

Abstract
Background
Infection or stimulation of the innate immune system by nonspecific microbial antigens is thought to educate the immune system to respond appropriately to allergens, preventing allergy.
Objective
To determine the immunologic pathways that might explain how infection/microbial exposure inhibits allergic sensitization.
Methods
Immunologic studies of non-antigen-specific functions of CD8 memory cells, their maturation in vivo, and their effects in a mouse asthma model, to test the hypothesis that CD8 memory is shaped by innate immunity in a way that can inhibit allergic disease.
Results
We found that CD8 memory T-cell (CD8 Tm) populations bridge innate and adaptive immunity by responding to either antigen or cytokines alone. CD8 Tm populations partially subvert the clonal selection process by activating their neighbors through induction of dendritic cell IL-12. Stimulation of innate or acquired immunity in the lung or gut causes expansion/maturation of CD8 Tm populations, which provide an early source of cytokines, enhance TH1 immunity, and inhibit allergic sensitization and airway inflammation/hyperresponsiveness in a non-antigen-specific fashion.
Conclusion
CD8 T-cell–mediated immune memory is long-lived and can retain its capacity for rapid cytokine release in a nonantigen-specific fashion. This novel type of memory enhances TH1 over TH2 immunity and prevents allergic sensitization after exposure to environmental antigens or infection.
Keywords: CD8 T cell, immunologic memory, innate immunity, allergy, TH1/TH2, dendritic cell, IL-12
Interaction between the innate and adaptive immune system is critical for establishing appropriate immune responses to newly encountered antigens and pathogens. In allergic disease, inappropriate immunity, skewed toward the TH2 phenotype, results in immunopathology triggered by allergen exposure. Cells of the innate system provide an early source of IFN-γ and other cytokines on first exposure to antigen, with IFN-γ central for the TH1 component of the response.1 Atopic patients are deficient in IL-12 after allergen challenge,2 and the immature phenotype of respiratory dendritic cells (DCs) contributes to poor IL-12 synthesis.3 Thus, early IFN-γ production is likely to be a limiting factor in ensuring a balanced response to inhaled antigens. Innate cells providing such DC-activating signals include NK cells, NKT cells, and γδ T cells. It is becoming apparent that CD8 T cells, normally considered a part of adaptive immunity, also participate in innate immune responses.
It has been proposed that exposure to various microbes nonspecifically suppresses allergy,4 increasing incidences of allergic diseases where infectious diseases are rare. Because early life events predict development of allergic disease,5 nonspecific stimuli must have a long-lasting effect on adaptive responses to unrelated allergens. Memory lymphocytes with defined antigenic specificity emerge through the process of clonal selection. However, non–antigen-specific, innate immunity lacks immunologic memory because the receptors involved do not generate diverse specificities, and the cells are relatively short-lived. Therefore, epidemiologic evidence and data from rodent models suggest there must be nonantigen-specific or innate memory that develops in response to environmental antigens and pathogen-associated molecular patterns (PAMPs).
In the absence of Toll-like receptor (TLR) agonists, CD8 T cells may be essential for the TH1 component of immunity,6 and CD8 cells, triggered by cytokines alone, rapidly produce IFN-γ.7 Expression of nuclear factor of activated T cells c2 (NFATc2) in CD8 memory populations drives IFN-γ and suppresses TH2 and TH17 responses in the lung.8 The ability of respiratory syncytial virus infection to prevent airway inflammation in C57BL/6 mice is linked to CD8 IFN-γ secretion.9 Here we have investigated whether TH1-promoting nonspecific responsiveness is provided by resting CD8 memory T-cell (CD8 Tm) populations, providing an immunologic pathway linking infection with prevention of allergic disease.
METHODS
Mice
C57BL/6 (B6) mice (6-8 weeks) were from Harlan UK (Oxford, United Kingdom [UK]). OT-1 and OT-2 T-cell receptor (TCR) transgenic mice (B6 background) were bred in our facility. CD8+ OT-1 and CD4+ OT-2 T cells recognize ovalbumin peptides SIINFEKL (SII) and ISQAVHAAHAEINEAGR (ISQ), respectively. Protocols complied with UK Home Office regulations and were approved by our Animal Welfare Committee.
Cell isolations and T-cell:DC assays
For antibodies/reagents, see this article’s Online Repository at www.jacionline.org. Serum IgE was measured by ELISA10 and IL-12 p70 by ELISA kit (R&D Systems, Abingdon, UK). DCs were purified from spleens, CD8 naive/memory T cells from lymph node/spleen, and lymphocytes from liver and lung (see this article’s supplementary Methods text in the Online Repository). Proliferation was measured by 3H-thymidine or carboxyfluorescein diacetate succinimidyl ester (CFSE) labeling, and T-cell:DC cocultures were as detailed in this article’s supplementary Methods text in the Online Repository.
Eimeria vermiformis infection experiments
B6 mice were infected 28 days after birth with 10 to 1000 Eimeria vermiformis sporulated oocysts in sterile water by oral gavage. Three and 17 days later, some groups were allergen-sensitized with 10 μg ovalbumin absorbed to 1.8 μg alum intraperitoneally, and 29 days later challenged with 1 dose of 100 μg ovalbumin in PBS intranasally. Splenocytes were analyzed for CD8 Tm activity on day 31. In some experiments, ovalbumin sensitization was not started until day 24 postinfection, and mice were killed on day 52. There was no change in spleen cellularity or proportions of CD8 cells in infected animals.
CD8 T-cell depletion and adoptive transfer
Mice were depleted of CD8 T cells (>99%) as detailed in this article’s supplementary Methods text in the Online Repository. For transfer of CD8 memory, mice were injected intravenously with 5 × 106 OT-1 CD8 cells, or PBS alone, and immunized on the same day with 50 μg SII peptide + 25 μg anti-CD40 subcutaneously. SII-specific CD8 cells were identified with H-2Kb-SII pentamer/CD8 staining.
T-cell–dependent peptide-induced airway inflammation model
B6 mice were adoptively transferred with 5 × 106 OT-2 CD4 cells intravenously, sensitized with 10 μg ISQ/2 mg alum intraperitoneally on day 0, and challenged daily with 5 μg ISQ in 50 μL PBS intranasally on days 10 to 14. On day 15, lung resistance and compliance (resistance-lung and compliance-dynamic) were measured as described.11 Data are expressed as percentage of baseline measurements. Bronchoalveolar lavage (BAL) inflammatory cells were counted by flow cytometry essentially as described,12 with addition of fluorescein isothiocyanate–Gr-1 antibody to identify neutrophils positively. BAL cells were also stained for T1/ST2/CD3/CD4 to enumerate TH2 CD4 cells.
Statistics and data analysis
Nonparametric (Mann-Whitney) tests were used to compare results from groups of mice. Values of P < .05, P < .01, and P < .005 were considered significant and are indicated in Figs 1 through 5. Flow cytometry results are shown with mean percent staining ± SEM from 3 independent experiments.
FIG 1.

IL-12 + IL-18 induces non–antigen-specific, rapid IFN-γ producing effectors. A, Proliferation of CD8 cells separated into CD44lo and CD44hi fractions and cultured with cytokines as indicated. B, CD44hi CD8 Tms were labeled with CFSE before 3 days of culture in IL-12 ± IL-18 as indicated, washed, and restimulated with IL-12 + IL-18 before IFN-γ flow cytometry. C, CFSE-labeled OT-1 CD8 T lymphocytes were stimulated with IL-12 + IL-18 or antigen (SII peptide) and stained for CD25 after 3 days. D, Fresh CD8 Tms (unprimed) or CD8 Tms expanded for 4 days in IL-12 + IL-18 (effectors) were stimulated for 5 or 15 hours with IL-12/IL-18/monensin before IFN-γ staining. E, CD40L staining after priming/restimulation with cytokines: CD8 T lymphocytes cultured with IL-12 + IL-18 or anti-CD3/CD28 for 5 days were restimulated as initially. N = 3 in all panels.
FIG 5.

CD8 Tms inhibit allergic airway disease mediated entirely by CD4 T cells in a nonspecific fashion. CD8b-depleted or control (IgG) mice were given 5 × 106 OT-2 CD4 transgenic cells, immunized with ISQ peptide/alum and challenged intranasally with ISQ on days 10 to 14. A, Serum total IgE levels. B, Numbers of eosinophils, neutrophils, and T cells in BAL fluid on day 15. C, Numbers of BAL T1/ST2+ CD4+ TH2 cells. D, Airway reactivity to methacholine; all n = 6. E-H, Mice were injected with PBS, SII/anti-CD40 vaccine, and OT-1 CD8 T lymphocytes as in Fig 4, C. E, Day 14 ovalbumin-specific CD8 T lymphocytes in blood. Mice were sensitized and challenged with ISQ/OT-2 cells as in A, starting 7 days after CD8 transfer. BAL cell counts (F), TH2 cells (G), and airway reactivity (H) are shown after 15 days (n = 6). *P < .05; **P < .01.
RESULTS
Cytokine-stimulated CD8 Tms release early IFN-γ and develop into innate effectors
CD8 memory cells produced IFN-γ immediately after stimulation through the TCR or by stimulation with cytokines alone (see this article’s Fig E1 in the Online Repository at www.jacionline.org). Only CD44hi memory T cells produced early IFN-γ, and these were present in both lymphoid and nonlymphoid tissues such as lung and liver (Fig E1, A). In the absence of TCR ligation, a combination of IL-12 and IL-18, and to a lesser extent IL-12 + IL-15, induced rapid IFN-γ (Fig E1, B), and this cytokine-induced IFN-γ was similar in both central and effector memory CD8 cells (Fig E1, C). IFN-γ secretion kinetics were dramatically delayed in naive (CD44lo) compared with memory (CD44hi) fractions with either stimulus (Fig E1, D). Counterstaining for DX5, NKG2A/C/E, and αβ/γδ TCR suggested all CD8 IFN-γ+ cells were conventional αβ T cells and not NKT cells (not shown).
Proliferation assays of purified naive and memory CD8 cells (Fig 1, A) showed that IL-12 and IL-18 acted synergistically, mainly on memory cells, to induce proliferation. Progeny of IL-12/IL-18–induced cell division retained the capacity for early IFN-γ production (Fig 1, B), because restimulation of CFSE-labeled cells after 3 days with cytokines induced very high levels of IFN-γ in divided but not unresponsive cells. Proliferating cells also expressed CD25 after 3 to 4 cell divisions (Fig 1, C), indicating full activation and effector development. This was confirmed by kinetic analysis of IFN-γ production (Fig 1, D; n = 3). Freshly isolated CD8 Tms, when stimulated with IL-12 + IL-18, produced IFN-γ at low frequency, with similar levels detected after 5 or 15 hours. After 3 days of priming in IL-12/IL-18, however, restimulated CD8 Tms were nearly all high-level IFN-γ+ by 5 hours, but staining had declined by 15 hours. Thus, IL-12/IL-18 induced blastlike cells that released IFN-γ with the kinetics and quantities characteristic of T effectors. TNF-α, but not IL-4 or IL-10, was detectable from CD8 Tms, but only after prolonged (24-hour) stimulation with both IL-12 and IL-18 (not shown). Priming/restimulation with IL-12 + IL-18 also induced expression of CD40 ligand, another critical IL-12-inducing signal, albeit at lower levels than in conventional TCR-induced effectors (Fig 1, E). Furthermore, perforin expression, indicative of cytotoxic function, was induced in CD8 Tms after IL-12/IL-18 culture (see this article’s Fig E2 in the Online Repository at www.jacionline.org).
CD8 Tms stimulate DC IL-12 and respond to TLR agonist–activated DCs
Because CD8 Tms were a major source of early IFN-γ, we hypothesized that collaboration between CD8 Tms, DCs, and inflammatory signals is critical for initiating DC IL-12 production. Purified CD8 Tms from OT-1 mice were stimulated with splenic DCs and SIINFEKL (SII) peptide, an irrelevant peptide (RGY-VYQGL), or whole ovalbumin ± cytosine-phosphate-guanosine (CpG) DNATLR-9 agonist (Fig 2, A; n = 3). Memory CD8 T lymphocytes stimulated secretion of IL-12 p70 from DCs, but very low levels were detected in naive CD8 cell cocultures. This was dependent on cognate interaction because only the antigenic peptide induced IL-12. Low-level IL-12 could also be elicited by cross-presented whole ovalbumin; this was enhanced by CpG. Neutralizing antibodies were then used to determine mechanisms of IL-12 induction (Fig 2, B; n = 4). Anti–IFN-γ consistently blocked IL-12 secretion induced by CD8 Tms; anti-CD40L had no significant effect. Mice were then depleted of CD8 T cells by using anti-CD8ß antibody (see Fig E3 in the Online Repository at www.jacionline.org) and injected 24 hours later with staphylococcal enterotoxin B (SEB) superantigen, which induces circulating IL-12 within 4 hours. Depletion of CD8 T cells significantly reduced the release of early IL-12 (Fig 2, C; n = 7), confirming a role for CD8 Tms in innate immunity.
FIG 2.

Bidirectional stimulation between CD8 Tms and DCs. A and B, IL-12 induction by OT-1 naive/memory CD8 T lymphocytes stimulated with DCs + irrelevant peptide (RGY), relevant peptide (SII), or whole ovalbumin ± CpG (TLR9 ligand). “No T cells” and “no DC” cultures contained ovalbumin + CpG. In B, antibodies to CD40L/IFN-γ were added to peptide + CpG cultures. IL-12p70 in 24-hour supernatants from 3 (A) or 4 (B) experiments. C, CD8 T lymphocytes enhance IL-12 in vivo. Mice were depleted of CD8 T lymphocytes, injected with 75 μg SEB or PBS, and serum IL-12p70 was measured 4 hours later (n = 7). D, CD8 Tms proliferate in response to TLR ligands if DCs are present. CD44hi/CD44lo CD8 T lymphocytes were stimulated ± DC/TLR agonists. Mean proliferation ± SD of triplicates is shown; 1 of 3 similar experiments. E, CpG treatment expands and primes lung CD8 Tms. Lung or mediastinal lymph node cells from mice treated 3 and 6 days previously with 10 μg CpG intranasally (right) or PBS (left) were CD44-labeled and stimulated with anti-CD3/CD28 or IL-12/IL-18. IFN-γ staining in gated CD8 T lymphocytes is shown (n = 3). in, Intranasal; LN, lymph node.
We asked whether activation of DCs induced corresponding activation of CD8 Tms in a noncognate, antigen-independent fashion. CD8 T lymphocytes stimulated with TLR agonists, without antigen, failed to proliferate. However, if DCs were added, high levels of proliferation were induced by TLR-9 agonist CpG and TLR-1/2 agonist Pam3CysK4, but not TLR-3 agonist poly I:C (Fig 2, D; n = 3), with highest proliferation in the memory fraction. To test whether TLR agonists alone prime CD8 Tms in vivo, we challenged mice intranasally with CpG alone (Fig 2, E; n = 3). This primed CD8 Tms for heightened rapid IFN-γ production, most dramatically in lung T cells but also in draining lymph nodes. Bidirectional activation signals can therefore occur between CD8 Tms and DCs during innate/early immune responses.
Infection enhances the responsiveness of CD8 Tms
We predicted that microbial exposure, resulting in DC activation, would prime a non-antigen-specific response in CD8 cells, amplifying IL-12 secretion. To test this using a live infection, we used Eimeria, an intracellular protozoan parasite that infects intestinal epithelia, causes coccidiosis, and is commonly contracted in early life. Infection of mice with E vermiformis significantly enhanced early IFN-γ in splenic CD8 Tms when assessed by using IL-12/IL-18 stimulation (Fig 3, A; n = 16). This effect was apparent at 31 and 52 days after infection, although parasites are cleared in these animals by day 16 (not shown). This suggested a lasting effect of infection on CD8 memory. Mean IFN-γ triggered by anti-CD3/CD28 appeared higher in infected mice, but this did not reach statistical significance in all groups. Although most mice were also immunized with ovalbumin, ovalbumin treatment had no significant effect on CD8 IFN-γ production. The degree of CD8 IFN-γ enhancement was dependent on the challenge dose of Eimeria oocysts (Fig 3, B; n = 8). Cells stimulated with Eimeria extract failed to stain positive for IFN-γ (not shown), suggesting the enhanced response was not a result of restimulation of Eimeria antigen-specific CD8 Tms.
FIG 3.
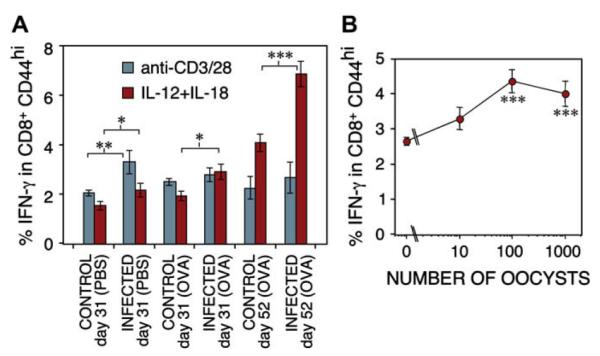
Infection with a gut parasite results in enhanced maturity of CD8 Tm populations. A, B6 mice were infected orally with 1000 E vermiformis oocysts on day 0, and subsequently immunized and challenged with ovalbumin (OVA) or PBS. Spleen cells harvested on day 31 or 52 were stimulated with anti-CD3/CD28 or IL-12 + IL-18. Percent IFN-γ+ cells within CD8+CD44hi populations shown (n = 16). B, As in A, but different doses of E vermiformis oocysts were used; IFN-γ from day 31 CD8 T lymphocytes stimulated with IL-12 + IL-18 is shown (n = 8). *P < .05; **P < .01; ***P < .005.
TCR triggering activates T-cell:T-cell activation via DCs
Because CD8 Tms elicited cytokines from DCs and DC-cytokines stimulate CD8 Tms, we postulated that TCR-driven CD8 Tms would activate bystander, non–antigen-specific CD8 Tm. TCR transgenic cells were stimulated with peptide + DCs and labeled, nontransgenic bystander cells (Fig 4, A; n = 3). Nontransgenic cells produced no IFN-γ when stimulated with peptide + DC alone (not shown). However, antigen-triggered OT-1 CD8 Tm activated neighboring nonspecific CD8 Tm to produce IFN-γ. Addition of anti–IL-12/anti–IL-18 partially inhibited the bystander response, which was greater than that induced by IL-12/IL-18 alone, suggesting other DC-cytokines are involved. To demonstrate such bystander activation in vivo, we transferred nonspecific CFSE-labeled CD8 cells into B6 or OT-1 animals (Fig 4, B; n = 3) and challenged with SII. Cell division was observed in B6 CD8 cells when resident in an OT-1 mouse, but only if surrounding T cells were activated with SII. The same cells transferred to a nontransgenic mouse immunized with peptide failed to proliferate, demonstrating that B6 cell division was dependent on OT-1 T-cell activation and not the appearance of antigen-specific cells or presence of contaminating endotoxin in the antigen preparation. Furthermore, analysis of Vα2 and Vβ5 TCR elements, which are associated with ovalbumin-specific responses (Fig 4, B, lower panels), showed that bystander-activated cells were not enriched in either Vα2+ or Vβ5+ populations. Therefore, the response was not antigen-specific.
FIG 4.

CD8 Tms activate each other, resulting in imprinting of nonspecific populations by specific immunity. A, OT-1 CD8 Tms were mixed with CFSE-labeled nonspecific (B6) CD8 Tms and DCs ± SII peptide overnight. Percent IFN-γ+ in SII-specific (CFSE−) and bystander (CFSE+) populations is shown (n = 3). B, CFSE-labeled nonspecific CD8 T lymphocytes were injected intravenously into OT-1 mice immunized with PBS/SII or into B6 mice given peptide as control. Bystander CD8 division (CFSE dilution) in lymph nodes after 3 days is shown (upper panels; n = 3). Vα2/Vβ5 TCR staining in bystander-activated group is shown in lower panels. C, An antigen-specific CD8 response leaves an imprint on non–antigen-specific CD8 Tms. B6 mice given PBS or OT-1 cells were immunized with SII + anti-CD40 (vaccine) as indicated. Left panel, Percent SII-specific CD8 in blood. Right panel, IFN-γ staining in gated non–antigen-specific (Pentamer−) CD8 T lymphocytes (n = 4). *P < .05; ***P < .005. ip, Intraperitoneal; iv, intravenous; LN, lymph node; SC, subcutaneous; Tm, memory T lymphocyte.
Antigen-specific CD8 responses leave an imprint on non–antigen-specific CD8 Tm populations
The predicted consequence of bystander activation was that non–antigen-specific memory would accompany the development of antigen-specific CD8 memory, providing a conduit for regulation of unrelated immune responses. To test this, we induced strong in vivo CD8 responses entirely directed to SII peptide (Fig 4, C; n = 4). This allowed us to identify all antigen-specific CD8 T lymphocytes using MHC pentamer staining. OT-1 CD8 T lymphocytes, which all stain positive with MHC pentamer (not shown), were transferred to B6 recipients given SII in anti-CD40 adjuvant, resulting in strong expansion of SII-specific CD8 T lymphocytes. Control mice given peptide/anti-CD40 vaccine without OT-1 cells failed to respond. After the antigen-specific response had declined, we analyzed each group for early IFN-γ production in non-antigen-specific CD8 T lymphocytes (MHC pentamer–negative), using strict gating. Activation of nonspecific CD8 Tms was significantly enhanced in mice that had previously had a strong ovalbumin-specific response compared with controls. This effect was most dramatic in lymph nodes but was also apparent in spleen. Thus, antigen-specific CD8 responses result in a lasting imprint of enhanced maturity on nonspecific populations.
Non–antigen-specific CD8 Tms suppress allergic airway responses and IgE secretion induced by a neoantigen
Finally, we asked whether nonspecific CD8 Tms inhibit allergic airway disease by using 2 distinct approaches. First, we used anti-CD8b depletion (as in Fig 2, D). To confirm that CD8 memory was the critical factor, we also induced defined SII-specific CD8 memory pools in B6 recipients using OT-1 cells and peptide vaccine as in Fig 4, C. Because allergen-specific CD8 T lymphocytes are known to suppress allergic responses, we designed a model in which airway inflammation was entirely dependent on CD4 cells directed to a single class II–restricted peptide (ISQ). This excluded antigen-specific effects because there was no cross-reactivity between responses to the 2 peptides (not shown). OT-2 CD4 cells were transferred into B6 mice (predepleted of CD8 T lymphocytes or given control IgG 24 hours previously), sensitized with ISQ peptide/alum, and repeatedly challenged intranasally with ISQ. CD8b depletion greatly enhanced IgE levels (Fig 5, A; n = 6). Challenge of control mice recruited T cells into the airways, but few eosinophils or neutrophils (Fig 5, B). CD8β depletion increased eosinophil numbers with no significant change in neutrophils or T cells. Data from BAL CD4+ TH2 analysis using the T1/ST2 TH2 marker (Fig 5, C) were not quite significant. Finally, anaesthetized mice were challenged with methacholine aerosols and bronchoconstriction was assessed (Fig 5, D), demonstrating significantly enhanced airway responsiveness in CD8β-depleted mice.
To determine effects of transferred memory, mice were given either OT-1 CD8 T lymphocytes + SII vaccine or vaccine alone as in Fig 4, C, and sensitized with ISQ in Fig 5, A-C, 2 weeks later (Fig 5, E-H; n = 6). Only mice receiving OT-1 CD8 T lymphocytes developed significant SII-specific CD8 Tms (Fig 5, E). IgE data are omitted because injection of anti-CD40, even in naive mice, causes a large increase in IgE levels. After rechallenge, eosinophilia and T-cell infiltration of airways were observed, with few neutrophils (Fig 5, F). Eosinophilia, T1/ST2+ TH2 infiltration (Fig 5, G), and airway reactivity (Fig 5, H) were all significantly suppressed by transfer of the OT-1 population, implying that CD8 memory would regulate allergic asthma in a nonspecific fashion. Our results are consistent with the theory that mature CD8 memory increases the availability of IL-12 from DCs early in responses to neoantigens, resulting in inhibition of TH2 development via immune deviation. How this might explain the role of microbial exposure in allergy is illustrated in Fig 6.
FIG 6.
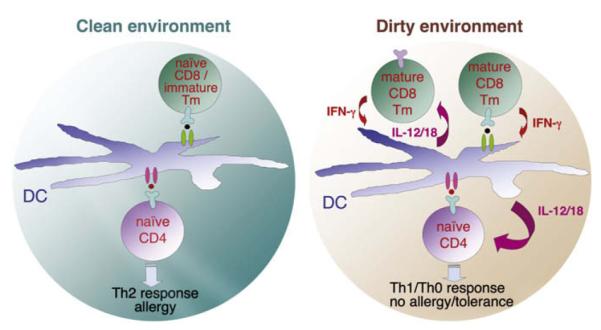
Model for regulation of allergy before and after antigenic/microbial exposure. Pink circle represents a newly encountered allergen peptide. Lack of mature CD8 Tms in a clean environment results in low innate phase cytokines and TH2 development. In a dirty environment, primed CD8 Tms are stimulated by cross-reactivity with allergen or via bystander activation and inhibit TH2 responses by eliciting TH1-promoting cytokines. Tm, Memory T lymphocyte.
DISCUSSION
A central dogma of immunology has been that T-cell responses are controlled by 2 signals: signal 1 (TCR/antigen) and signal 2/3 (costimulatory molecules/cytokines) induced by danger signals and PAMPs. It has been assumed that only signal 1 results in immunologic memory, whereas signals 2 and 3 determine the nature and longevity of the response. In this study, we asked whether it is possible that signals 2/3 also result in a form of immunologic memory, not for specific antigen, but for the amount of environmental danger experienced or the intensity of previous immune responses. If true, this would explain a key proposal of the hygiene hypothesis: that dangerous environments strengthen the immune system in a non–antigen-specific fashion, leading to potent TH1 immunity and little allergy.
CD8 Tms possess unique characteristics that allow them to bridge innate and adaptive immunity. They are activated by cytokines alone in the absence of TCR triggering. They proliferate and secrete cytokines IFN-γ and TNF-a in response to IL12 + IL-187,13 (see Fig E1), or IL-12 + IL-15. CD8 Tms are long-lived because of self-replication in response to IL-1514 and migrate to peripheral tissues. Non–antigen-specific division of CD8 Tms is enhanced by IL-12.15 Our data indicate these cells participate in early activation of tissue DCs, enhancing IL-12 secretion. Although NK/NKT cells and γδ T cells can also perform this function, they are innate cells that do not mediate long-lived memory. Here we showed that CD8 T cells were partially required for early IL-12 release after SEB injection. It is likely that NKT cells may have provided another stimulus in this system because they can express Vβ7/8, which bind SEB.16 CD8 Tms are a major source of early IFN-γ after LPS challenge.17 By contrast, CD4 T-cell stimulation with TLR ligands may enhance survival but does not induce proliferation.18 Like CD8 Tms, established TH1 effector cells respond to IL-12 + IL-18 by producing IFN-γ.19 However, TH1 effectors generate poor memory in experimental models, perhaps because CD4 Tms do not respond to IL-15.20 Bystander effects of CD4 cells are therefore likely to be short-lived in comparison with CD8 T lymphocytes.
The existence of early-activated CD8 T lymphocytes was first demonstrated when CD8 cells from rats immunized with antigen with no adjuvant were found to express a lectin-binding activation marker within 24 hours.21 The responder frequency of 50% indicated an innate type of response, and depletion of early-activated cells with ricin resulted in potent IgE responses. Although ricin in castor bean dust clearly acts similarly in human beings,22 we have found that such a response cannot be induced in pathogen-free animals, suggesting that infection sensitizes CD8 populations. A similar effect may cause differences between immune responsiveness of human beings raised in hygienic or pathogen-rich environments. This might also explain the severe inflammatory reactions to superagonistic CD28 antibody seen in human beings but not laboratory animals.23
Our concept of innate, or nonspecific, memory is clearly supported by the observation that CD8 Tms expanded by cytokines alone release IFN-γ with the kinetics and quantity of effector T cells. This priming effect is a key feature of antigen-specific effector activities. Expression of CD40L on cytokine-restimulated effectors would provide another IL-12–inducing signal to DCs. Chronic exposure to PAMPs may amplify maturation of CD8 Tms through this priming/restimulation mechanism, resulting in robust innate memory and propensity for TH1 responses. Our in vitro IL-12 induction model confirmed the importance of rapid IFN-γ secretion for synthesis of IL-12 by DCs. For the first time, we showed that CD8 Tms, triggered via their TCR, activate bystander, non-antigen-specific CD8 Tms to amplify the early response, and this can occur in vivo, resulting in expansion of bystander cells. This pathway defies the principles of clonal selection, because T cells are activating each other, via DCs, without presenting antigen to each other. The idea that specific immunity could leave a weaker imprint on nonspecific bystander populations was confirmed by inducing a defined peptide-specific CD8 response and then demonstrating the imprint (enhanced rapid IFN-γ) in CD8 Tms, which we could confirm were not antigen-induced and were present several weeks after the initial response (Fig 4, C). Therefore, viral infection in early life could increase CD8 Tm maturity globally. This would increase the likelihood of TH1 immunity to a neoantigen.
The ability of nonspecific CD8 Tms to inhibit eosinophilic airway inflammation, lung TH2 responses, and airway hyperreactivity is consistent with other studies. CD8 T lymphocytes transferred directly into the airways suppressed allergic airway inflammation,24 and IL-15 transgenic mice, which have enhanced CD8 memory, are resistant to airway hyperreactivity.25 We found nonspecific CD8 T lymphocytes also inhibit IgE production. Because CD8 Tms can reside for long periods in the lung,26 they are in an ideal position to influence the earliest events in T-cell priming to inhaled allergens. CD8 Tms expanded in IL-12/IL-18 retain their TCRs (not shown), which would allow them to respond rapidly to neoantigens via TCR cross-reactivity. Antigen-specific CD8 responses to inhaled allergen are rapid and suppress eosinophilia.27 A combination of antigen-specific and nonspecific CD8 Tms, therefore, is probably involved in prevention of asthma.
The nature of gut flora may have a dominant role in shaping nonspecific memory because of the large bacterial load present at the gastrointestinal mucosa. Helminth infections are known to suppress allergy despite stimulating TH2 immunity.28 As we show here, Eimeria infection, which induces CD4 TH1-associated immunity, primes CD8 Tms for enhanced responsiveness. Recent data also indicate that Eimeria infection suppresses allergic airway inflammation after allergen challenge (D. Gibbons, S. Haque, A. Smith, and A. Hayday, unpublished data, September 2008). We therefore propose that increased maturity of CD8 Tms built up through extended antigenic experience reflects an innate memory of the levels of danger/infectious burden encountered. Development of this memory primes for rapid elimination of pathogens but also educates the immune system to respond appropriately to allergens to avoid allergic disease.
Supplementary Material
Clinical implications: Lack of infection in early life could result in delayed maturation of CD8 Tms, resulting in increased incidence of allergy/asthma. Boosting immune memory could therefore prevent sensitization.
Acknowledgments
Disclosure of potential conflict of interest: A. Noble has received research support from Asthma UK Charity. D. L. Gibbons has received research support from the National Institute of Allergy and Infectious Diseases. A. L. Smith has received research support from the Biotechnology & Biological Sciences Research Council and the Department for Environment, Food and Rural Affairs. The rest of the authors have declared that they have no conflict of interest.
Supported by the Medical Research Council and Asthma UK. C.M.L. is supported by Wellcome Trust Senior Fellowship 057704.
We thank Mike Kemeny for helpful discussions and Lesley Smyth and Giovanna Lombardi for OT-1/OT-2 cells.
Abbreviations used
- BAL
Bronchoalveolar lavage
- CD8 Tm
CD8 memory T cell
- DC
Dendritic cell
- NK
Natural killer
- PAMP
Pathogen-associated molecular pattern
- SEB
Staphylococcal enterotoxin B
- TCR
T-cell receptor
- TLR
Toll-like receptor
REFERENCES
- 1.Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, et al. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol. 2002;3:549–57. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]
- 2.van der Pouw Kraan TC, Boeije LC, de Groot ER, Stapel SO, Snijders A, Kapsenberg ML, et al. Reduced production of IL-12 and IL-12-dependent IFN-γ release in patients with allergic asthma. J Immunol. 1997;158:5560–5. [PubMed] [Google Scholar]
- 3.Constant SL, Lee KS, Bottomly K. Site of antigen delivery can influence T cell priming: pulmonary environment promotes preferential Th2-type differentiation. Eur J Immunol. 2000;30:840–7. doi: 10.1002/1521-4141(200003)30:3<840::AID-IMMU840>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 4.Strachan DP. Hay fever, hygiene, and household size. BMJ. 1989;299:1259–60. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Upham JW, Holt PG. Environment and development of atopy. Curr Opin Allergy Clin Immunol. 2005;5:167–72. doi: 10.1097/01.all.0000162310.79555.ed. [DOI] [PubMed] [Google Scholar]
- 6.Mailliard RB, Egawa S, Cai Q, Kalinska A, Bykovskaya SN, Lotze MT, et al. Complementary dendritic cell-activating function of CD8+ and CD4+ T cells: helper role of CD8+ T cells in the development of T helper type 1 responses. J Exp Med. 2002;195:473–83. doi: 10.1084/jem.20011662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lertmemongkolchai G, Cai G, Hunter CA, Bancroft GJ. Bystander activation of CD8+ T cells contributes to the rapid production of IFN-γ in response to bacterial pathogens. J Immunol. 2001;166:1097–105. doi: 10.4049/jimmunol.166.2.1097. [DOI] [PubMed] [Google Scholar]
- 8.Karwot R, Maxeiner JH, Schmitt S, Scholtes P, Hausding M, Lehr HA, et al. Protective role of nuclear factor of activated T cells 2 in CD8+ long-lived memory T cells in an allergy model. J Allergy Clin Immunol. 2008;121:992–9. doi: 10.1016/j.jaci.2007.12.1172. [DOI] [PubMed] [Google Scholar]
- 9.Smitt JJ, Boon L, Lukacs NW. Respiratory virus-induced regulation of asthma-like responses in mice depends upon CD8 T cells and interferon-γ production. Am J Pathol. 2007;171:1944–51. doi: 10.2353/ajpath.2007.070578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noble A, Leggat JA, Inderberg EM. CD8+ immunoregulatory cells in the graft-versus-host reaction: CD8 T cells activate dendritic cells to secrete interleukin-12/interleukin-18 and induce T helper 1 autoantibody. Immunology. 2003;109:476–86. doi: 10.1046/j.1365-2567.2003.01687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kearley J, Barker JE, Robinson DS, Lloyd CM. Resolution of airway inflammation and hyperreactivity after in vivo transfer of CD4+CD25+ regulatory T cells is interleukin 10 dependent. J Exp Med. 2005;202:1539–47. doi: 10.1084/jem.20051166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Rijt LS, Kuipers H, Vos N, Hijdra D, Hoogsteden HC, Lambrecht BN. A rapid flow cytometric method for determining the cellular composition of bronchoalveolar lavage fluid cells in mouse models of asthma. J Immunol Methods. 2004;288:111–21. doi: 10.1016/j.jim.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 13.Tough DF, Zhang X, Sprent J, Sun S, Tough D, Hwang I. An IFN-γ-dependent pathway controls stimulation of memory phenotype CD8+ T cell turnover in vivo by IL-12, IL-18, and IFN-γ. J Immunol. 2001;166:6007–11. doi: 10.4049/jimmunol.166.10.6007. [DOI] [PubMed] [Google Scholar]
- 14.Murali-Krishna K, Lau LL, Sambhara S, Lemonnier F, Altman J, Ahmed R. Persistence of memory CD8 T cells in MHC class I-deficient mice. Science. 1999;286:1377–81. doi: 10.1126/science.286.5443.1377. [DOI] [PubMed] [Google Scholar]
- 15.Kieper WC, Prlic M, Schmidt CS, Mescher MF, Jameson SC. IL-12 enhances CD8 T cell homeostatic expansion. J Immunol. 2001;166:5515–21. doi: 10.4049/jimmunol.166.9.5515. [DOI] [PubMed] [Google Scholar]
- 16.Habu Y, Shinomiya N, Kinoshita M, Matsumoto A, Kawabata T, Seki S. Mice deficient in Vß8+NKT cells are resistant to experimental hepatitis but are partially susceptible to generalised Shwartzman reaction. Clin Exp Med. 2007;7:30–8. doi: 10.1007/s10238-007-0122-2. [DOI] [PubMed] [Google Scholar]
- 17.Kambayashi T, Assarsson E, Lukacher AE, Ljunggren HG, Jensen PE. Memory CD8+ T cells provide an early source of IFN-γ. J Immunol. 2003;170:2399–408. doi: 10.4049/jimmunol.170.5.2399. [DOI] [PubMed] [Google Scholar]
- 18.Gelman AE, Zhang J, Choi Y, Turka LA. Toll-like receptor ligands directly promote activated CD4+ T cell survival. J Immunol. 2004;172:6065–73. doi: 10.4049/jimmunol.172.10.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang J, Murphy TL, Ouyang W, Murphy KM. Induction of interferon-g production in Th1 CD4+ T cells: evidence for two distinct pathways for promoter activation. Eur J Immunol. 1999;29:548–55. doi: 10.1002/(SICI)1521-4141(199902)29:02<548::AID-IMMU548>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 20.Homann D, Teyton L, Oldstone MB. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat Med. 2001;7:913–9. doi: 10.1038/90950. [DOI] [PubMed] [Google Scholar]
- 21.Diaz-Sanchez D, Lee TH, Kemeny DM. Ricin enhances IgE responses by inhibiting a subpopulation of early-activated IgE regulatory CD8+ T cells. Immunology. 1993;78:226–36. [PMC free article] [PubMed] [Google Scholar]
- 22.Thorpe SC, Kemeny DM, Panzani R, Lessof MH. Allergy to castor bean, I: its relationship to sensitization to common inhalant allergens (atopy) J Allergy Clin Immunol. 1988;82:62–6. doi: 10.1016/0091-6749(88)90052-8. [DOI] [PubMed] [Google Scholar]
- 23.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–28. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 24.Marsland BJ, Harris NL, Camberis M, Kopf M, Hook SM, Le Gros G. Bystander suppression of allergic airway inflammation by lung resident memory CD8+ T cells. Proc Natl Acad Sci U S A. 2004;101:6116–21. doi: 10.1073/pnas.0401582101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishimitsu R, Nishimura H, Yajima T, Watase T, Kawauchi H, Yoshikai Y. Overexpression of IL-15 in vivo enhances Tc1 response, which inhibits allergic inflammation in a murine model of asthma. J Immunol. 2001;166:1991–2001. doi: 10.4049/jimmunol.166.3.1991. [DOI] [PubMed] [Google Scholar]
- 26.Hogan RJ, Cauley LS, Ely KH, Cookenham T, Roberts AD, Brennan JW, et al. Long-term maintenance of virus-specific effector memory CD8+ T cells in the lung airways depends on proliferation. J Immunol. 2002;169:4976–81. doi: 10.4049/jimmunol.169.9.4976. [DOI] [PubMed] [Google Scholar]
- 27.Wells JW, Cowled CJ, Giorgini A, Kemeny DM, Noble A. Regulation of allergic airway inflammation by class I-restricted allergen presentation and CD8 T-cell infiltration. J Allergy Clin Immunol. 2007;119:226–34. doi: 10.1016/j.jaci.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 28.Wilson MS, Taylor MD, Balic A, Finney CA, Lamb JR, Maizels RM. Suppression of allergic airway inflammation by helminth-induced regulatory T cells. J Exp Med. 2005;202:1199–212. doi: 10.1084/jem.20042572. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.