

Abstract
The Pediatric HIV/AIDS Cohort Study’s Surveillance Monitoring of ART Toxicities Study is a prospective cohort study conducted at 22 US sites between 2007 and 2011 that was designed to evaluate the safety of in utero antiretroviral drug exposure in children not infected with human immunodeficiency virus who were born to mothers who were infected. This ongoing study uses a “trigger-based” design; that is, initial assessments are conducted on all children, and only those meeting certain thresholds or “triggers” undergo more intensive evaluations to determine whether they have had an adverse event (AE). The authors present the estimated rates of AEs for each domain of interest in the Surveillance Monitoring of ART Toxicities Study. They also evaluated the efficiency of this trigger-based design for estimating AE rates and for testing associations between in utero exposures to antiretroviral drugs and AEs. The authors demonstrate that estimated AE rates from the trigger-based design are unbiased after correction for the sensitivity of the trigger for identifying AEs. Even without correcting for bias based on trigger sensitivity, the trigger approach is generally more efficient for estimating AE rates than is evaluating a random sample of the same size. Minor losses in efficiency when comparing AE rates between persons exposed and unexposed in utero to particular antiretroviral drugs or drug classes were observed under most scenarios.
Keywords: antiretroviral, asymptotic relative efficiency, HIV-exposed, infants, outcome-dependent sampling, safety, surveillance
Widespread use of antiretroviral drug regimens for the prevention of mother-to-child transmission of human immunodeficiency virus (HIV) and for treatment of HIV-infected pregnant women has contributed to a substantial reduction in the number of HIV-infected infants, particularly in resource-rich countries (1–4). However, concerns remain about potential adverse consequences of in utero exposure to antiretroviral drugs, particularly the possibility of an increased risk of mitochondrial dysfunction (5–8). Nucleoside reverse transcriptase inhibitors inhibit mitochondrial DNA polymerase gamma, and drugs in other classes have also been implicated in mitochondrial DNA dysfunction (9, 10). Although overt mitochondrial dysfunction is rare, its clinical consequences are severe. The evidence regarding mitochondrial toxicity in HIV-exposed but uninfected children remains equivocal; several studies have not observed associations between in utero antiretroviral exposures and mitochondrial dysfunction in uninfected infants (11–15), whereas others have suggested potential associations with nucleoside reverse transcriptase inhibitors either alone or in combination (7, 8, 16–18). The expanding use of newer antiretroviral agents intensifies the need to monitor the safety of in utero exposure to antiretroviral drugs.
One complexity in designing studies to evaluate the risk of mitochondrial dysfunction associated with in utero antiretroviral drug exposure is the difficulty in confirming that diagnosis, given that mitochondrial dysfunction manifests with nonspecific symptoms and/or conditions and can only be definitively diagnosed via biopsy or other invasive means. Manifestations of mitochondrial dysfunction include hearing and language impairment, neurologic disorders, delayed neurodevelopment, metabolic abnormalities, and cardiomyopathy. The intensity of evaluations needed to identify potential cases of mitochondrial dysfunction is at odds with the need to maintain a high retention rate, which is a particular challenge in an uninfected population (19). Because it is expected that severe adverse events (AEs) will be relatively rare in this population, it is neither cost-effective nor beneficial to participants to conduct extensive and potentially invasive assessments on every enrolled child (20, 21).
As a result of the need to monitor the safety of such prenatal antiretroviral drug exposures in a cost-effective manner, we established a surveillance system to prospectively monitor HIV-uninfected infants born to HIV-infected women for signs of mitochondrial toxicity. The Surveillance Monitoring of ART Toxicities (SMARTT) Study, initiated by the Pediatric HIV/AIDS Cohort Study, was designed with a streamlined set of assessments for all children and more intensive evaluations only for those meeting certain triggers in domains of interest relevant to the risk of mitochondrial dysfunction or other potential AEs of prenatal antiretroviral drug exposure. We present an overview of the design of the SMARTT Study and summarize the estimated rates of AEs for each domain of interest. We also consider the performance of a trigger-based design in terms of bias and efficiency for estimating AE rates and testing associations between in utero exposure to antiretroviral drugs and AEs. On the basis of current guidelines (4), almost all HIV-infected women in high-resource settings receive some antiretroviral drugs during pregnancy, and most receive a combination of antiretroviral drugs referred to as highly active antiretroviral therapy (HAART), thereby precluding comparisons with HIV-infected women who had not taken antiretroviral drugs. However, given the large number of potential combination regimens, identification of individual antiretroviral drugs or combinations of antiretroviral drugs associated with AEs may help guide future treatment recommendations.
MATERIALS AND METHODS
Description of protocol and study population
The Pediatric HIV/AIDS Cohort Study’s SMARTT Study opened to participating sites in the United States and Puerto Rico in March 2007 and includes 2 cohorts: the static cohort of children 1–12 years of age at study entry who were previously enrolled in the Pediatric AIDS Clinical Trials Group 219C Late Outcome Study (22) or the Women and Infants Transmission Study (WITS) (23) or for whom detailed information was available on maternal antiretroviral drug use during gestation; and the dynamic cohort of HIV-infected women and their infants enrolled between 22 weeks of gestation and 72 hours after delivery. The static cohort completed enrollment in 2010, but the dynamic cohort is ongoing. The SMARTT Study protocol was approved by human subject research review boards at each of the participating sites and by the Harvard School of Public Health (the data coordinating center of the Pediatric HIV/AIDS Cohort Study). Staff at the local sites obtained written informed consent from the parents or legal guardians of the enrolled children.
At the time of study entry, we obtained the participants’ medical histories, including clinical diagnoses and dates of prenatal antiretroviral drug use. We also collected information on birth characteristics (gestational age, birth weight, and mode of delivery) and maternal HIV disease characteristics both early during pregnancy (first or second trimester, if available) and before delivery, including plasma HIV RNA concentrations (viral load), absolute CD4+ lymphocyte cell counts, and the percentage of CD4+ lymphocytes. We obtained overall and trimester-specific information on substance use during pregnancy, including use of alcohol, tobacco, marijuana, opiates, and other substances, as described in greater detail elsewhere (24). The children’s caregivers completed questionnaires on household composition, educational and income levels, past psychiatric or substance use diagnoses, and other information related to family environment. Mothers or caregivers also completed an assessment of cognitive functioning.
After enrollment in this ongoing study, investigators follow up with children and their mothers or caregivers at annual study visits. We collect information on any changes in household situations, along with new diagnoses or illnesses. A complete physical examination is conducted, including anthropometric assessments (height, weight, body mass index (weight (kg)/height (m)2), and skinfold measurement), and point-of-care capillary blood lactate assessments are obtained (25). Neurodevelopmental, hearing, and language assessments are conducted at specific ages of interest.
The study team defined potential AEs of exposure to antiretroviral drugs during pregnancy in multiple domains for the SMARTT Study based on expert input and existing literature; these included metabolic, growth, neurologic, neurodevelopment, language, hearing, and laboratory abnormalities. For each domain, we established a study trigger based on laboratory, clinical, or screening tests that could be easily conducted without invasive assessments. For example, for children 2 years of age or older, a metabolic trigger was defined as a child’s exceeding the age- and sex-specific 95th percentile for body mass index (or for weight-for-length, as appropriate). A complete list of the study triggers is provided in Table 1. In general, triggers were defined to achieve high sensitivity for identifying potential AEs because it was desirable to capture all children who might have AEs related to antiretroviral medication. This strategy for defining triggers with high sensitivity at the expense of lower specificity was felt to be appropriate for a safety surveillance study.
Table 1.
Trigger Definitions in Domains of Interest for the Surveillance Monitoring of Antiretroviral Treatment Toxicities Study Conducted by the Pediatric HIV/AIDS Cohort Study, 2007 to the Present
| Trigger | Criteria for Meeting Trigger | Criteria for Defining Adverse Event |
| Impaired growth | At any age: (1) Weight less than the third percentile based on Centers for Disease Control and Prevention growth charts adjusted for age and sex or on growth charts for premature births given gestational age, (2) a drop in weight or length z score (>1.3 SDs) over 6 or more months, or (3) triceps skinfold thickness or mid-upper arm circumference less than the fifth percentile. For children 3 years of age or older, height less than the third percentile is added to above criteria. | Growth failure based on meeting growth trigger at 2 study visits (consecutive or not) and verified by specialist consultation |
| Metabolic abnormality | Body mass index or weight-for-length above the 95th percentile among participants 2 years of age or older | Abnormal fasting lipids defined by total cholesterol greater than 220 mg/dL, LDL greater than 130 mg/dL or HDL less than 40 mg/dL, or triglycerides above 110 mg/dL for children 0–9 years of age and above 150 for children 10 years of age or older; or insulin resistance defined by a HOMA score greater than 2.5 for children less than 8 years of age or at Tanner Stage 1 or a HOMA score above 4.0 for children over 8 years of age |
| Neurologic diagnosis | Febrile or afebrile seizure disorder, microcephaly, or other neurologic diagnosis | Confirmed neurologic diagnosis based on pediatric neurology consultation and review by SMARTT Review Panel, including verification of head circumference more than 2 SDs below the age-specific mean for defining microcephaly at age 6 months or older |
| Neurodevelopmental impairment | Score more than 2 SDs below the population norm on tests conducted within age groups, specifically, Bayley III (ages 1 and 3 years): mental or motor score less than 76 and one other domain less than 85; WPPSI-III (age 5 years): full-scale IQ less than 70; and WISC-IV (age 7–16 years): full-scale IQ less than 70 | Confirmation of neurodevelopmental functioning scores below the age-specific threshold along with exclusion of other alternative etiology based on pediatric neurologist consultation and review by SMARTT Review Panel |
| Laboratory toxicity | Grade 3 or higher value for target hematologic or clinical chemistry measurement confirmed by repeat assessment | Confirmation of laboratory abnormality without alternative etiology based on review by SMARTT Review Panel |
| Elevated venous lactate | Two repeated point-of-care lactate measures above 3 mmol/L | Venous lactate level above 3 mmol/L |
| Language impairment | Language test score more than 2 SDs below population norms; tests conducted at specific ages | Confirmation of language scores below the age-specific threshold along with exclusion of other alternative etiology based on review by SMARTT Review Panel |
| Hearing impairment | Abnormal newborn hearing screen or mixed or sensorineural hearing loss for non-newborns | Confirmation of sensorineural or mixed hearing loss based on full audiologic examination followed by confirmation by otolaryngologist, with no alternative explanation for hearing loss |
Abbreviations: HDL, high density lipoprotein; HOMA, homeostatic model assessment; LDL, low density lipoprotein; SD, standard deviation; SMARTT, Surveillance Monitoring for ART Toxicities; WISC, Wechsler Intelligence Scale for Children; WPPSI, Wechsler Preschool and Primary Scale of Intelligence.
We requested that children who met the study trigger for a particular domain undergo more detailed prespecified assessments or be evaluated by an appropriate specialist. For example, children meeting the metabolic trigger were asked to provide fasting blood samples and were defined as metabolic cases if they had abnormal lipid levels or evidence of insulin resistance (Table 1). The results of the trigger-based work-up were reviewed by the SMARTT Study Review Panel, a group of clinical, epidemiologic, and statistical experts who developed case definitions for AEs in each of the target domains. The SMARTT Study Review Panel then followed these strict guidelines when determining whether each subject who met a trigger met the corresponding case definition for the various AEs.
Statistical implications of trigger-based study design
Implications for estimation of AE rates.
Although the trigger-based design is cost-effective and allows the most detailed evaluations to be conducted on persons at highest risk of an AE, it has implications for the statistical estimation of AE rates. The true prevalence, pAE, of an AE in a particular domain can be expressed as the sum of probabilities of an AE either with or without meeting a trigger:
where T is the event of meeting a trigger for that domain. Using standard rules of probability, this can be re-expressed as a sum of probabilities of having an AE conditional on whether one does or does not meet the trigger:
| (1) |
In practice, the implication of using a trigger-based design is that only AEs in persons who meet the domain-specific trigger are observed. This is illustrated in Figure 1 for a hypothetical study of 1,000 subjects, of whom 100 (10%) meet a trigger and 30 (30% of persons meeting a trigger) have the defined AE. The shaded boxes in Figure 1 indicate outcomes that are not observable because the children not meeting a trigger would not have undergone the additional evaluations necessary to determine whether they have an AE. Assuming that the sensitivity of the trigger is high (or equivalently, that 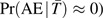 ), the AE probability can be approximated by just the first term in equation 1 above:
), the AE probability can be approximated by just the first term in equation 1 above:
| (2) |
For the hypothetical study in Figure 1, the prevalence of AEs for this domain would be approximated as pu = 0.03 (= 0.10 × 0.30). AE rates approximated under the assumption of high sensitivity will generally underestimate the true AE rate. However, if the sensitivity of the trigger for the AE, S = Pr(T|AE), is known or can be estimated based on prior data, then an adjusted prevalence, pa, can be calculated as:
| (3) |
For the hypothetical example shown in Figure 1, the sensitivity of the trigger is S = 30/40 = 0.75, so pa = 0.03/0.75 = 0.04. If the sensitivity is known, then the adjusted prevalence above will be unbiased for the true AE rate. The adjusted prevalence may be biased if the wrong sensitivity is used, and such bias could result in either an underestimation or an overestimation of the true AE rate.
Figure 1.

Hypothetical example of a study with 1,000 subjects and a trigger rate of 10% for a particular adverse event (AE). Shaded boxes show unobserved outcomes.
Although the estimated AE rate may be slightly biased if the sensitivity of the trigger for the AE is unknown, it will typically have greater precision than that from a study design that includes a randomly selected subset of the same size. In general, if the adjusted prevalence, Pa, can be estimated using a known or unbiased estimate of the sensitivity, then the asymptotic relative efficiency (ARE) of the trigger-based design relative to the random subset design, given the same sample size on which AEs are evaluated, is ARE = 1/pT, where pT = Pr(T). If the sensitivity is unknown and the unadjusted prevalence pu is used as in equation 2, then comparisons of the 2 approaches can be made via the mean squared error (MSE):
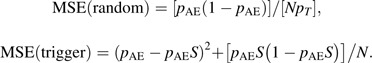 |
The ratio of MSEs for random versus trigger designs, given a fixed sensitivity S, can be expressed as:
| (4) |
The MSE ratios for the random subset approach versus the trigger approach are shown in Figure 2A as a function of trigger sensitivity and trigger rate, assuming a fixed sample size of 1,000 and an AE rate of pAE = 0.04. MSE ratios greater than 1 indicate greater efficiency of the trigger-based design (i.e., smaller MSE). Even when the unadjusted prevalence is biased, the trigger design often yields AE event rates that have smaller MSEs for most reasonable scenarios in this range of sample sizes. With higher trigger probability (pT > 0.3) and lower sensitivity, the random subset design begins to perform as well as the trigger-based design.
Figure 2.

Efficiency of a trigger-based study design versus random subset design for estimating the rate of adverse events (AEs) (A) and for estimating log odds ratio (OR) (B), based on sample sizes of 1,000 and a true adverse event rate of 0.04. Shown is the ratio of the mean squared error (MSE) of the random subset design to the trigger-based design, with values above 1indicating greater efficiency of the trigger-based design.
Implications for estimating exposure effects on AE rates.
The primary objective of the SMARTT Study is to evaluate the association between maternal antiretroviral drug use during pregnancy and AEs in HIV-exposed but uninfected children. As noted previously, the widespread use of antiretroviral drugs during pregnancy makes comparison with an unexposed population impossible; only 4% of mothers in the SMARTT Study were unexposed, and these mothers tended to be different by other measures that could possibly be associated with outcomes. Thus, primary comparisons are typically either between children exposed to a specific antiretroviral drug and children who were unexposed or based on the timing of exposure, such as first trimester use of HAART versus initiation HAART later in the pregnancy. For these comparisons, crude associations can be estimated using relative risks, and given the rarity of AEs in HIV-uninfected children, with odds ratios. If the sensitivity of the trigger is assumed to be the same for both exposed and unexposed participants, then the extent of relative underestimation in the unadjusted prevalence estimates pu,E and (with subscript E indicating exposed and unexposed) would be the same for both groups. Thus, the estimated relative risk from the trigger design, RRtrig, would be unbiased for the true relative risk, RR:
However, the efficiency of the estimated relative risk will decrease as the sensitivity of the trigger decreases. The ARE of the estimated relative risk based on the trigger-based design as compared with a design in which all AEs are identified (i.e., trigger sensitivity S = 1.0) can be written as:
where
and
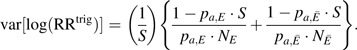 |
In practice, when event rates are low, this ARE can be approximated by the sensitivity of the trigger, that is, ARE is approximately equal to sensitivity (see Table 2).
Table 2.
Bias and Efficiency When Estimating Effects of Exposure on Adverse Event Rates Using a Trigger-based Design as a Function of Trigger Sensitivity in Identifying Adverse Eventsa
| No. of Subjects | RR | True AE Rate (Exposed) | Sensitivity | Expected AE Rates |
OR | Bias (LogOR) | SE (LogOR) | Mean Squared Error | SE (logRR) | ARE for RR | |
| pu,E | |||||||||||
| 1,000 | 1.5 | 0.060 | 0.75 | 0.045 | 0.030 | 1.524 | −0.005 | 0.340 | 0.115 | 0.327 | 0.741 |
| 0.85 | 0.051 | 0.034 | 1.527 | −0.003 | 0.320 | 0.102 | 0.307 | 0.844 | |||
| 0.95 | 0.057 | 0.038 | 1.530 | −0.001 | 0.303 | 0.092 | 0.289 | 0.948 | |||
| 1.00 | 0.060 | 0.040 | 1.532 | 0.000 | 0.296 | 0.088 | 0.282 | 1.000 | |||
| 2.0 | 0.080 | 0.75 | 0.060 | 0.030 | 2.064 | −0.011 | 0.323 | 0.104 | 0.310 | 0.740 | |
| 0.85 | 0.068 | 0.034 | 2.073 | −0.007 | 0.304 | 0.092 | 0.290 | 0.843 | |||
| 0.95 | 0.076 | 0.038 | 2.082 | −0.002 | 0.288 | 0.083 | 0.274 | 0.947 | |||
| 1.00 | 0.080 | 0.040 | 2.087 | 0.000 | 0.282 | 0.079 | 0.266 | 1.000 | |||
| 2.5 | 0.100 | 0.75 | 0.075 | 0.030 | 2.622 | −0.017 | 0.312 | 0.098 | 0.299 | 0.739 | |
| 0.85 | 0.085 | 0.034 | 2.639 | −0.010 | 0.294 | 0.087 | 0.280 | 0.842 | |||
| 0.95 | 0.095 | 0.038 | 2.657 | −0.003 | 0.279 | 0.078 | 0.264 | 0.947 | |||
| 1.00 | 0.100 | 0.040 | 2.667 | 0.000 | 0.273 | 0.074 | 0.257 | 1.000 | |||
| 2,000 | 1.5 | 0.060 | 0.75 | 0.045 | 0.030 | 1.524 | −0.005 | 0.240 | 0.058 | 0.231 | 0.741 |
| 0.85 | 0.051 | 0.034 | 1.527 | −0.003 | 0.226 | 0.051 | 0.217 | 0.844 | |||
| 0.95 | 0.057 | 0.038 | 1.530 | −0.001 | 0.214 | 0.046 | 0.205 | 0.948 | |||
| 1.00 | 0.060 | 0.040 | 1.532 | 0.000 | 0.209 | 0.044 | 0.199 | 1.000 | |||
| 2.0 | 0.080 | 0.75 | 0.060 | 0.030 | 2.064 | −0.011 | 0.228 | 0.052 | 0.219 | 0.740 | |
| 0.85 | 0.068 | 0.034 | 2.073 | −0.007 | 0.215 | 0.046 | 0.205 | 0.843 | |||
| 0.95 | 0.076 | 0.038 | 2.082 | −0.002 | 0.204 | 0.042 | 0.194 | 0.947 | |||
| 1.00 | 0.080 | 0.040 | 2.087 | 0.000 | 0.199 | 0.040 | 0.188 | 1.000 | |||
| 2.5 | 0.100 | 0.75 | 0.075 | 0.030 | 2.622 | −0.017 | 0.221 | 0.049 | 0.211 | 0.739 | |
| 0.85 | 0.085 | 0.034 | 2.639 | −0.010 | 0.208 | 0.043 | 0.198 | 0.842 | |||
| 0.95 | 0.095 | 0.038 | 2.657 | −0.003 | 0.197 | 0.039 | 0.187 | 0.947 | |||
| 1.00 | 0.100 | 0.040 | 2.667 | 0.000 | 0.193 | 0.037 | 0.182 | 1.000 | |||
Abbreviations: AE, adverse events; ARE, asymptotic relative efficiency; OR, odds ratio; pu,E, unadjusted prevalence estimates for persons exposed; , unadjusted prevalence estimates for persons not exposed; RR, relative risk; SE, standard error.
Assuming a background AE rate of 4% and an exposure rate of 50%.
Adjusted associations can be evaluated by fitting a logistic regression model for the probability of an AE as a function of a vector of antiretroviral exposure X of interest, with appropriate control for a vector of p covariates, Z = (Z1, …, Zp), using the following standard modeling framework:
| (5) |
For the purposes of illustration, consider a single exposure, X, representing maternal use of HAART during the first trimester of pregnancy. If a model is fitted to include all participants evaluated for the trigger (whether they meet it or not), the estimated odds ratio will tend to be biased slightly toward the null relative to a design including AE evaluations on the full cohort because of the underestimation of AE rates using pu,E and . For example, if the true event rate in the unexposed was 0.04 and the relative risk was 2, then the true odds ratio would be 2.087, but the expected odds ratio using a trigger-based design with sensitivity S = 0.75 would be 2.064. In addition, the variance of the estimated parameter β1 = log(OR) would be slightly inflated because of the smaller expected counts of persons with AEs. Table 2 provides a summary of the bias, asymptotic standard error, and MSE for estimating the parameter β1 = log(OR) using a trigger-based design (as a function of the trigger sensitivity) for true relative risks of 1.5, 2, and 2.5 and sample sizes of 1,000 or 2,000, assuming a background AE rate in unexposed persons of 0.04 and a 50% rate of exposure. The assumption of a 50% exposure rate is based on the approximate percentage of HIV-infected women with first-trimester HAART exposure among persons with any HAART during pregnancy, whereas a 4% event rate represents a relatively rare event, such as neurodevelopmental delay, for which the power to detect associations is desired. Also provided is the asymptotic standard error for the relative risk under a trigger design along with the AREs for based on the trigger design relative to the full cohort.
Although the efficiency of estimating the relative risk and odds ratio is decreased when using a trigger-based design, the loss in efficiency using the random subset design is typically substantially greater. This is because the denominator Nrandom = pT N for the random subset design is a small percentage of the total sample size N, based on the percent meeting the trigger. As a result, the variance for is [1/pT] times greater for the random subset design than for the full cohort and is substantially higher than the variance for the trigger-based design. For example, if relative risk = 2, pAE = 0.04, and S = 0.75, the asymptotic standard errors for are 0.20 and 0.23 for the full cohort and trigger-based design, respectively, but 0.36–0.63 for the random subset design when pT ranges from 0.1 to 0.3. The random subset design thus provides unbiased estimates of exposure effects, but those estimates are highly inefficient. The ratios of the MSEs for estimating β1 based on the trigger-based design versus the random subset design decrease as the trigger rate increases but are consistently above 2 for all scenarios considered; a representative illustration is provided in Figure 2B.
Implications for testing exposure effects on AE rates.
The fact that variances for the estimated log(OR) may be inflated may impact power for testing associations of AEs with exposure. The power for a hypothesis test of H0: β1 = 0 in the logistic regression model given in equation 5 is illustrated in Figure 3 for a sample size of 1,500 and alternative hypothesis relative risk = 2, indicating slight losses in power for a trigger-based study design as the sensitivity of the trigger decreases. In contrast, the power for detecting exposure effects based on the random subset design is substantially lower. A summary of power for testing exposure effect hypotheses at α = 0.05 is provided in Table 3 for background AE rates of 0.03–0.05 and sample sizes of 1,000–2,000. Power calculations were based on inverting a large-sample test for difference in 2 proportions using the total sample size for the full cohort design and the effective sample size Nrandom = pT N for the random subset design, and assuming true AE rates pA,E and for the full cohort and random subset designs but unadjusted AE rates pu,E and for the trigger designs based on trigger sensitivity. All power calculations were confirmed using PASS (Number Cruncher Statistical Systems, Kaysville, Utah). Because our focus is on rare AEs, even the full cohort design requires a sample size of at least 1,500 subjects to provide at least 80% power for detecting an exposure effect of a relative risk of 2 when the background rate is only 3%. The trigger-based design would require an increase in sample size from 1,500 to approximately 2,000 to maintain 80% power under this scenario if the trigger sensitivity was 0.75. However, the random subset design would require a total sample size of 5,000 subjects if the trigger rate was pT = 0.3 and even greater sample sizes for pT < 0.3.
Figure 3.

Power for detecting an exposure effect based on a trigger-based design versus a full cohort design and random subset design as a function of sensitivity of the trigger assuming a background adverse event (AE) rate in the unexposed of 4%, n = 1,500, and relative risk = 2.
Table 3.
Power for Detecting Effects of Exposure on Adverse Event Rates Using a Trigger-based Design as Compared With a Full Cohort Design and Random Subset Designa
| AE Rate (Unexposed) | True OR | No. of Subjects | Power for Full Cohort | Power for Random Subset Design as a Function of Trigger Rate Pr(T) |
Power for Trigger Design as a Function of Trigger Sensitivity S |
|||
| Pr(T) = 0.3 | Pr(T) = 0.2 | S = 0.75 | S = 0.85 | S = 0.95 | ||||
| 0.03 | 2.064 | 1,000 | 0.629 | 0.239 | 0.174 | 0.504 | 0.557 | 0.606 |
| 1,500 | 0.801 | 0.335 | 0.239 | 0.675 | 0.731 | 0.779 | ||
| 2,000 | 0.900 | 0.425 | 0.304 | 0.796 | 0.845 | 0.884 | ||
| 0.04 | 2.087 | 1,000 | 0.760 | 0.307 | 0.220 | 0.629 | 0.686 | 0.737 |
| 1,500 | 0.904 | 0.431 | 0.307 | 0.801 | 0.850 | 0.888 | ||
| 2,000 | 0.965 | 0.541 | 0.391 | 0.900 | 0.933 | 0.956 | ||
| 0.05 | 2.111 | 1,000 | 0.852 | 0.375 | 0.267 | 0.731 | 0.786 | 0.832 |
| 1,500 | 0.958 | 0.521 | 0.375 | 0.884 | 0.921 | 0.948 | ||
| 2,000 | 0.989 | 0.643 | 0.475 | 0.954 | 0.974 | 0.985 | ||
Abbreviations: AE, adverse event; OR, odds ratio.
For background AE rates of 0.03–0.05 and a relative risk of 2, assuming an exposure rate of 50%.
Application to the SMARTT Study.
Characteristics of study population.
A total of 2,279 subjects enrolled into the SMARTT Study at 22 sites between March 2007 and December 2010. Background characteristics are summarized in Table 4; 70% were black and 33% self-reported being of Hispanic ethnicity. There was a high rate of both preterm births (<37 completed weeks of gestation; 21%), and children born at low birth weight (<2,500 g; 19%). Use of illicit drugs was less common than previously reported among HIV-infected pregnant women, with only 3% of mothers reporting use of cocaine or opiates during pregnancy. Overall, 78% of mothers received HAART during pregnancy, with 37% reporting use in the first trimester; further details on maternal antiretroviral drug exposures have been reported elsewhere (26). The median follow-up as of December 2010 was 33.6 months in the static cohort and 19.2 months in the dynamic cohort. Retention rates were high, with 95% remaining in the study as of December 2010.
Table 4.
Characteristics of 2,279 Infants and Children Enrolled in the US-based Surveillance Monitoring for ART Toxicities Study Between 2007 and 2010
| Characteristic | Static Cohort (n = 1,240) |
Dynamic Cohort (n = 1,039) |
Total (N = 2,279) |
|||||
| No. | % | Median (IQR) | No. | % | No. | % | Median (IQR) | |
| Female | 597 | 48 | 489 | 50 | 1,086 | 49 | ||
| Age in years at entry | 4.1 (2.0–7.0) | 0 | 0.9 (0–4.5) | |||||
| Racea | ||||||||
| Black | 788 | 69 | 715 | 72 | 1,503 | 70 | ||
| White or other | 359 | 31 | 275 | 28 | 634 | 30 | ||
| Hispanic/Latino ethnicity | 432 | 35 | 316 | 30 | 748 | 33 | ||
| Birth characteristicsb | ||||||||
| Preterm birth (gestation <37 weeks) | 238 | 22 | 193 | 21 | 431 | 21 | ||
| Low birth weight (<2500 g) | 225 | 19 | 183 | 19 | 408 | 19 | ||
| Maternal health status prior to labor and deliveryc | ||||||||
| Viral load above 1,000 copies/mL | 213 | 21 | 138 | 15 | 351 | 18 | ||
| CD4+ lymphocyte count less than 250 cells/mm3 | 163 | 15 | 165 | 18 | 328 | 16 | ||
| % of CD4+ lymphocytes less than 25% | 298 | 29 | 289 | 32 | 587 | 30 | ||
| Maternal substance use during pregnancyd | ||||||||
| Any alcohol use | 72 | 7 | 92 | 10 | 164 | 8 | ||
| Any tobacco use | 197 | 19 | 170 | 18 | 367 | 18 | ||
| Illicit drug use (marijuana, cocaine, or opiates) | 86 | 8 | 87 | 9 | 173 | 9 | ||
| Hard drug use (cocaine or opiates) | 31 | 3 | 26 | 3 | 57 | 3 | ||
| Maternal antiretroviral use during pregnancye | ||||||||
| Any antiretroviral use | 1,118 | 96 | 856 | 97 | 1,974 | 96 | ||
| HAART use | 845 | 73 | 747 | 85 | 1,592 | 78 | ||
| HAART use in the first trimester | 379 | 33 | 368 | 42 | 747 | 37 | ||
| Protease inhibitor use | 735 | 63 | 712 | 81 | 1,447 | 71 | ||
| Nonnucleoside reverse transcriptase inhibitor use | 216 | 18 | 94 | 11 | 310 | 15 | ||
| Zidovudine plus lamivudine | 876 | 75 | 615 | 70 | 1,491 | 73 | ||
Abbreviations: HAART, highly active antiretroviral therapy; IQR, interquartile range.
A total of 142 subjects did not report their race and 4 did not report their ethnicity.
Data on gestational age at birth were missing for 238 children and data on birth weight were missing for 143.
Data on maternal viral load were missing for 321 subjects, data on maternal CD4+ lymphocyte count were missing for 282 subjects, and data on maternal percentage of CD4+ lymphocytes were missing for 347 subjects. Maternal health status measures were the latest available before labor or delivery.
Data on maternal substance use during pregnancy were missing for 291 subjects, primarily because of nonenrollment of mothers in the static cohort.
Data on maternal antiretroviral drug use during pregnancy were missing for 228 women; all percentages in the table were calculated among those for whom we had data.
The numbers of SMARTT participants who met each of the trigger and case criteria for each AE are shown in Table 5. Of 2,177 subjects with a study visit, 900 (41%) met at least 1 study trigger. The triggers met most often were the metabolic trigger (27%) and the growth trigger (16%). Based on the percentage meeting the AE criteria among children meeting the trigger (45% and 37%, respectively), the resulting estimated AE prevalences were 12.1% and 5.8% (equation 2).
Table 5.
Numbers of Infants and Children Meeting Adverse Event Case Definitions in Domains of Interest for the Surveillance Monitoring for ART Toxicities Study and Estimated Prevalence of Adverse Events Between 2007 and 2010
| Case Description | No. of Evaluated | Effective Sample Sizea | Meeting Trigger |
AE Rate in those Meeting Trigger and With AE Evaluations |
Estimated AE Prevalence | 95% CI | Adjusted AE Prevalence Assuming an 80% Trigger Sensitivity | 95% CI | ||
| No. of Subjects | % | No. of Subjects/Total | % | |||||||
| Impaired growth | 2,238 | 1,275 | 351 | 15.6 | 74/200 | 37.0 | 5.80 | 4.58, 7.23 | 7.29 | 5.93, 8.86 |
| Metabolic abnormality | 1,364 | 934 | 365 | 26.8 | 113/250 | 45.2 | 12.10 | 10.08, 14.36 | 15.12 | 12.86, 17.56 |
| Neurologic diagnoses | 2,171 | 2,082 | 122 | 5.6 | 77/117 | 65.8 | 3.70 | 2.93, 4.60 | 4.61 | 3.75, 5.60 |
| Neurodevelopmental impairment | 1,101 | 1,101 | 39 | 3.5 | 39/39 | 100 | 3.54 | 2.53, 4.81 | 4.43 | 3.31, 5.84 |
| Laboratory toxicity (other than lactate) | 2,149 | 1,322 | 13 | 0.6 | 5/8 | 62.5 | 0.38 | 0.12, 0.88 | 0.45 | 0.17, 0.99 |
| Elevated lactate | 1,980 | 1,681 | 106 | 5.4 | 39/90 | 43 | 2.32 | 1.65, 3.16 | 2.90 | 2.16, 3.84 |
| Severe language impairment | 1,050 | 1,050 | 114 | 10.9 | 114/114 | 100 | 10.86 | 9.04, 12.90 | 13.62 | 11.60, 15.84 |
| Sensorineural or mixed hearing loss | 940 | 745 | 29 | 3.1 | 2/23 | 8.70 | 0.27 | 0.03, 0.97 | 0.34 | 0.05, 1.02 |
Abbreviation: AE, adverse event; CI, confidence interval.
Effective sample size calculated based on percentage meeting trigger with complete follow-up to evaluate AE applied to the total number of participants evaluated for the trigger.
Unadjusted prevalence estimates for AEs ranged from less than 1% to 12% but are likely to be underestimations of the true AE rates for the reasons noted previously. We also present adjusted AE rates for each domain under the assumption of 80% sensitivity of the trigger for the AE, with 95% confidence intervals calculated using the effective sample size based on the percentage of participants with completed AE assessment among persons meeting the trigger. If external information on trigger sensitivity is available, then the adjusted prevalence would form the most appropriate basis for comparison to other populations, such as children born to women without HIV-infection.
DISCUSSION
We present a trigger-based study design that is useful for evaluation of rare AEs in children exposed in utero to antiretroviral drugs. This design allows invasive or expensive evaluations to be limited to participants who are at the highest risk of experiencing an AE and most likely to benefit from identification of such a problem. It is also more cost-effective than performing detailed evaluations on the whole cohort and reduces site and patient burden. This study design provides a basis for estimating AE rates that are unbiased after correction for trigger sensitivity. The study design also offers several advantages in terms of efficiency and increased power for detecting exposure effects as compared with a study design in which detailed evaluations are conducted on a random subset of enrolled participants.
The ultimate goal of the SMARTT Study is to evaluate the safety of antiretroviral drugs used by HIV-infected mothers. Our results indicate that reasonable power can be obtained when comparing uncommon AEs (4% or higher) between persons exposed to certain antiretroviral combinations or drug classes and those who are unexposed or between persons initiating HAART early in pregnancy versus those who begin later in pregnancy. As newer agents become more widely used, focused monitoring is warranted to evaluate safety in exposed infants; for example, both tenofovir disoproxil fumarate and emtricitabine were approved relatively recently but are now used by almost 40% of pregnant women (26). For certain agents of particular concern, such as efavirenz, which has been contraindicated during pregnancy because of its teratogenic risk (27), the limited percentage of infants exposed in utero (<5%) may limit the power of the trigger-based design to detect associations with rare AEs. Other combinations of interest, such as zidovudine plus lamivudine, are still widely used but have been suggested to increase the risk of mitochondrial dysfunction (18); our design would provide adequate power to detect differences in rare AEs, such as impaired growth between persons exposed (73%) versus persons not exposed to zidovudine plus lamivudine. Finally, although an ideal comparison group may be infants who were exposed to HIV but not antiretroviral drugs, this comparison group no longer exists in high-resource settings.
The trigger-based design results in a slight loss of power as compared with a full cohort design and is therefore most suitable for outcomes that cannot be easily determined in all enrolled participants; thus, it may not be appropriate for outcomes such as congenital anomalies, preterm birth, or intrauterine growth retardation. Many postmarketing studies for reviewing drug safety use spontaneous reporting approaches (19–21, 28, 29), but the relatively small percentage of women exposed to antiretroviral drugs in the general population along with the wide range of domains of interest makes reliance on traditional voluntary reporting methods untenable in this setting. Trigger tools have been widely used to identify AEs and medication errors in hospital settings in both adults (30–32) and children (33, 34) and have identified substantially higher AE rates than have standard hospital-based reporting systems (35, 36). In a recent review of medication safety assessment methods, use of a trigger tool was found to be the most effective and labor-efficient method, although voluntary “incident reporting” better identified high-severity AEs (37).
The trigger-based design has some similarities to designs based on outcome-dependent sampling, which have also been shown to be cost-efficient (38). However, in outcome-dependent sampling approaches, one typically over-samples from known cases and then retrospectively determines exposure levels (e.g., the familiar case-control design). In the trigger-based design, one selects observations for evaluation of the outcome within a group expected to have a higher risk of the outcome based on an auxiliary variable (39). Schildcrout and Rathouz (40) describe a design for longitudinally assessed binary outcomes in which some participants are selected for follow-up based on referral for psychiatric outcomes (high risk), whereas other participants come from a group of nonreferred children (low risk). They propose a semiparametric modeling approach based on generalized estimating equations under the assumption that the analyst can specify the prevalence of the outcome (similar to specifying the sensitivity of the trigger in our approach).
One limitation of our approach is that the ARE and power calculations provided here assume that the trigger sensitivity is known without error. Extensions of these calculations are required to account for random variability in the trigger sensitivity, particularly if it is estimated from past studies or concurrent data. For example, the trigger-based design we describe could potentially be modified by including a random sample from the entire cohort without regard to trigger status, thus allowing estimation of the trigger sensitivity. If known or estimated trigger sensitivities are unavailable, we recommend considering a range of possible sensitivities to allow evaluation of the impact on estimation and testing of exposure effects. In summary, use of a trigger-based design shows great promise as a cost-efficient strategy for monitoring rare AEs or diagnoses that can only be identified using invasive or expensive assessments and provides a foundation for future analyses evaluating associations with exposures.
Acknowledgments
Author affiliations: Center for Biostatistics in AIDS Research, Harvard School of Public Health, Boston, Massachusetts (Paige L. Williams, George R. Seage III, Raymond Griner, Katherine Tassiopoulos, Cenk Yildirim, Yanling Huo, Denise L. Jacobson); Department of Biostatistics, Harvard School of Public Health, Boston, Massachusetts (Paige L. Williams); Department of Epidemiology, Harvard School of Public Health, Boston, Massachusetts (George R. Seage III, Katherine Tassiopoulos); Tulane University Health Sciences Center, New Orleans, Louisiana (Russell B. Van Dyke); Eunice Kennedy Shriver National Institute of Child Health and Human Development, Bethesda, Maryland (George K. Siberry, Lynne M. Mofenson); National Vaccine Program Office, Office of the Assistant Secretary for Health, Office of the Secretary, Department of Health and Human Services, Washington, DC (Jennifer S. Read); and University of Illinois, Chicago, Illinois (Kenneth Rich).
The Pediatric HIV/AIDS Cohort Study was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, with co-funding from the National Institute of Allergy and Infectious Diseases, the National Institute on Drug Abuse, the National Institute of Mental Health, the National Institute of Deafness and Other Communication Disorders, the National Heart, Lung, and Blood Institute, the National Institute of Neurological Disorders and Stroke, and the National Institute on Alcohol Abuse and Alcoholism, through cooperative agreements with the Harvard University School of Public Health (U01 HD052102-04) (Principal Investigator: George Seage; Project Director: Julie Alperen) and the Tulane University School of Medicine (U01 HD052104-01) (Principal Investigator: Russell Van Dyke; Co-Principal Investigator: Kenneth Rich; Project Director: Patrick Davis). Data management services were provided by Frontier Science and Technology Research Foundation (Principal Investigator: Suzanne Siminski), and regulatory services and logistical support were provided by Westat, Inc (Principal Investigator: Julie Davidson).
The authors thank the individuals and institutions involved in the conduct of Pediatric HIV/AIDS Cohort Study’s Surveillance Monitoring of ART Toxicities Study. The authors recognize the contributions of the members of the Surveillance Monitoring of ART Toxicities Study Review Panel: Pim Brouwers, Lucy Civitello, Marilyn Crain, Carol Elgie, Rohan Hazra, Kenneth Rich, George Seage, George Siberry, Russell Van Dyke, Paige Williams, and Cenk Yildirim.
The following institutions, clinical site investigators and staff participated in conducting Pediatric HIV/AIDS Cohort Study’s Surveillance Monitoring of ART Toxicities Study in 2010, in alphabetical order: Baylor College of Medicine, Houston, Texas: William Shearer, Norma Cooper, and Lynette Harris; Bronx-Lebanon Hospital Center, New York, New York: Murli Purswani, Emma Stuard, and Anna Cintron; Children’s Diagnostic & Treatment Center, Fort Lauderdale, Florida: Ana Puga, Dia Cooley, and Doyle Patton; The Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania: Richard Rutstein, Carol Vincent, and Nancy Silverman; Children’s Memorial Hospital, Chicago, Illinois: Ram Yogev, Kathleen Malee, Scott Hunter, and Eric Cagwin; Jacobi Medical Center, New York, New York: Andrew Wiznia, Marlene Burey, and Molly Nozyce; School of Medicine, New York University, New York, New York: William Borkowsky, Sandra Deygoo, and Helen Rozelman; St. Jude Children’s Research Hospital, Memphis, Tennessee: Katherine Knapp, Kim Allison, and Patricia Garvie; Department of Pediatrics, San Juan Hospital, San Juan, Puerto Rico: Midnela Acevedo-Flores, Lourdes Angeli-Nieves, and Vivian Olivera; SUNY Downstate Medical Center, Brooklyn, New York: Hermann Mendez, Ava Dennie, and Susan Bewley; Stony Brook University, Stony Brook, New York: Sharon Nachman, Margaret Oliver, and Helen Rozelman; Tulane University Health Sciences Center, New Orleans, Louisiana: Russell Van Dyke, Karen Craig, and Patricia Sirois; University of Alabama at Birmingham, Birmingham, Alabama: Marilyn Crain, Newana Beatty, and Dan Marullo; University of California, San Diego, La Jolla, California: Stephen Spector, Jean Manning, and Sharon Nichols; University of Colorado Denver Health Sciences Center, Denver, Colorado: Elizabeth McFarland, Emily Barr, and Robin McEvoy; University of Florida, Jacksonville, Florida: Mobeen Rathore, Kathleen Thoma, and Ann Usitalo; University of Illinois at Chicago, Chicago, Illinois: Kenneth Rich, Delmyra Turpin, and Renee Smith; University of Maryland, Baltimore, Baltimore, Maryland: Douglas Watson, LaToya Stubbs, and Rose Belanger; University of Medicine and Dentistry of New Jersey, Newark, New Jersey: Arry Dieudonne, Linda Bettica, and Susan Adubato; University of Miami, Miami, Florida: Gwendolyn Scott, Erika Lopez, and Elizabeth Willen; University of Southern California, Los Angeles, California: Toinette Frederick, Mariam Davtyan, and Maribel Mejia; and Puerto Rico Medical Center, University of Puerto Rico, San Juan, Puerto Rico: Zoe Rodriguez, Ibet Heyer, and Nydia Scalley Trifilio.
The conclusions and opinions expressed in this article are those of the authors and do not necessarily reflect those of the National Institutes of Health or the Department of Health and Human Services.
Conflict of interest: none declared.
Glossary
Abbreviations
- AE
adverse event
- ARE
asymptotic relative efficiency
- HAART
highly active antiretroviral therapy
- HIV
human immunodeficiency virus
- MSE
mean squared error
- SMARTT
Surveillance Monitoring of ART Toxicities
References
- 1.Townsend CL, Cortina-Borja M, Peckham CS, et al. Low rates of mother-to-child transmission of HIV following effective pregnancy interventions in the United Kingdom and Ireland, 2000–2006. AIDS. 2008;22(8):973–981. doi: 10.1097/QAD.0b013e3282f9b67a. [DOI] [PubMed] [Google Scholar]
- 2.Cooper ER, Charurat M, Mofenson L, et al. Combination antiretroviral strategies for the treatment of pregnant HIV-1-infected women and prevention of perinatal HIV-1 transmission. Women and Infants’ Transmission Study Group. J Acquir Immune Defic Syndr. 2002;29(5):484–494. doi: 10.1097/00126334-200204150-00009. [DOI] [PubMed] [Google Scholar]
- 3.Suksomboon N, Poolsup N, Ket-aim S. Systematic review of the efficacy of antiretroviral therapies for reducing the risk of mother-to-child transmission of HIV infection. J Clin Pharm Ther. 2007;32(3):293–311. doi: 10.1111/j.1365-2710.2007.00825.x. [DOI] [PubMed] [Google Scholar]
- 4.Public Health Service recommendations for use of antiretroviral drugs in pregnant HIV-1-infected women for maternal health and interventions to reduce perinatal HIV-1 transmission in the United States. MMWR Morb Mortal Wkly Rep. 1998;47(RR-2):1–38. [PubMed] [Google Scholar]
- 5.European Collaborative Study. Exposure to antiretroviral therapy in utero or early life: the health of uninfected children born to HIV-infected women. J Acquir Immune Defic Syndr. 2003;32(4):380–387. doi: 10.1097/00126334-200304010-00006. [DOI] [PubMed] [Google Scholar]
- 6.Thorne C, Newell ML. Safety of agents used to prevent mother-to-child transmission of HIV: is there any cause for concern? Drug Saf. 2007;30(3):203–213. doi: 10.2165/00002018-200730030-00004. [DOI] [PubMed] [Google Scholar]
- 7.Blanche S, Tardieu M, Rustin P, et al. Persistent mitochondrial dysfunction and perinatal exposure to antiretroviral nucleoside analogues. Lancet. 1999;354(9184):1084–1089. doi: 10.1016/S0140-6736(99)07219-0. [DOI] [PubMed] [Google Scholar]
- 8.Barret B, Tardieu M, Rustin P, et al. Persistent mitochondrial dysfunction in HIV-1-exposed but uninfected infants: clinical screening in a large prospective cohort. French Perinatal Cohort Study Group. AIDS. 2003;17(12):1769–1785. doi: 10.1097/00002030-200308150-00006. [DOI] [PubMed] [Google Scholar]
- 9.Gerschenson M, Brinkman K. Mitochondrial dysfunction in AIDS and its treatment. Mitochondrion. 2004;4(5-6):763–777. doi: 10.1016/j.mito.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 10.Montaner JS, Côté HC, Harris M, et al. Mitochondrial toxicity in the era of HAART: evaluating venous lactate and peripheral blood mitochondrial DNA in HIV-infected patients taking antiretroviral therapy. J Acquir Immune Defic Syndr. 2003;34(suppl 1):S85–S90. doi: 10.1097/00126334-200309011-00013. [DOI] [PubMed] [Google Scholar]
- 11.Nucleoside exposure in the children of HIV-infected women receiving antiretroviral drugs: absence of clear evidence for mitochondrial disease in children who died before 5 years of age in five United States cohorts. J Acquir Immune Defic Syndr. 2000;25(3):261–268. doi: 10.1097/00126334-200011010-00009. [DOI] [PubMed] [Google Scholar]
- 12.Lindegren ML, Rhodes P, Gordon L, et al. Drug safety during pregnancy and in infants. Lack of mortality related to mitochondrial dysfunction among perinatally HIV-exposed children in pediatric HIV surveillance. Perinatal Safety Review Working Group; State and Local Health Department HIV/AIDS Surveillance Programs. Ann N Y Acad Sci. 2000;918(1):222–235. [PubMed] [Google Scholar]
- 13.Bulterys M, Nesheim S, Abrams EJ, et al. Lack of evidence of mitochondrial dysfunction in the offspring of HIV-infected women: retrospective review of perinatal exposure to antiretroviral drugs in the Perinatal AIDS Collaborative Transmission Study. Perinatal Safety Review Working Group. Ann N Y Acad Sci. 2000;918(1):212–221. doi: 10.1111/j.1749-6632.2000.tb05491.x. [DOI] [PubMed] [Google Scholar]
- 14.Dominguez K, Bertolli J, Fowler M, et al. Lack of definitive severe mitochondrial signs and symptoms among deceased HIV-uninfected and HIV-indeterminate children < or = 5 years of age, Pediatric Spectrum of HIV Disease Project (PSD), USA. PSD Consortium; Perinatal Safety Review Working Group. Ann N Y Acad Sci. 2000;918(1):236–246. doi: 10.1111/j.1749-6632.2000.tb05493.x. [DOI] [PubMed] [Google Scholar]
- 15.Mofenson LM, Munderi P. Safety of antiretroviral prophylaxis of perinatal transmission for HIV-infected pregnant women and their infants. J Acquir Immune Defic Syndr. 2002;30(2):200–215. doi: 10.1097/00042560-200206010-00010. [DOI] [PubMed] [Google Scholar]
- 16.Lipshultz SE, Shearer WT, Thompson B, et al. Cardiac effects of antiretroviral therapy in HIV-negative infants born to HIV-positive mothers: NHLBI CHAART-1 (National Heart, Lung, and Blood Institute Cardiovascular Status of HAART Therapy in HIV-Exposed Infants and Children Cohort Study) J Am Coll Cardiol. 2011;57(1):76–85. doi: 10.1016/j.jacc.2010.08.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poirier MC, Divi RL, Al-Harthi L, et al. Long-term mitochondrial toxicity in HIV-uninfected infants born to HIV-infected mothers. Women and Infants Transmission Study (WITS) Group. J Acquir Immune Defic Syndr. 2003;33(2):175–183. doi: 10.1097/00126334-200306010-00010. [DOI] [PubMed] [Google Scholar]
- 18.Brogly SB, Ylitalo N, Mofenson LM, et al. In utero nucleoside reverse transcriptase inhibitor exposure and signs of possible mitochondrial dysfunction in HIV-uninfected children. AIDS. 2007;21(8):929–938. doi: 10.1097/QAD.0b013e3280d5a786. [DOI] [PubMed] [Google Scholar]
- 19.Williams PL, Van Dyke R, Eagle M, et al. Association of site-specific and participant-specific factors with retention of children in a long-term pediatric HIV cohort study. PACTG 219C Team. Am J Epidemiol. 2008;167(11):1375–1386. doi: 10.1093/aje/kwn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glasser SP, Salas M, Delzell E. Importance and challenges of studying marketed drugs: what is a phase IV study? Common clinical research designs, registries, and self-reporting systems. J Clin Pharmacol. 2007;47(9):1074–1086. doi: 10.1177/0091270007304776. [DOI] [PubMed] [Google Scholar]
- 21.Andrews E, Dombeck M. The role of scientific evidence of risks and benefits in determining risk management policies for medications. Pharmacoepidemiol Drug Saf. 2004;13(9):599–608. doi: 10.1002/pds.899. [DOI] [PubMed] [Google Scholar]
- 22.Brogly SB, Abzug MJ, Watts DH, et al. Birth defects among children born to human immunodeficiency virus-infected women: pediatric AIDS clinical trials protocols 219 and 219C. Pediatr Infect Dis J. 2010;29(8):721–727. doi: 10.1097/INF.0b013e3181e74a2f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheon AR, Fox HE, Rich KC, et al. The Women and Infants Transmission Study (WITS) of maternal-infant HIV transmission: study design, methods and baseline data. J Womens Health. 1996;5(1):69–78. [Google Scholar]
- 24.Tassiopoulos K, Read JS, Brogly S, et al. Substance use in HIV-infected women during pregnancy: self-report versus meconium analysis. AIDS Behav. 2010;14(6):1269–1278. doi: 10.1007/s10461-010-9705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kiragga AK, Ocama P, Reynolds SJ, et al. Validation of a portable hand-held lactate analyzer for determination of blood lactate in patients on antiretroviral therapy in Uganda. J Acquir Immune Defic Syndr. 2008;49(5):564–566. doi: 10.1097/QAI.0b013e31817e6391. [DOI] [PubMed] [Google Scholar]
- 26.Griner R, Williams PL, Read JS, et al. In utero and postnatal exposure to antiretrovirals among HIV-exposed but uninfected children in the United States. Pediatric HIV-AIDS Cohort Study. AIDS Patient Care STDs. 2011;25(7):385–394. doi: 10.1089/apc.2011.0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Panel on Treatment of HIV-Infected Pregnant Women and Prevention of Perinatal Transmission. Washington, DC: Department of Health and Human Services; 2010. Recommendations for use of antiretroviral drugs in pregnant HIV-1-infected women for maternal health and interventions to reduce perinatal HIV transmission in the United States. ( http://aidsinfo.nih.gov/ContentFiles/PerinatalGL.pdf). (Accessed April 13, 2011) [Google Scholar]
- 28.Hauben M, Zhou X. Quantitative methods in pharmacovigilance: focus on signal detection. Drug Saf. 2003;26(3):159–186. doi: 10.2165/00002018-200326030-00003. [DOI] [PubMed] [Google Scholar]
- 29.Stricker BH, Psaty BM. Detection, verification, and quantification of adverse drug reactions. BMJ. 2004;329(7456):44–47. doi: 10.1136/bmj.329.7456.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rozich JD, Haraden CR, Resar RK. Adverse drug event trigger tool: a practical methodology for measuring medication related harm. Qual Saf Health Care. 2003;12(3):194–200. doi: 10.1136/qhc.12.3.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Resar RK, Rozich JD, Classen D. Methodology and rationale for the measurement of harm with trigger tools. Qual Saf Health Care. 2003;12(suppl 2):ii39–ii45. doi: 10.1136/qhc.12.suppl_2.ii39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Resar RK, Rozich JD, Simmonds T, et al. A trigger tool to identify adverse events in the intensive care unit. Jt Comm J Qual Patient Saf. 2006;32(10):585–590. doi: 10.1016/s1553-7250(06)32076-4. [DOI] [PubMed] [Google Scholar]
- 33.Sharek PJ, Horbar JD, Mason W, et al. Adverse events in the neonatal intensive care unit: development, testing, and findings of an NICU-focused trigger tool to identify harm in North American NICUs. Pediatrics. 2006;118(4):1332–1340. doi: 10.1542/peds.2006-0565. [DOI] [PubMed] [Google Scholar]
- 34.Dickerman MJ, Jacobs BR, Vinodrao H, et al. Recognizing hypoglycemia in children through automated adverse-event detection. Pediatrics. 2011;127(4):e1035–e1041. doi: 10.1542/peds.2009-3432. [DOI] [PubMed] [Google Scholar]
- 35.Takata GS, Mason W, Taketomo C, et al. Development, testing, and findings of a pediatric-focused trigger tool to identify medication-related harm in US children’s hospitals. Pediatrics. 2008;121(4):e927–e935. doi: 10.1542/peds.2007-1779. [DOI] [PubMed] [Google Scholar]
- 36.Ferranti J, Horvath MM, Cozart H, et al. Reevaluating the safety profile of pediatrics: a comparison of computerized adverse drug event surveillance and voluntary reporting in the pediatric environment. Pediatrics. 2008;121(5):e1201–e1207. doi: 10.1542/peds.2007-2609. [DOI] [PubMed] [Google Scholar]
- 37.Meyer-Massetti C, Cheng CM, Schwappach DL, et al. Systematic review of medication safety assessment methods. Am J Health Syst Pharm. 2011;68(3):227–240. doi: 10.2146/ajhp100019. [DOI] [PubMed] [Google Scholar]
- 38.Zhou H, Chen J, Rissanen TH, et al. Outcome-dependent sampling: an efficient sampling and inference procedure for studies with a continuous outcome. Epidemiology. 2007;18(4):461–468. doi: 10.1097/EDE.0b013e31806462d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Wu Y, Zhou H. Outcome- and auxiliary-dependent subsampling and its statistical inference. J Biopharm Stat. 2009;19(6):1132–1150. doi: 10.1080/10543400903243025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schildcrout JS, Rathouz PJ. Longitudinal studies of binary response data following case-control and stratified case-control sampling: design and analysis. Biometrics. 2010;66(2):365–373. doi: 10.1111/j.1541-0420.2009.01306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]