animal models and are scheduled to enter human clinical trials within the next year [147,148].
Acknowledgement
This work was supported by: NIH NIMGS GRANT # 8P20GM103421-09 (formerly P20RR017695).
References
- 1.Gorlich D, Mattaj IW. Nucleocytoplasmic transport. Science. 1996;271:1513–1518. doi: 10.1126/science.271.5255.1513. [DOI] [PubMed] [Google Scholar]
- 2.Pante N, Aebi U. Toward the molecular details of the nuclear pore complex. J Struct Biol. 1994;113:179–189. doi: 10.1006/jsbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 3.Simos G, Hurt EC. Nucleocytoplasmic transport: factors and mechanisms. FEBS Lett. 1995;369:107–112. doi: 10.1016/0014-5793(95)00674-x. [DOI] [PubMed] [Google Scholar]
- 4.Rout MP, Wente SR. Pores for thought: nuclear pore complex proteins. Trends Cell Biol. 1994;4:357–365. doi: 10.1016/0962-8924(94)90085-x. [DOI] [PubMed] [Google Scholar]
- 5.Goldberg MW, Allen TD. Structural and functional organization of the nuclear envelope. Curr Opin Cell Biol. 1995;7:301–309. doi: 10.1016/0955-0674(95)80083-2. [DOI] [PubMed] [Google Scholar]
- 6.Doye V, Hurt E. From nucleoporins to nuclear pore complexes. Curr Opin Cell Biol. 1997;9:401–411. doi: 10.1016/s0955-0674(97)80014-2. [DOI] [PubMed] [Google Scholar]
- 7.Davis LI. The nuclear pore complex. Annu Rev Biochem. 1995;64:865–896. doi: 10.1146/annurev.bi.64.070195.004245. [DOI] [PubMed] [Google Scholar]
- 8.Chadrin A, Hess B, San Roman M, Gatti X, Lombard B, Loew D, Barral Y, Palancade B, Doye V. Pom33, a novel transmembrane nucleoporin required for proper nuclear pore complex distribution. J Cell Biol. 2010;189:795–811. doi: 10.1083/jcb.200910043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerace L, Ottaviano Y, Kondor-Koch C. Identification of a major polypeptide of the nuclear pore complex. J Cell Biol. 1982;95:826–837. doi: 10.1083/jcb.95.3.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hallberg E, Wozniak RW, Blobel G. An integral membrane protein of the pore membrane domain of the nuclear envelope contains a nucleoporin-like region. J Cell Biol. 1993;122:513–521. doi: 10.1083/jcb.122.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mansfeld J, Guttinger S, Hawryluk-Gara LA, Pante N, Mall M, Galy V, Haselmann U, Muhlhausser P, Wozniak RW, Mattaj IW, Kutay U, Antonin W. The conserved transmembrane nucleoporin NDC1 is required for nuclear pore complex assembly in vertebrate cells. Mol Cell. 2006;22:93–103. doi: 10.1016/j.molcel.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 12.Stavru F, Hulsmann BB, Spang A, Hartmann E, Cordes VC, Gorlich D. NDC1: a crucial membrane-integral nucleoporin of metazoan nuclear pore complexes. J Cell Biol. 2006;173:509–519. doi: 10.1083/jcb.200601001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Terry LJ, Wente SR. Flexible gates: dynamic topologies and functions for FG nucleoporins in nucleocytoplasmic transport. Eukaryot Cell. 2009;8:1814–1827. doi: 10.1128/EC.00225-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walde S, Kehlenbach RH. The Part and the Whole: functions of nucleoporins in nucleocytoplasmic transport. Trends Cell Biol. 2010;20:461–469. doi: 10.1016/j.tcb.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 15.Wente SR, Rout MP. The nuclear pore complex and nuclear transport. Cold Spring Harb Perspect Biol. 2010;2:a000562. doi: 10.1101/cshperspect.a000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nigg EA. Nucleocytoplasmic transport: signals, mechanisms and regulation. Nature. 1997;386:779–787. doi: 10.1038/386779a0. [DOI] [PubMed] [Google Scholar]
- 17.Izaurralde E, Lewis J, Gamberi C, Jarmolowski A, McGuigan C, Mattaj IW. A cap-binding protein complex mediating U snRNA export. Nature. 1995;376:709–712. doi: 10.1038/376709a0. [DOI] [PubMed] [Google Scholar]
- 18.Mattaj IW, Englmeier L. Nucleocytoplasmic transport: the soluble phase. Annu Rev Biochem. 1998;67:265–306. doi: 10.1146/annurev.biochem.67.1.265. [DOI] [PubMed] [Google Scholar]
- 19.Fried H, Kutay U. Nucleocytoplasmic transport: taking an inventory. Cell Mol Life Sci. 2003;60:1659–1688. doi: 10.1007/s00018-003-3070-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerace L. Nuclear export signals and the fast track to the cytoplasm. Cell. 1995;82:341–344. doi: 10.1016/0092-8674(95)90420-4. [DOI] [PubMed] [Google Scholar]
- 21.Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–1060. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 22.Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M, Nishida E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature. 1997;390:308–311. doi: 10.1038/36894. [DOI] [PubMed] [Google Scholar]
- 23.Stade K, Ford CS, Guthrie C, Weis K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell. 1997;90:1041–1050. doi: 10.1016/s0092-8674(00)80370-0. [DOI] [PubMed] [Google Scholar]
- 24.Melchior F, Gerace L. Two-way trafficking with Ran. Trends Cell Biol. 1998;8:175–179. doi: 10.1016/s0962-8924(98)01252-5. [DOI] [PubMed] [Google Scholar]
- 25.Moore MS. Ran and nuclear transport. J Biol Chem. 1998;273:22857–22860. doi: 10.1074/jbc.273.36.22857. [DOI] [PubMed] [Google Scholar]
- 26.Wozniak RW, Rout MP, Aitchison JD. Karyopherins and kissing cousins. Trends Cell Biol. 1998;8:184–188. doi: 10.1016/s0962-8924(98)01248-3. [DOI] [PubMed] [Google Scholar]
- 27.Rexach M, Blobel G. Protein import into nuclei: association and dissociation reactions involving transport substrate, transport factors, and nucleoporins. Cell. 1995;83:683–692. doi: 10.1016/0092-8674(95)90181-7. [DOI] [PubMed] [Google Scholar]
- 28.Izaurralde E, Kutay U, von Kobbe C, Mattaj IW, Gorlich D. The asymmetric distribution of the constituents of the Ran system is essential for transport into and out of the nucleus. EMBO J. 1997;16:6535–6547. doi: 10.1093/emboj/16.21.6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kutay U, Bischoff FR, Kostka S, Kraft R, Gorlich D. Export of importin alpha from the nucleus is mediated by a specific nuclear transport factor. Cell. 1997;90:1061–1071. doi: 10.1016/s0092-8674(00)80372-4. [DOI] [PubMed] [Google Scholar]
- 30.Kutay U, Izaurralde E, Bischoff FR, Mattaj IW, Gorlich D. Dominant-negative mutants of importin-beta block multiple pathways of import and export through the nuclear pore complex. EMBO J. 1997;16:1153–1163. doi: 10.1093/emboj/16.6.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arts GJ, Kuersten S, Romby P, Ehresmann B, Mattaj IW. The role of exportin-t in selective nuclear export of mature tRNAs. EMBO J. 1998;17:7430–7441. doi: 10.1093/emboj/17.24.7430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coutavas E, Ren M, Oppenheim JD, D'Eustachio P, Rush MG. Characterization of proteins that interact with the cell-cycle regulatory protein Ran/TC4. Nature. 1993;366:585–587. doi: 10.1038/366585a0. [DOI] [PubMed] [Google Scholar]
- 33.Bischoff FR, Krebber H, Smirnova E, Dong W, Ponstingl H. Co-activation of RanGTPase and inhibition of GTP dissociation by Ran-GTP binding protein RanBP1. EMBO J. 1995;14:705–715. doi: 10.1002/j.1460-2075.1995.tb07049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohtsubo M, Okazaki H, Nishimoto T. The RCC1 protein, a regulator for the onset of chromosome condensation locates in the nucleus and binds to DNA. J Cell Biol. 1989;109:1389–1397. doi: 10.1083/jcb.109.4.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adachi Y, Yanagida M. Higher order chromosome structure is affected by cold-sensitive mutations in a Schizosaccharomyces pombe gene crm1+ which encodes a 115-kD protein preferentially localized in the nucleus and its periphery. J Cell Biol. 1989;108:1195–1207. doi: 10.1083/jcb.108.4.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toda T, Shimanuki M, Saka Y, Yamano H, Adachi Y, Shirakawa M, Kyogoku Y, Yanagida M. Fission yeast pap1-dependent transcription is negatively regulated by an essential nuclear protein, crm1. Mol Cell Biol. 1992;12:5474–5484. doi: 10.1128/mcb.12.12.5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishi K, Yoshida M, Fujiwara D, Nishikawa M, Horinouchi S, Beppu T. Leptomycin B targets a regulatory cascade of crm1, a fission yeast nuclear protein, involved in control of higher order chromosome structure and gene expression. J Biol Chem. 1994;269:6320–6324. [PubMed] [Google Scholar]
- 38.Turi TG, Webster P, Rose JK. Brefeldin A sensitivity and resistance in Schizosaccharomyces pombe. Isolation of multiple genes conferring resistance. J Biol Chem. 1994;269:24229–24236. [PubMed] [Google Scholar]
- 39.Kumada K, Yanagida M, Toda T. Caffeine-resistance in fission yeast is caused by mutations in a single essential gene, crm1+ Mol Gen Genet. 1996;250:59–68. doi: 10.1007/BF02191825. [DOI] [PubMed] [Google Scholar]
- 40.Toone WM, Kuge S, Samuels M, Morgan BA, Toda T, Jones N. Regulation of the fission yeast transcription factor Pap1 by oxidative stress: requirement for the nuclear export factor Crm1 (Exportin) and the stress-activated MAP kinase Sty1/Spc1. Genes Dev. 1998;12:1453–1463. doi: 10.1101/gad.12.10.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fornerod M, van Baal S, Valentine V, Shapiro DN, Grosveld G. Chromosomal localization of genes encoding CAN/Nup214-interacting proteins--human CRM1 localizes to 2p16, whereas Nup88 localizes to 17p13 and is physically linked to SF2p32. Genomics. 1997;42:538–540. doi: 10.1006/geno.1997.4767. [DOI] [PubMed] [Google Scholar]
- 42.Fornerod M, van Deursen J, van Baal S, Reynolds A, Davis D, Murti KG, Fransen J, Grosveld G. The human homologue of yeast CRM1 is in a dynamic subcomplex with CAN/Nup214 and a novel nuclear pore component Nup88. EMBO J. 1997;16:807–816. doi: 10.1093/emboj/16.4.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Askjaer P, Jensen TH, Nilsson J, Englmeier L, Kjems J. The specificity of the CRM1-Rev nuclear export signal interaction is mediated by RanGTP. J Biol Chem. 1998;273:33414–33422. doi: 10.1074/jbc.273.50.33414. [DOI] [PubMed] [Google Scholar]
- 44.Neville M, Stutz F, Lee L, Davis LI, Rosbash M. The importin-beta family member Crm1p bridges the interaction between Rev and the nuclear pore complex during nuclear export. Curr Biol. 1997;7:767–775. doi: 10.1016/s0960-9822(06)00335-6. [DOI] [PubMed] [Google Scholar]
- 45.Pollard VW, Malim MH. The HIV-1 Rev protein. Annu Rev Microbiol. 1998;52:491–532. doi: 10.1146/annurev.micro.52.1.491. [DOI] [PubMed] [Google Scholar]
- 46.Fischer U, Huber J, Boelens WC, Mattaj IW, Luhrmann R. The HIV-1 Rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell. 1995;82:475–483. doi: 10.1016/0092-8674(95)90436-0. [DOI] [PubMed] [Google Scholar]
- 47.Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signal for rapid export of proteins from the nucleus. Cell. 1995;82:463–473. doi: 10.1016/0092-8674(95)90435-2. [DOI] [PubMed] [Google Scholar]
- 48.Huber J, Cronshagen U, Kadokura M, Marshallsay C, Wada T, Sekine M, Luhrmann R. Snurportin1, an m3G-cap-specific nuclear import receptor with a novel domain structure. EMBO J. 1998;17:4114–4126. doi: 10.1093/emboj/17.14.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paraskeva E, Izaurralde E, Bischoff FR, Huber J, Kutay U, Hartmann E, Luhrmann R, Gorlich D. CRM1-mediated recycling of snurportin 1 to the cytoplasm. J Cell Biol. 1999;145:255–264. doi: 10.1083/jcb.145.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monecke T, Guttler T, Neumann P, Dickmanns A, Gorlich D, Ficner R. Crystal structure of the nuclear export receptor CRM1 in complex with Snurportin1 and RanGTP. Science. 2009;324:1087–1091. doi: 10.1126/science.1173388. [DOI] [PubMed] [Google Scholar]
- 51.Dong X, Biswas A, Suel KE, Jackson LK, Martinez R, Gu H, Chook YM. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature. 2009;458:1136–1141. doi: 10.1038/nature07975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wohlwend D, Strasser A, Dickmanns A, Ficner R. Structural basis for RanGTP independent entry of spliceosomal U snRNPs into the nucleus. J Mol Biol. 2007;374:1129–1138. doi: 10.1016/j.jmb.2007.09.065. [DOI] [PubMed] [Google Scholar]
- 53.Mitrousis G, Olia AS, Walker-Kopp N, Cingolani G. Molecular basis for the recognition of snurportin 1 by importin beta. J Biol Chem. 2008;283:7877–7884. doi: 10.1074/jbc.M709093200. [DOI] [PubMed] [Google Scholar]
- 54.Gorlich D, Pante N, Kutay U, Aebi U, Bischoff FR. Identification of different roles for RanGDP and RanGTP in nuclear protein import. EMBO J. 1996;15:5584–5594. [PMC free article] [PubMed] [Google Scholar]
- 55.Guttler T, Madl T, Neumann P, Deichsel D, Corsini L, Monecke T, Ficner R, Sattler M, Gorlich D. NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nat Struct Mol Biol. 2010;17:1367–1376. doi: 10.1038/nsmb.1931. [DOI] [PubMed] [Google Scholar]
- 56.Kutay U, Guttinger S. Leucine-rich nuclear-export signals: born to be weak. Trends Cell Biol. 2005;15:121–124. doi: 10.1016/j.tcb.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 57.Cook A, Bono F, Jinek M, Conti E. Structural biology of nucleocytoplasmic transport. Annu Rev Biochem. 2007;76:647–671. doi: 10.1146/annurev.biochem.76.052705.161529. [DOI] [PubMed] [Google Scholar]
- 58.Engelsma D, Bernad R, Calafat J, Fornerod M. Supraphysiological nuclear export signals bind CRM1 independently of RanGTP and arrest at Nup358. EMBO J. 2004;23:3643–3652. doi: 10.1038/sj.emboj.7600370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Waldmann I, Spillner C, Kehlenbach RH. The nucleoporin-like protein NLP1 (hCG1) promotes CRM1-dependent nuclear protein export. J Cell Sci. 2012;125:144–154. doi: 10.1242/jcs.090316. [DOI] [PubMed] [Google Scholar]
- 60.Callanan M, Kudo N, Gout S, Brocard M, Yoshida M, Dimitrov S, Khochbin S. Developmentally regulated activity of CRM1/XPO1 during early Xenopus embryogenesis. J Cell Sci. 2000;113:451–459. doi: 10.1242/jcs.113.3.451. [DOI] [PubMed] [Google Scholar]
- 61.Collier S, Chan HY, Toda T, McKimmie C, Johnson G, Adler PN, O'Kane C, Ashburner M. The Drosophila embargoed gene is required for larval progression and encodes the functional homolog of schizosaccharomyces Crm1. Genetics. 2000;155:1799–1807. doi: 10.1093/genetics/155.4.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Collier S, Gubb D. Drosophila tissue polarity requires the cell-autonomous activity of the fuzzy gene, which encodes a novel transmembrane protein. Development. 1997;124:4029–4037. doi: 10.1242/dev.124.20.4029. [DOI] [PubMed] [Google Scholar]
- 63.Brinkley BR. Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol. 2001;11:18–21. doi: 10.1016/s0962-8924(00)01872-9. [DOI] [PubMed] [Google Scholar]
- 64.Sluder G, Hinchcliffe EH. Control of centrosome reproduction: the right number at the right time. Biol Cell. 1999;91:413–427. [PubMed] [Google Scholar]
- 65.Wang W, Budhu A, Forgues M, Wang XW. Temporal and spatial control of nucleophosmin by the Ran-Crm1 complex in centrosome duplication. Nat Cell Biol. 2005;7:823–830. doi: 10.1038/ncb1282. [DOI] [PubMed] [Google Scholar]
- 66.Morris SW, Kirstein MN, Valentine MB, Dittmer K, Shapiro DN, Look AT, Saltman DL. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1995;267:316–317. doi: 10.1126/science.267.5196.316-b. [DOI] [PubMed] [Google Scholar]
- 67.Redner RL, Rush EA, Faas S, Rudert WA, Corey SJ. The t(5;17) variant of acute promyelocytic leukemia expresses a nucleophosmin-retinoic acid receptor fusion. Blood. 1996;87:882–886. [PubMed] [Google Scholar]
- 68.Yoneda-Kato N, Look AT, Kirstein MN, Valentine MB, Raimondi SC, Cohen KJ, Carroll AJ, Morris SW. The t(3;5)(q25.1;q34) of myelodysplastic syndrome and acute myeloid leukemia produces a novel fusion gene, NPM-MLF1. Oncogene. 1996;12:265–275. [PubMed] [Google Scholar]
- 69.Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, La Starza R, Diverio D, Colombo E, Santucci A, Bigerna B, Pacini R, Pucciarini A, Liso A, Vignetti M, Fazi P, Meani N, Pettirossi V, Saglio G, Mandelli F, Lo-Coco F, Pelicci PG, Martelli MF. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 70.Narod SA, Foulkes WD. BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer. 2004;4:665–676. doi: 10.1038/nrc1431. [DOI] [PubMed] [Google Scholar]
- 71.Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, Pasini B, Radice P, Manoukian S, Eccles DM, Tang N, Olah E, Anton-Culver H, Warner E, Lubinski J, Gronwald J, Gorski B, Tulinius H, Thorlacius S, Eerola H, Nevanlinna H, Syrjakoski K, Kallioniemi OP, Thompson D, Evans C, Peto J, Lalloo F, Evans DG, Easton DF. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72:1117–1130. doi: 10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Easton DF, Ford D, Bishop DT. Breast and ovarian cancer incidence in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Am J Hum Genet. 1995;56:265–271. [PMC free article] [PubMed] [Google Scholar]
- 73.Deng CX. Roles of BRCA1 in centrosome duplication. Oncogene. 2002;21:6222–6227. doi: 10.1038/sj.onc.1205713. [DOI] [PubMed] [Google Scholar]
- 74.Hsu LC, White RL. BRCA1 is associated with the centrosome during mitosis. Proc Natl Acad Sci USA. 1998;95:12983–12988. doi: 10.1073/pnas.95.22.12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Parvin JD, Sankaran S. The BRCA1 E3 ubiquitin ligase controls centrosome dynamics. Cell Cycle. 2006;5:1946–1950. doi: 10.4161/cc.5.17.3208. [DOI] [PubMed] [Google Scholar]
- 76.Starita LM, Machida Y, Sankaran S, Elias JE, Griffin K, Schlegel BP, Gygi SP, Parvin JD. BRCA1-dependent ubiquitination of gamma-tubulin regulates centrosome number. Mol Cell Biol. 2004;24:8457–8466. doi: 10.1128/MCB.24.19.8457-8466.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brodie KM, Henderson BR. Characterization of BRCA1 Protein Targeting, Dynamics, and Function at the Centrosome: a role for the nuclear export signal, Crm1, and Aurora A kinase. J Biol Chem. 2012;287:7701–7716. doi: 10.1074/jbc.M111.327296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fabbro M, Rodriguez JA, Baer R, Henderson BR. BARD1 induces BRCA1 intranuclear foci formation by increasing RING-dependent BRCA1 nuclear import and inhibiting BRCA1 nuclear export. J Biol Chem. 2002;277:21315–21324. doi: 10.1074/jbc.M200769200. [DOI] [PubMed] [Google Scholar]
- 79.Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol. 2012;83:1021–1032. doi: 10.1016/j.bcp.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Altieri DC. The case for survivin as a regulator of microtubule dynamics and cell-death decisions. Curr Opin Cell Biol. 2006;18:609–615. doi: 10.1016/j.ceb.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 81.Lens SM, Vader G, Medema RH. The case for Survivin as mitotic regulator. Curr Opin Cell Biol. 2006;18:616–622. doi: 10.1016/j.ceb.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 82.Verdecia MA, Huang H, Dutil E, Kaiser DA, Hunter T, Noel JP. Structure of the human anti-apoptotic protein survivin reveals a dimeric arrangement. Nat Struct Biol. 2000;7:602–608. doi: 10.1038/76838. [DOI] [PubMed] [Google Scholar]
- 83.Chantalat L, Skoufias DA, Kleman JP, Jung B, Dideberg O, Margolis RL. Crystal structure of human survivin reveals a bow tie-shaped dimer with two unusual alpha-helical extensions. Mol Cell. 2000;6:183–189. [PubMed] [Google Scholar]
- 84.Knauer SK, Bier C, Habtemichael N, Stauber RH. The Survivin-Crm1 interaction is essential for chromosomal passenger complex localization and function. EMBO Rep. 2006;7:1259–1265. doi: 10.1038/sj.embor.7400824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rodriguez JA, Span SW, Ferreira CG, Kruyt FA, Giaccone G. CRM1-mediated nuclear export determines the cytoplasmic localization of the antiapoptotic protein Survivin. Exp Cell Res. 2002;275:44–53. doi: 10.1006/excr.2002.5492. [DOI] [PubMed] [Google Scholar]
- 86.Stauber RH, Rabenhorst U, Rekik A, Engels K, Bier C, Knauer SK. Nucleocytoplasmic shuttling and the biological activity of mouse survivin are regulated by an active nuclear export signal. Traffic. 2006;7:1461–1472. doi: 10.1111/j.1600-0854.2006.00486.x. [DOI] [PubMed] [Google Scholar]
- 87.Engelsma D, Rodriguez JA, Fish A, Giaccone G, Fornerod M. Homodimerization antagonizes nuclear export of survivin. Traffic. 2007;8:1495–1502. doi: 10.1111/j.1600-0854.2007.00629.x. [DOI] [PubMed] [Google Scholar]
- 88.Wang H, Holloway MP, Ma L, Cooper ZA, Riolo M, Samkari A, Elenitoba-Johnson KS, Chin YE, Altura RA. Acetylation directs survivin nuclear localization to repress STAT3 oncogenic activity. J Biol Chem. 2010;285:36129–36137. doi: 10.1074/jbc.M110.152777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Riolo MT, Cooper ZA, Holloway MP, Cheng Y, Bianchi C, Eakirevich E, Ma L, Chin YE, Altura RA. HDAC6 deacetylates survivin for its nuclear export in breast cancer. J Biol Chem. 2012 Feb 9; doi: 10.1074/jbc.M111.308791. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ruchaud S, Carmena M, Earnshaw WC. The chromosomal passenger complex: one for all and all for one. Cell. 2007;131:230–231. doi: 10.1016/j.cell.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 91.Vagnarelli P, Earnshaw WC. Chromosomal passengers: the four-dimensional regulation of mitotic events. Chromosoma. 2004;113:211–222. doi: 10.1007/s00412-004-0307-3. [DOI] [PubMed] [Google Scholar]
- 92.Klein UR, Nigg EA, Gruneberg U. Centromere targeting of the chromosomal passenger complex requires a ternary subcomplex of Borealin, Survivin, and the N-terminal domain of INCENP. Mol Biol Cell. 2006;17:2547–2558. doi: 10.1091/mbc.E05-12-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rodriguez JA, Lens SM, Span SW, Vader G, Medema RH, Kruyt FA, Giaccone G. Subcellular localization and nucleocytoplasmic transport of the chromosomal passenger proteins before nuclear envelope breakdown. Oncogene. 2006;25:4867–4879. doi: 10.1038/sj.onc.1209499. [DOI] [PubMed] [Google Scholar]
- 94.Rannou Y, Troadec MB, Petretti C, Hans F, Dutertre S, Dimitrov S, Prigent C. Localization of aurora A and aurora B kinases during interphase: role of the N-terminal domain. Cell Cycle. 2008;7:3012–3020. doi: 10.4161/cc.7.19.6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kelly RJ, Lopez-Chavez A, Citrin D, Janik JE, Morris JC. Impacting tumor cell-fate by targeting the inhibitor of apoptosis protein survivin. Mol Cancer. 2011;10:35. doi: 10.1186/1476-4598-10-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Altieri DC. Survivin and IAP proteins in cell-death mechanisms. Biochem J. 2010;430:199–205. doi: 10.1042/BJ20100814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- 98.Li F, Yang J, Ramnath N, Javle MM, Tan D. Nuclear or cytoplasmic expression of survivin: what is the significance? Int J Cancer. 2005;114:509–512. doi: 10.1002/ijc.20768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Knauer SK, Kramer OH, Knosel T, Engels K, Rodel F, Kovacs AF, Dietmaier W, Klein-Hitpass L, Habtemichael N, Schweitzer A, Brieger J, Rodel C, Mann W, Petersen I, Heinzel T, Stauber RH. Nuclear export is essential for the tumor-promoting activity of survivin. FASEB J. 2007;21:207–216. doi: 10.1096/fj.06-5741com. [DOI] [PubMed] [Google Scholar]
- 100.Chan KS, Wong CH, Huang YF, Li HY. Survivin withdrawal by nuclear export failure as a physiological switch to commit cells to apoptosis. Cell Death Dis. 2010;1:e57. doi: 10.1038/cddis.2010.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mahotka C, Liebmann J, Wenzel M, Suschek CV, Schmitt M, Gabbert HE, Gerharz CD. Differential subcellular localization of functionally divergent survivin splice variants. Cell Death Differ. 2002;9:1334–1342. doi: 10.1038/sj.cdd.4401091. [DOI] [PubMed] [Google Scholar]
- 102.Sampath J, Pelus LM. Alternative splice variants of survivin as potential targets in cancer. Curr Drug Discov Technol. 2007;4:174–191. doi: 10.2174/157016307782109652. [DOI] [PubMed] [Google Scholar]
- 103.Knauer SK, Bier C, Schlag P, Fritzmann J, Dietmaier W, Rodel F, Klein-Hitpass L, Kovacs AF, Doring C, Hansmann ML, Hofmann WK, Kunkel M, Brochhausen C, Engels K, Lippert BM, Mann W, Stauber RH. The survivin isoform survivin-3B is cytoprotective and can function as a chromosomal passenger complex protein. Cell Cycle. 2007;6:1502–1509. [PubMed] [Google Scholar]
- 104.Hengst L, Dulic V, Slingerland JM, Lees E, Reed SI. A cell cycle-regulated inhibitor of cyclin-dependent kinases. Proc Natl Acad Sci USA. 1994;91:5291–5295. doi: 10.1073/pnas.91.12.5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massague J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- 106.Slingerland JM, Hengst L, Pan CH, Alexander D, Stampfer MR, Reed SI. A novel inhibitor of cyclin-Cdk activity detected in transforming growth factor beta-arrested epithelial cells. Mol Cell Biol. 1994;14:3683–3694. doi: 10.1128/mcb.14.6.3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ. The p21(Cip1) and p27(Kip1) CDK 'inhibitors' are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18:1571–1583. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Slingerland J, Pagano M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol. 2000;183:10–17. doi: 10.1002/(SICI)1097-4652(200004)183:1<10::AID-JCP2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 109.Millard SS, Yan JS, Nguyen H, Pagano M, Kiyokawa H, Koff A. Enhanced ribosomal association of p27(Kip1) mRNA is a mechanism contributing to accumulation during growth arrest. J Biol Chem. 1997;272:7093–7098. doi: 10.1074/jbc.272.11.7093. [DOI] [PubMed] [Google Scholar]
- 110.Connor MK, Kotchetkov R, Cariou S, Resch A, Lupetti R, Beniston RG, Melchior F, Hengst L, Slingerland JM. CRM1/Ran-mediated nuclear export of p27(Kip1) involves a nuclear export signal and links p27 export and proteolysis. Mol Biol Cell. 2003;14:201–213. doi: 10.1091/mbc.E02-06-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rodier G, Montagnoli A, Di Marcotullio L, Coulombe P, Draetta GF, Pagano M, Meloche S. p27 cytoplasmic localization is regulated by phosphorylation on Ser10 and is not a prerequisite for its proteolysis. EMBO J. 2001;20:6672–6682. doi: 10.1093/emboj/20.23.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Boehm M, Yoshimoto T, Crook MF, Nallamshetty S, True A, Nabel GJ, Nabel EG. A growth factor-dependent nuclear kinase phosphorylates p27(Kip1) and regulates cell cycle progression. EMBO J. 2002;21:3390–3401. doi: 10.1093/emboj/cdf343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ishida N, Hara T, Kamura T, Yoshida M, Nakayama K, Nakayama KI. Phosphorylation of p27Kip1 on serine 10 is required for its binding to CRM1 and nuclear export. J Biol Chem. 2002;277:14355–14358. doi: 10.1074/jbc.C100762200. [DOI] [PubMed] [Google Scholar]
- 114.Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1) Cell. 1996;85:721–732. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- 115.Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- 116.Larrea MD, Wander SA, Slingerland JM. p27 as Jekyll and Hyde: regulation of cell cycle and cell motility. Cell Cycle. 2009;8:3455–3461. doi: 10.4161/cc.8.21.9789. [DOI] [PubMed] [Google Scholar]
- 117.Wander SA, Zhao D, Slingerland JM. p27: a barometer of signaling deregulation and potential predictor of response to targeted therapies. Clin Cancer Res. 2011;17:12–18. doi: 10.1158/1078-0432.CCR-10-0752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Catzavelos C, Bhattacharya N, Ung YC, Wilson JA, Roncari L, Sandhu C, Shaw P, Yeger H, Morava-Protzner I, Kapusta L, Franssen E, Pritchard KI, Slingerland JM. Decreased levels of the cell-cycle inhibitor p27Kip1 protein: prognostic implications in primary breast cancer. Nat Med. 1997;3:227–230. doi: 10.1038/nm0297-227. [DOI] [PubMed] [Google Scholar]
- 119.Tan P, Cady B, Wanner M, Worland P, Cukor B, Magi-Galluzzi C, Lavin P, Draetta G, Pagano M, Loda M. The cell cycle inhibitor p27 is an independent prognostic marker in small (T1a,b) invasive breast carcinomas. Cancer Res. 1997;57:1259–1263. [PubMed] [Google Scholar]
- 120.Nohara T, Ryo T, Iwamoto S, Gon G, Tanigawa N. Expression of cell-cycle regulator p27 is correlated to the prognosis and ER expression in breast carcinoma patients. Oncology. 2001;60:94–100. doi: 10.1159/000055303. [DOI] [PubMed] [Google Scholar]
- 121.Aylon Y, Oren M. p53: guardian of ploidy. Mol Oncol. 2011;5:315–323. doi: 10.1016/j.molonc.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bieging KT, Attardi LD. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol. 2012;22:97–106. doi: 10.1016/j.tcb.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Soussi T. TP53 mutations in human cancer: database reassessment and prospects for the next decade. Adv Cancer Res. 2011;110:107–139. doi: 10.1016/B978-0-12-386469-7.00005-0. [DOI] [PubMed] [Google Scholar]
- 124.Cai X, Liu X. Inhibition of Thr-55 phosphorylation restores p53 nuclear localization and sensitizes cancer cells to DNA damage. Proc Natl Acad Sci USA. 2008;105:16958–16963. doi: 10.1073/pnas.0804608105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Freedman DA, Levine AJ. Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol Cell Biol. 1998;18:7288–7293. doi: 10.1128/mcb.18.12.7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.van der Watt PJ, Leaner VD. The nuclear exporter, Crm1, is regulated by NFY and Sp1 in cancer cells and repressed by p53 in response to DNA damage. Biochim Biophys Acta. 2011;1809:316–326. doi: 10.1016/j.bbagrm.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 127.Stommel JM, Marchenko ND, Jimenez GS, Moll UM, Hope TJ, Wahl GM. A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J. 1999;18:1660–1672. doi: 10.1093/emboj/18.6.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang Y, Xiong Y. A p53 amino-terminal nuclear export signal inhibited by DNA damage-induced phosphorylation. Science. 2001;292:1910–1915. doi: 10.1126/science.1058637. [DOI] [PubMed] [Google Scholar]
- 129.Lohrum MA, Woods DB, Ludwig RL, Balint E, Vousden KH. C-terminal ubiquitination of p53 contributes to nuclear export. Mol Cell Biol. 2001;21:8521–8532. doi: 10.1128/MCB.21.24.8521-8532.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nie L, Sasaki M, Maki CG. Regulation of p53 nuclear export through sequential changes in conformation and ubiquitination. J Biol Chem. 2007;282:14616–14625. doi: 10.1074/jbc.M610515200. [DOI] [PubMed] [Google Scholar]
- 131.Stewart D, Ghosh A, Matlashewski G. Involvement of nuclear export in human papillomavirus type 18 E6-mediated ubiquitination and degradation of p53. J Virol. 2005;79:8773–8783. doi: 10.1128/JVI.79.14.8773-8783.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103:311–320. doi: 10.1016/s0092-8674(00)00122-7. [DOI] [PubMed] [Google Scholar]
- 133.Burgess AW, Faux MC, Layton MJ, Ramsay RG. Wnt signaling and colon tumorigenesis--a view from the periphery. Exp Cell Res. 2011;317:2748–2758. doi: 10.1016/j.yexcr.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 134.Minde DP, Anvarian Z, Rudiger SG, Maurice MM. Messing up disorder: how do missense mutations in the tumor suppressor protein APC lead to cancer? Mol Cancer. 2011;10:101. doi: 10.1186/1476-4598-10-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Benchabane H, Ahmed Y. The adenomatous polyposis coli tumor suppressor and Wnt signaling in the regulation of apoptosis. Adv Exp Med Biol. 2009;656:75–84. doi: 10.1007/978-1-4419-1145-2_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 137.Neufeld KL, Nix DA, Bogerd H, Kang Y, Beckerle MC, Cullen BR, White RL. Adenomatous polyposis coli protein contains two nuclear export signals and shuttles between the nucleus and cytoplasm. Proc Natl Acad Sci USA. 2000;97:12085–12090. doi: 10.1073/pnas.220401797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Henderson BR, Fagotto F. The ins and outs of APC and beta-catenin nuclear transport. EMBO Rep. 2002;3:834–839. doi: 10.1093/embo-reports/kvf181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Eleftheriou A, Yoshida M, Henderson BR. Nuclear export of human beta-catenin can occur independent of CRM1 and the adenomatous polyposis coli tumor suppressor. J Biol Chem. 2001;276:25883–25888. doi: 10.1074/jbc.M102656200. [DOI] [PubMed] [Google Scholar]
- 140.Tickenbrock L, Cramer J, Vetter IR, Muller O. The coiled coil region (amino acids 129-250) of the tumor suppressor protein adenomatous polyposis coli (APC). Its structure and its interaction with chromosome maintenance region 1 (Crm-1) J Biol Chem. 2002;277:32332–32338. doi: 10.1074/jbc.M203990200. [DOI] [PubMed] [Google Scholar]
- 141.Henderson BR. Nuclear-cytoplasmic shuttling of APC regulates beta-catenin subcellular localization and turnover. Nat Cell Biol. 2000;2:653–660. doi: 10.1038/35023605. [DOI] [PubMed] [Google Scholar]
- 142.Sansom O. Tissue-specific tumour suppression by APC. Adv Exp Med Biol. 2009;656:107–118. [PubMed] [Google Scholar]
- 143.Anderson CB, Neufeld KL, White RL. Subcellular distribution of Wnt pathway proteins in normal and neoplastic colon. Proc Natl Acad Sci USA. 2002;99:8683–8688. doi: 10.1073/pnas.122235399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hamamoto T, Gunji S, Tsuji H, Beppu T. Leptomycins A and B, new antifungal antibiotics. I. Taxonomy of the producing strain and their fermentation, purification and characterization. J Antibiot (Tokyo) 1983;36:639–645. doi: 10.7164/antibiotics.36.639. [DOI] [PubMed] [Google Scholar]
- 145.Yoshida M, Nishikawa M, Nishi K, Abe K, Horinouchi S, Beppu T. Effects of leptomycin B on the cell cycle of fibroblasts and fission yeast cells. Exp Cell Res. 1990;187:150–156. doi: 10.1016/0014-4827(90)90129-x. [DOI] [PubMed] [Google Scholar]
- 146.Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, Yoshida M, Horinouchi S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci USA. 1999;96:9112–9117. doi: 10.1073/pnas.96.16.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.London CA, Barnard S, Kisseberth W, Plamondon L, Shacham S, Kauffman M. Preliminary results of a phase I study of the novel CRM1 inhibitors KPT-276 and KPT-335 in dogs with spontaneous cancer. Blood (abstract) 2012 [Google Scholar]
- 148.Landesman Y, Senapedis W, Sanint-Martin J-R, Kashyap T, Plamondon L, Sandanayaka V, Shechter S, Froim D, McCauley D, Kauffman M, Shacham S. Pharmacokinetic (PK)/pharmacodynamic (PD) and efficacy relationship of selective inhibitors of nuclear export (KPT-SINE) Blood (abstract) 2012 [Google Scholar]
- 149.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Simossis VA, Heringa J. PRALINE: a multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res. 2005;33:W289–294. doi: 10.1093/nar/gki390. [DOI] [PMC free article] [PubMed] [Google Scholar]