

Abstract
Glutamatergic synapses in early postnatal development transiently express calcium-permeable AMPA receptors (CP-AMPARs). Although these GluA2-lacking receptors are essential and are elevated in response to brain-derived neurotrophic factor (BDNF), little is known regarding molecular mechanisms that govern their expression and synaptic insertion. Here we show that BDNF-induced GluA1 translation in rat primary hippocampal neurons requires the activation of mammalian target of rapamycin (mTOR) via calcium calmodulin-dependent protein kinase kinase (CaMKK). Specifically, BDNF-mediated phosphorylation of threonine 308 (T308) in AKT, a known substrate of CaMKK and an upstream activator of mTOR-dependent translation, was prevented by (1) pharmacological inhibition of CaMKK with STO-609, (2) overexpression of a dominant-negative CaMKK, or (3) short hairpin-mediated knockdown of CaMKK. GluA1 surface expression induced by BDNF, as assessed by immunocytochemistry using an extracellular N-terminal GluA1 antibody or by surface biotinylation, was impaired following knockdown of CaMKK or treatment with STO-609. Activation of CaMKK by BDNF requires transient receptor potential canonical (TRPC) channels as SKF-96365, but not the NMDA receptor antagonist d-APV, prevented BDNF-induced GluA1 surface expression as well as phosphorylation of CaMKI, AKTT308, and mTOR. Using siRNA we confirmed the involvement of TRPC5 and TRPC6 subunits in BDNF-induced AKTT308 phosphorylation. The BDNF-induced increase in mEPSC was blocked by IEM-1460, a selected antagonist of CP-AMPARs, as well as by the specific repression of acute GluA1 translation via siRNA to GluA1 but not GluA2. Together these data support the conclusion that newly synthesized GluA1 subunits, induced by BDNF, are readily incorporated into synapses where they enhance the expression of CP-AMPARs and synaptic strength.
Introduction
During early postnatal development, excitatory glutamatergic synapses have been shown to transiently express calcium-permeable AMPA receptors (CP-AMPARs) before expression of NMDA receptors (Kumar et al., 2002; Eybalin et al., 2004; Ho et al., 2007). These CP-AMPARs lack the critical GluA2 subunit that renders AMPARs calcium impermeable (Hollmann et al., 1991; Sommer et al., 1991; Verdoorn et al., 1991; Seeburg et al., 1998). The transient nature of CP-AMPAR expression is thought to play an important role in Ca2+ entry during synapse maturation. These receptors are also thought to mediate plasticity within developing networks (Rozov and Burnashev, 1999; Liu and Cull-Candy, 2000; Zhu et al., 2000; Aizenman et al., 2002; Ho et al., 2007).
Interestingly, although the majority of mature CA1 hippocampal synapses predominantly lack CP-AMPARs, some paradigms that induce NMDAR-dependent LTP result in their transient synaptic incorporation (Plant et al., 2006; Lu et al., 2007; Guire et al., 2008; Fortin et al., 2010). The trafficking of these CP-AMPARs is dependent upon calcium/calmodulin-dependent protein kinase kinase (CaMKK) and its downstream effector CaMKI (Guire et al., 2008; Fortin et al., 2010). CaMKK has also been postulated as a “master” regulator of homeostatic plasticity (Goold and Nicoll, 2010). In fact, CaMKK activity is linked to many Ca2+-dependent neuronal processes, including gene transcription and neuronal development, as well as synaptic plasticity (Wayman et al., 2008).
Brain-derived neurotrophic factor (BDNF) is synthesized and released at glutamatergic nerve terminals, where it plays an important role in neuronal development by regulating protein synthesis (Kang and Schuman, 1996; Alsina et al., 2001; Bramham and Messaoudi, 2005). BDNF has been shown to increase the translation of hundreds of proteins isolated from synaptoneurosomes (Liao et al., 2007). BDNF binding to TrkB receptors induces activation of PI3K, which in turn activates AKT and the subsequent phosphorylation and activation of mammalian target of rapamycin complex 1 (mTORC1) (Hoeffer and Klann, 2010) to promote cap-dependent translation (Hay and Sonenberg, 2004).
Although BDNF upregulates the total cellular levels of AMPAR subunits GluA1–GluA4 (Narisawa-Saito et al., 1999; Schratt et al., 2004; Caldeira et al., 2007; Slipczuk et al., 2009), only GluA1 traffics to the membrane (Caldeira et al., 2007; Nakata and Nakamura, 2007). However, little is known regarding the molecular mechanisms governing GluA1 synthesis and subsequent membrane insertion. The membrane incorporation of GluA1 following BDNF appears to be associated with Ca2+ flux induced by the activation of transient receptor potential canonical (TRPC) channels (Nakata and Nakamura, 2007). Our laboratory has previously demonstrated that Ca2+ flux through TRPC5 channels can activate CaMKK (Davare et al., 2009), and thus CaMKK may play a role in mediating these effects of BDNF. In the present study, we demonstrate that CaMKK contributes to the translation and surface expression of newly synthesized GluA1, induced by BDNF. We show that CaMKK controls mTOR activation following BDNF via phosphorylation of AKT at T308, which was mediated by TRPC5-containing and TRPC6-containing TRPC channels. Furthermore, we provide direct evidence demonstrating that BDNF results in enhanced synaptic strength via the synaptic delivery of newly translated GluA1 subunits as CP-AMPARs.
Materials and Methods
Pharmacological reagents.
d-(-)-2-Amino-5-phosphonopentanoic acid (d-APV), 1,8-naphthoylene benzimidazole-3-carboxylic acid (STO-609), N,N,N,-trimethyl-5-[(tricyclo[3.3.1.13,7]dec-1-ylmethyl)amino]-1-pentanaminiumbromide hydrobromide (IEM-1460), strychnidin-10-one hydrochloride (strychnine), (2R,3S,4S)-2-([4-methoxyphenyl]methyl)-3,4-pyrrolidinediol 3-acetate (anisomycin), 6-imino-3-(4-methoxyphenyl)-1(6H)-pyridazinebutanoic acid hydrobromide (gabazine), and octahydro-12-(hydroxymethyl)-2-imino-5,9:7,10a-dimethan-o-10aH-[1,3]dioxocino[6,5-d]pyrimidine-4,7,10,11,12-pentol (TTX) were purchased from Tocris Bioscience. 1-[β-[3-(4-Methoxyphenyl)propoxy]-4-methoxyphenethyl]-1H-imidazole, HCl (SKF-96365) was purchased from Calbiochem. Rapamycin was purchased from LC Laboratories. Puromycin was purchased from Sigma-Aldrich. BDNF was purchased from Peprotech.
shRNA, siRNA, and plasmids.
The construction, validation, and specificity of the plasmid-based shRNA constructs for knockdown of CaMKK have been described previously (Wayman et al., 2006; Saneyoshi et al., 2008). Specific knockdown of TRPC3, TRPC5, and TRPC6 subunits was performed using previously described siRNA sequences targeting TRPC3 (40 nm) (Amaral and Pozzo-Miller, 2007a), TRPC5 (20 nm), and TRPC6 (40 nm) (Davare et al., 2009). For selective knockdown of GluA1 and GluA2 subunits, we used Stealth RNA interference (RNAi) (Invitrogen). siGluA1 and siGluA2 were custom synthesized targeting nucleotide starting position 608 in rat GluA1 (accession number NM_031608) and 1054 in rat GluA2 (accession number NM_017261) [targeting sequence for siGluA1: (sense) CGACCAUUACAAGUGGCAAACCUUU; targeting sequence for siGluA2: (sense) UACUUUAUCCCUUUCACAGUCCAGG]. siRNAs were kept in 40 μm stock at −20°C before use. siRNAs were validated for knockdown potency and specificity by cotransfection of wtGluA1 or wtGluA2 with 0.4 and 4 nm siRNAs for GluA1 or GluA2 in human embryonic kidney (HEK) cells. Dominant-negative CaMKK (dnCaMKK) and monomeric RFP-β-actin constructs have been described previously (Wayman et al., 2004, 2006; Saneyoshi et al., 2008). HA-tagged AKT was described previously (Yano et al., 1998).
Cell culture and neuronal transfection.
Hippocampal neurons were isolated from embryonic day 21 male and female Sprague Dawley rats as described previously (Wayman et al., 2006). After harvesting, neurons were plated on 12 mm glass coverslips coated with poly-l-lysine (Sigma-Aldrich; molecular weight 300,000) at a density of 4.0 × 104 cells per square centimeter or on coated plastic 35 mm wells at 4.5 × 105 cells per square centimeter. Neurons were maintained in Neurobasal media (Invitrogen) supplemented with B27 (Invitrogen) and 0.5 mm l-glutamine with 5 μm cytosine-d-arabinofuranoside added at 2 DIV. Neurons were cultured for 5–9 d before being transfected using LipofectAMINE 2000 (Invitrogen) and/or treated with indicated pharmacological reagents. DNA, transfection reagent, and transfection duration were optimized to minimize toxicity and maximize transfection efficiency (3–5% of neurons between 4 and 6 DIV).
HEK293 cells were obtained from ATCC and cultured in DMEM, 10% fetal bovine serum, penicillin/streptomycin, and l-glutamine at 37°C in 5% CO2, 95% air. HEK293 cells were transfected with plasmids indicated in figure legends with Lipofectamine 2000 according to manufacturer's protocols.
Immunocytochemistry.
Hippocampal neurons were fixed in 4% paraformaldehyde, 4% sucrose in PBS, and 50 mm HEPES, pH 7.5, at 37°C for 15 min. Neurons were rinsed three times for 10 min in PBS and blocked for 1 h in blocking buffer (PBS containing 3% BSA) at room temperature (22°C). Neurons were then stained for surface-expressed GluA1 using a rabbit anti-GluA1 N-terminal antibody (Calbiochem, 1:100), which recognizes amino acids 271–285 of rat GluA1, in blocking buffer overnight at room temperature and washed three times for 10 min in blocking buffer. Surface GluA1 was subsequently detected by incubating coverslips in blocking buffer containing anti-rabbit Alexa Fluor-488 (Invitrogen, 1:2000) for 40 min at room temperature. Hoechst dye (Molecular Probes) was also added to the secondary buffer to enable assessment of cell viability. Coverslips were then washed quickly in PBS and mounted onto slides using Elvanol mounting medium.
GluA1 surface biotinylation.
Biotinylation experiments were performed as previously described (Oh et al., 2006). Briefly, following treatments, neuronal cell cultures were washed with ice-cold ACSF, pH adjusted to 8.2, to fully protonate amine groups before biotinylation. After removing the wash buffer, cells were incubated with ice-cold ACSF, pH 8.2, containing 0.8 mg/ml EZ-Link Sulfo-NHS-LC-biotin (Thermo Scientific) for 30 min on a shaker at 4°C. Cells were then quenched with ACSF containing 50 mm Tris-HCl for 10 min on a shaker at 4°C. Cells were then flash frozen using liquid nitrogen and stored at −20°C before being subjected to a GluA1 pulldown the following day.
For GluA1 pulldowns, cells were thawed on ice and lyzed in ice-cold RIPA lysis buffer. Lysates were then centrifuged for 10 min at 14,000 rpm at 4°C to pellet insoluble material. GluA1 antibody (2 μg, Millipore) was added to the supernatant. Lysates containing GluA1 antibody and Protein A/G Sepharose beads (Invitrogen) were rotated at 4°C overnight and then the beads were washed two times with ice-cold lysis buffer. The bound biotinylated proteins were then immunoprecipitated with streptavidin to isolate the biotinylated surface fraction of GluA1 and extracted with 2× SDS sample buffer before being subjected to a Western blot (see below). Biotinylated GluA1 and total GluA1 (cell lysate) were detected with Streptavidin-IR-800 and anti-GluA1 antibody (1:100, Millipore). Blots were then quantified using software supplied with the Odyssey Infrared System (LI-COR Biosciences).
HA-AKT pulldowns.
Hippocampal neurons were rapidly harvested with ice-cold lysis buffer containing 1% Triton X-100, and (in mm) 50 HEPES, pH 7.4, 150 NaCl, and 25 MgCl2 that was supplemented just before use with complete protease inhibitor mixture (EDTA free, Roche) and complete phosphatase inhibitor mixture (Calbiochem). The homogenates were centrifuged at 15,000 × g for 10 min and supernatant was collected. Lysates were then incubated with HA antibodies overnight and then further incubated with Protein A/G Sepharose beads (Invitrogen) for a further 2 h at 4°C on a rotating shaker. Proteins bound to the beads were further washed three times in lysis buffer before being subjected to Western blot.
Antibodies and Western blotting.
Polyclonal rabbit GluA1 (AB1504) was purchased from Millipore. Polyclonal rabbit phospho-mTORS2448, phospho-AKTT308, phospho-AKTS473, mouse monoclonal phospho-Erk1Thr202/ERK2Tyr204, and rabbit monoclonal phospho-YB1S102 antibodies were purchased from Cell Signaling Technologies. Mouse monoclonal transferrin receptor antibody was purchased from Invitrogen. Antibody to phospho-CaMKIT177 was kindly provided by Dr. Naohito Nozaki (nnozaki@monoclo.jp). Monoclonal antibody directed against HA (clone 12CA5) and E7 tubulin were purchased from Developmental Studies Hybridoma Bank (University of Iowa, Iowa City). 9E10 antibody used to detect Myc protein was purified from hybridoma supernatant. Puromycin antibody was generously provided by Dr. Phillippe Pierre (Université de la Méditerranée).
Western blotting was performed as previously described (Davare et al., 2004). Briefly, equivalent amounts of protein were loaded on SDS PAGE and transferred to PVDF membranes (Millipore). Western blotting was performed with the indicated antibodies at the following dilutions: anti-pmTOR, 1:1000; anti-pAKTT308, 1:1000; anti-pAKTS473, 1:1000; anti-pErk 42/44, 1:1000; anti-pCaMKI, 1:100; anti-HA, 1:3000; anti-GluA1, 1:100; anti-GluA2, 1:1000; anti-pYB1, 1:1000; β-tubulin, 1:5000; and anti-puromycin, 1:1000. Western blot detection was performed using IR-700-conjugated and IR-800-conjugated secondary antibodies to the appropriate antibody species and the band pixel density quantification was performed using the Odyssey Infrared System (LI-COR Biosciences).
Electrophysiology.
Whole-cell voltage-clamp recordings were performed on cultured hippocampal neurons as described above using an Axopatch-200b amplifier (Molecular Devices). Cells were continuously perfused (1 ml·min−1) with normal ACSF that contained the following (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 5 HEPES, and 33 glucose; pH was adjusted to 7.3 using NaOH. Osmolarity was adjusted to 290 mosmol·l−1. To isolate miniature EPSCs, gabazine (10 μm), strychnine (3 μm), and tetrodotoxin (0.5 μm) were added to the external buffer to block GABAA receptor, glycine receptor, and Na channel activity, respectively. Patch electrodes were pulled from thin-walled borosilicate glass capillaries (tip resistance ranged from 4 to 6 MΩ) and filled with internal buffer solution that contained the following (in mm): 100 cesium methanesulfonate, 25 CsCl, 2 MgCl2, 4 Mg2+-ATP, 0.4 Na-GTP, 10 phosphocreatine, 0.4 EGTA, and 10 HEPES, pH 7.4 (284 mosmol·l−1). In some experiments, the internal buffer also included 2 nm siGluA1 or siGluA2. All experiments were performed at room temperature (22°C). Whole-cell recordings were only established after a high-resistance seal (>2 GΩ) was achieved. Only cells that had an input resistance of >150 MΩ and resting membrane potentials <−50 mV were considered for experiments. Resting membrane potentials were measured immediately upon breaking into whole-cell mode by setting the current to 0 pA. Cells were then voltage-clamped at a holding potential of −70 mV unless otherwise noted. Access resistance (Ra), monitored at the beginning and end of each experiment with small voltage pulses, typically ranged between 10 and 15 MΩ and was not compensated. Cells were rejected from analysis if Ra increased by >15% during the course of the experiment or if the input resistance fell below 150 MΩ.
Results
CaMKK is required for BDNF-induced synthesis and membrane trafficking of GluA1
While it is known that BDNF increases intracellular Ca2+ concentrations and increases synaptic strength, the cellular mechanisms linking these two events remain unclear. We have previously implicated CaMKK in NMDA-dependent synaptic delivery of GluA1-containing CP-AMPARs. Based on these observations, we wanted to determine whether CaMKK might also play a role in BDNF-mediated regulation of GluA1. We initially investigated whether CaMKK might play a role in BDNF-mediated increases in GluA1 protein by treating primary hippocampal neurons (7–9 DIV) with 50 ng/ml BDNF for increasing lengths of time (0–60 min) and examining cell lysates for total GluA1 and phosphorylated (active) CaMKI. Phosphorylation of CaMKI by CaMKK is required for its catalytic activity (Tokumitsu et al., 1995). BDNF induced a time-dependent increase in total GluA1 levels (Fig. 1A,B) consistent with previous reports (Schratt et al., 2004; Caldeira et al., 2007; Slipczuk et al., 2009). We also found that CaMKI phosphorylation was rapidly increased by BDNF (Fig. 1A,D), suggesting that hippocampal neurons activate CaMKK in response to BDNF. Importantly, the phosphorylation of CaMKI was found to precede the increase in total GluA1 protein, consistent with a possible role of CaMKK in mediating GluA1 synthesis. To initially assess whether CaMKK contributed to the BDNF-induced increases in total GluA1 protein, we treated hippocampal neurons with BDNF for 15 min in the absence or presence of STO-609, a selective pharmacological inhibitor of CaMKK (Tokumitsu et al., 2002). Pretreatment of hippocampal neurons with STO-609 (4 h) fully prevented the increase in GluA1 protein levels by BDNF (Fig. 1E). As a positive control for BDNF we probed the same cell lysates for phosphorylation of the mitogen-activated protein kinases p44/ERK1 and p42/ERK2. It is well established that BDNF activates the MAPK (mitogen-activated protein kinase) pathway (Kaplan and Miller, 2000). Consistent with previous reports, BDNF rapidly increased phosphorylation of p44/p42 (Fig. 1A,C), which was sensitive to the general tyrosine kinase inhibitor, k252a (200 nm; BDNF, 174.1 ± 8.2%; k252a, 108 ± 12.9%, compared with controls, respectively).
Figure 1.

BDNF-induced increase in total GluA1 protein requires CaMKK. A–D, Representative immunoblot of hippocampal cell lysates illustrating the increase in total GluA1 (A, B), phosphorylated ERK (p-ERK) (C), and CaMKK-mediated phosphorylation of CaMKI (p-CaMKI) (D) following increasing times of BDNF treatment. Error bars indicate SEM (n = 5 from 5 independent experiments). *p < 0.05 by one-way ANOVA and Tukey's post hoc test. E, Representative immunoblot (left) and quantification (4 independent experiments) of total GluA1 protein following 15 min of BDNF treatment in the absence or presence of 10 μm STO-609 (STO, CaMKK inhibitor) or 1 μm rapamycin (Rap, mTORC1 inhibitor). Error bars indicate SEM. *p < 0.05, **p < 0.01 by Student's t test. In A and E, β-tubulin is shown as loading control.
Given that BDNF is a potent modulator of mTOR-mediated cap-dependent translation (Takei et al., 2001), we also treated hippocampal neurons with 1 μm rapamycin to inhibit mTOR complex formation and thus mTOR activity. Consistent with previous studies, we found that pretreatment with rapamycin (30 min) inhibited the increase in total GluA1 protein by BDNF (Fig. 1E). These results are consistent with the hypothesis that BDNF increases the activity of CaMKK and most likely increases GluA1 protein levels by engaging the mTOR-dependent translational machinery.
In addition to increasing total GluA1 subunits, BDNF also specifically increases the number of GluA1 subunits on the plasma membrane (Caldeira et al., 2007). To directly assess surface expression of GluA1, surface membrane proteins in hippocampal neurons were biotinylated following BDNF treatment in the absence or presence of STO-609 or rapamycin. BDNF led to a twofold increase in the amount of biotinylated GluA1 compared with control (Fig. 2A). More importantly, the increase in surface GluA1 was fully prevented by STO-609 as well as rapamycin. These data demonstrate that the increased surface expression of GluA1 mediated by BDNF requires the activation of CaMKK as well as mTOR-dependent translation. Furthermore, it suggests that translational events are required for the increase in GluA1 surface expression.
Figure 2.
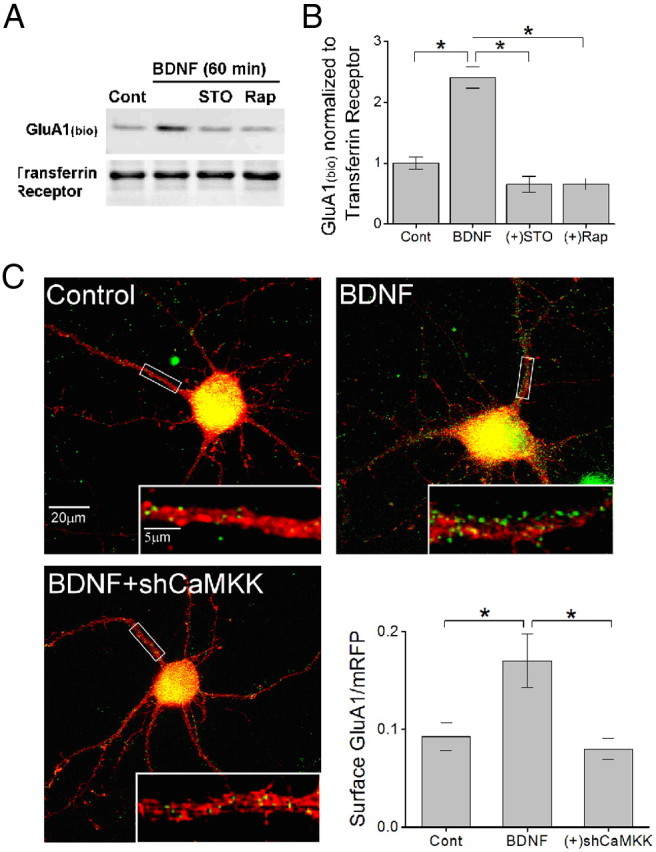
CaMKK mediates BDNF-induced increases in GluA1 surface expression. A, Representative immunoblot of biotinylated surface GluA1 (GluA1bio) and surface transferrin receptor following 60 min of BDNF treatment in the absence or presence of STO-609 or Rap as shown. B, Quantification of surface biotinylated GluA1 normalized to surface transferrin receptor for indicated conditions (n = 5 from 5 independent experiments). C, Representative immunofluorescence images of hippocampal primary neurons (10 DIV) transfected with monomeric RFP (red) and superimposed with surface GluA1 (pseudo-colored in green) for control. Neurons treated with 50 ng/ml BDNF plus or minus coexpression of short hairpins to α and β CaMKK (shCaMKK). Inset shows a magnification of the boxed segment of proximal dendrite. Lower right, Group data for surface GluA1 levels for indicated conditions (n = 8–12 neurons per coverslip from 3 independent experiments). Error bars indicate SEM *p < 0.05 by Student's t test.
To confirm the role of CaMKK in surface GluA1 mobilization as determined by biotinylation, we transfected hippocampal neurons with monomeric RFP (to visualize dendrites) and knocked down the expression of endogenous CaMKKα and CaMKKβ isoforms via short hairpins (shCaMKK) driven by the pmU6 promoter (Wayman et al., 2006; Saneyoshi et al., 2008). After 48 h of knockdown, neurons were treated with and without BDNF for 60 min and fixed for immunofluorescence examination. Surface GluA1 subunits were detected under nonpermeabilizing conditions using an epitope antibody directed toward the N terminus of GluA1 conjugated to Alexa 488. We found that BDNF led to a significant increase in the number of GluA1-positive puncta at the plasma membrane in agreement with Caldeira et al. (2007). More importantly, the increase in surface GluA1 was absent in neurons expressing shCaMKK (Fig. 2B).
BDNF-induced protein synthesis is mediated by CaMKK phosphorylation of T308 in ATK
To further investigate the role of CaMKK in BDNF-induced synthesis of GluA1, we tested whether CaMKK signaling was operating upstream or downstream of the mTOR complex. We have previously determined that CaMKK can directly phosphorylate AKT at one of its activation sites, T308 but not serine 473 (S473; Yano et al., 1998). Phosphorylation of AKT at both sites is important for its activation and subsequent activation of mTOR (Hoeffer and Klann, 2010). Direct activation of mTOR by AKT was assessed using a phospho-specific antibody directed toward S2448 (Memmott and Dennis, 2009), a phospho-site that correlates with increased mTORC1 activity and upregulated protein synthesis (Rosner et al., 2010). Figure 3 illustrates that mTOR is phosphorylated within 15 min of BDNF stimulation, and that this S2448 phosphorylation is blocked by treatment with STO-609. In addition, mTOR activation by BDNF was completely suppressed by rapamycin, which inhibits FKBP12 association with mTOR, thus preventing mTORC1 formation (Huang et al., 2003). Therefore, it is very likely that CaMKK lies upstream of the mTORC1 complex.
Figure 3.
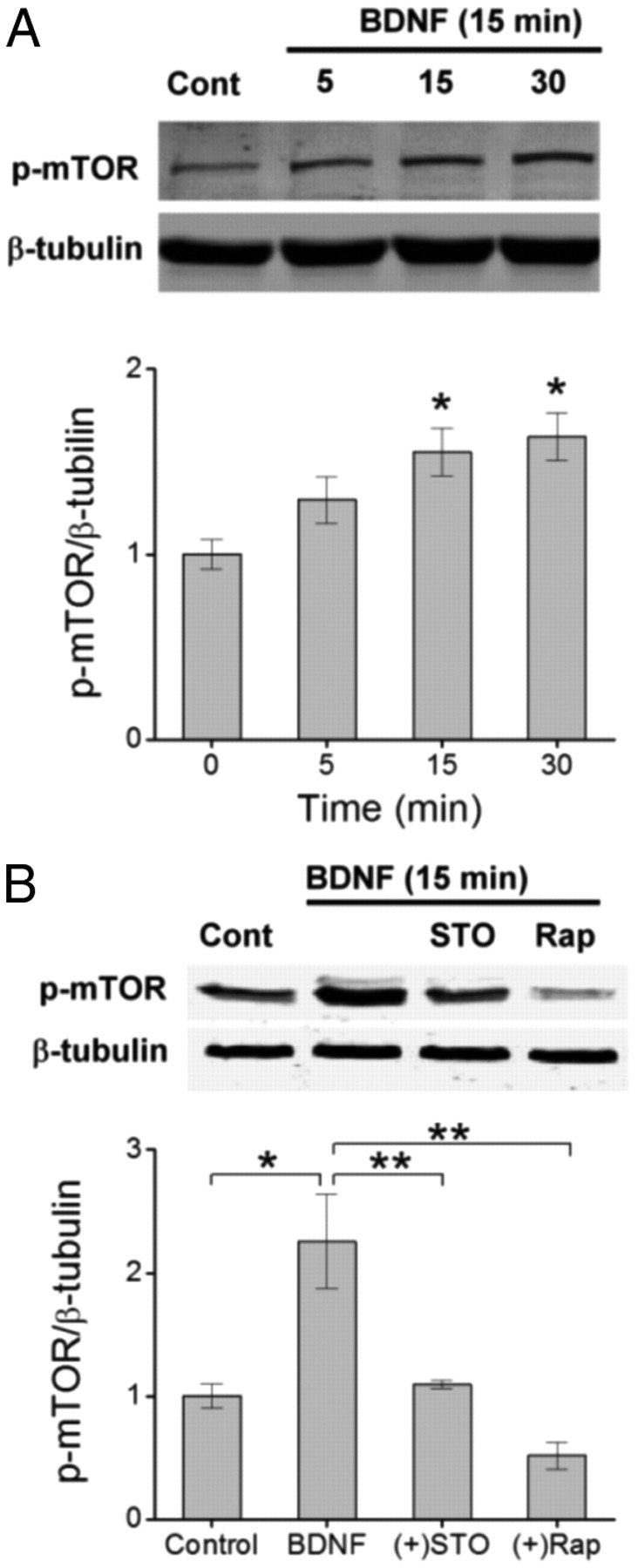
CaMKK functions upstream to activate mTOR. A, Top, Representative Western blot illustrating the time-dependent increase in phosphorylated mTOR (p-mTOR, S2448) following treatment with BDNF for the indicated times shown. Bottom, Group data for the ratio of p-mTOR to β-tubulin for each given time point. Error bars indicate SEM (n = 3 from 3 independent experiments). *p < 0.05 by one-way ANOVA and Tukey's post hoc test. B, Top, Immunoblot of p-mTOR levels following BDNF treatment for 15 min in the absence or presence of STO-609 (10 μm) or rapamycin (1 μm). Bottom, Group data for p-mTOR levels normalized to β-tubulin for indicated conditions. Error bars indicate SEM (n = 4 from 4 independent experiments). **p < 0.01 by Student's t test.
Although CaMKK can phosphorylate AKT at T308 but not S473 when expressed in COS-7 cells (Yano et al., 1998), we wanted to confirm that AKT was a substrate of CaMKK in neurons following BDNF stimulation. Using a phospho-specific antibody directed toward AKTT308, we probed neuronal cell lysates from primary hippocampal cultures that had been stimulated for 5 min with BDNF in the absence or presence of STO-609. BDNF significantly increased phosphorylation of endogenous AKT at T308 and this phosphorylation was sensitive to blockade by STO-609 (Fig. 4A). Phosphorylation at AKTT308 is important for its kinase activity (Leslie et al., 2001), so we tested whether inhibition of endogenous AKTT308 phosphorylation by STO-609 would alter the kinase activity of AKT in the presence of BDNF. The kinase activity of AKT was monitored using a phospho-specific antibody directed toward S102 of the Y-box binding protein YB1, a direct substrate of AKT (Evdokimova et al., 2006). Consistent with STO-609 inhibition of AKTT308 phosphorylation, STO-609 also suppressed phosphorylation of YB1S102 (Fig. 4A). Thus, CaMKK phosphorylation of AKTT308 appears to be a required step for its kinase activity and/or substrate specificity in neurons.
Figure 4.

BDNF-induced phosphorylation of AKT at T308, but not S473, requires CaMKK. A, Left, Representative Western blot illustrating the change in phosphorylation of AKT [p-AKT(T308)] and its substrate Y-box binding protein 1 (p-YB1) following 5 min of BDNF treatment in presence or absence of STO-609. Right, Group data for both p-AKT and p-YB1 normalized to β-tubulin (n = 4 from 4 independent experiments). B, C, Representative Western blots of immunoprecipitated HA-tagged AKT expressed or coexpressed with shCaMKK in hippocampal neurons treated with BDNF for 5 min and probed for phosphorylation at AKTT308 (B) or AKTS473 (C). Group data of p-AKT for T308 or S473 (n = 3 from 3 independent experiments) to total AKT for conditions indicated are shown below. Error bars indicate SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by Student's t test.
To further validate that AKTT308 was a CaMKK substrate in neurons, we coexpressed HA-AKT with and without shRNA against the two isoforms (α and β) of CaMKK. After 48 h, neurons were stimulated with BDNF for 5 min and then subjected to immunoprecipitation to isolate HA-AKT. Cell lysates were prepared, immunoblotted, and probed using phospho-specific antibodies directed toward AKTT308 and AKTS473. We found that while knockdown of CaMKK failed to alter AKTS473 phosphorylation (Fig. 4B), disruption of CaMKK signaling completely blocked the ability of BDNF to phosphorylate AKTT308 (Fig. 4C). Overexpression of dnCaMKK in the presence of BDNF elicited similar results (BNDF, 3.6 ± 0.2-fold over controls; dnCaMKK, 0.9 ± 0.2-fold over controls). These data support the conclusion that BDNF may in part regulate mTOR-dependent signaling via CaMKK phosphorylation of AKTT308. Phosphorylation of S473 in AKT (Fig. 4B) is presumably catalyzed by PI3-kinase activated by BDNF (Hoeffer and Klann, 2010).
TRPC channels, but not NMDA receptors, contribute to BDNF-mediated mTOR activation and membrane trafficking of GluA1
The enzymatic activity of CaMKK requires elevations in intracellular calcium. In hippocampal pyramidal neurons, BDNF is known to modulate the activities of NMDA receptors (Levine et al., 1998) as well as TRPC channels (Amaral and Pozzo-Miller, 2007b; Nakata and Nakamura, 2007), two well established calcium sources. To determine the calcium source required for the activation of CaMKK by BDNF, we pretreated cultures for 1 h with APV (25 μm) or SKF-96265 (30 μm) to block NMDA receptors and TRPC channels, respectively. Cultures were then subsequently stimulated with BDNF for 5 min in the presence of drug, lyzed, and subjected to Western blot analysis. BDNF-induced activation of CaMKK (i.e., p-CaMKI) as well as phosphorylation of AKT (T308) and mTOR were sensitive to blockade by SKF-96265 but not APV (Fig. 5A–D). These data indicate that TRPC channels are likely the main source of intracellular calcium induced by BDNF during this stage of development. This is consistent with the downstream calcium events associated with TrkB-phospholipase C-γ signaling via IP3 and TRPC that also contribute to BDNF-mediated AMPA receptor trafficking (Nakata and Nakamura, 2007).
Figure 5.
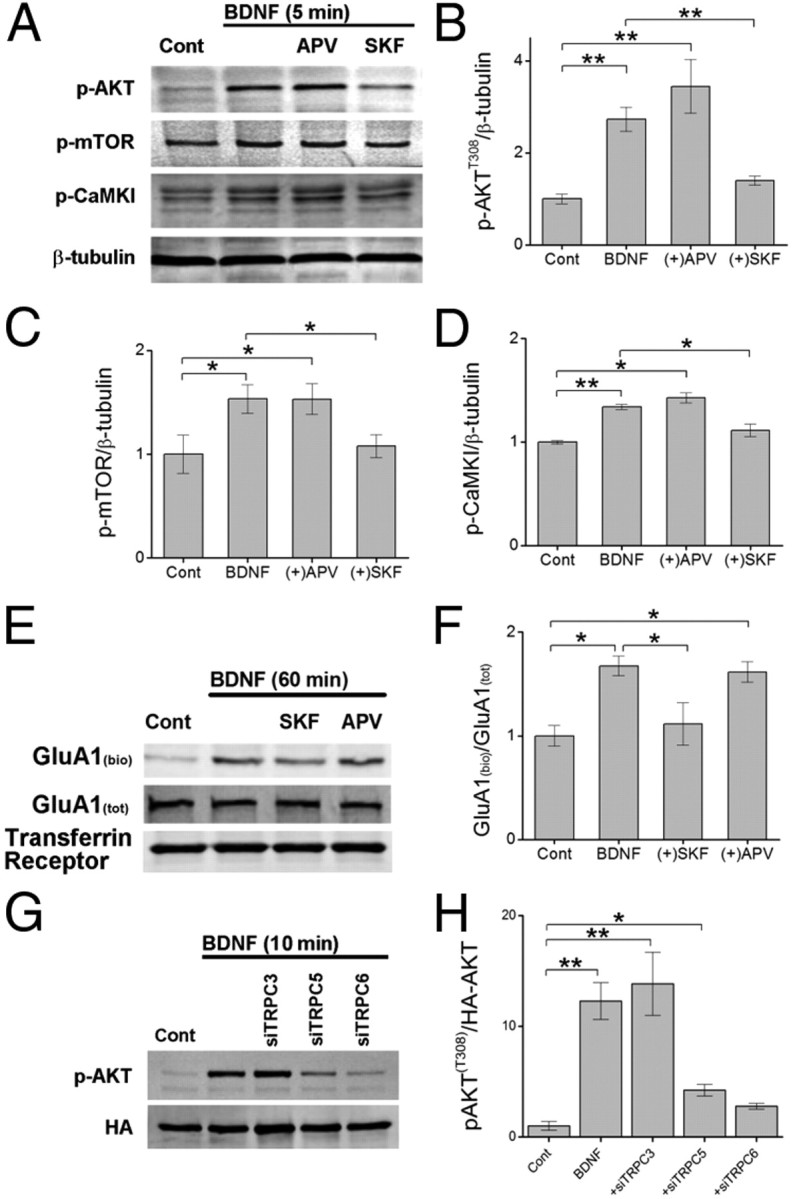
TRPC channels contribute to BDNF-mediated activation of CaMKK, the mTOR complex, and membrane trafficking of GluA1. A–D, Representative Western blot of hippocampal cell lysates illustrating the increase in phosphorylated AKT (T308), mTOR (S2448), and CaMKI (T177) following 5 min of BDNF treatment in the absence or presence of APV (25 μm or SKF-96265 (SKF, 30 μm) (A). Quantification of changes in total p-AKT (B), p-mTOR (C), and p-CaMKI (D) normalized to β-tubulin for indicated conditions. Error bars indicate SEM (n = 5 from 5 independent experiments). E, Representative Western blot of surface biotinylated GluA1 [GluA1(bio)], GluA1(tot), and transferrin receptor following 60 min of BDNF treatment in the presence or absence of SKF or APV. F, Mean data from four independent experiments for conditions shown. G, Neurons were transfected with HA-AKT alone or with siTRPC3, siTRPC5, or siTRPC6. After 72 h, neurons were treated with BDNF for 10 min before undergoing immunoprecipitation to obtain HA-AKT. Representative Western blot of pAKT (T308) and HA following BDNF treatment is shown for indicated conditions. H, Group data for conditions shown in E demonstrating loss of AKTT308 phosphorylation by BDNF in the presence of siTRPC5 or siTRPC6. *p < 0.05, **p < 0.01 by Student's t test.
To directly assess whether TRPC channels contribute to the surface delivery of GluA1, we performed surface biotinylation experiments in which neuronal cultures were treated with BDNF in the absence or presence of SKF-96265. While BDNF (60 min at 37°C) induced a significant increase in the amount of biotinylated GluA1, this increase was obviated in neurons treated with SKF-96265 (Fig. 5E,F). APV, which had no effect on AKT and mTOR activity, also failed to block the increase in surface GluA1 induced by BDNF. Together, these data demonstrate that TRPC channels likely play a major role in initiating mTOR-dependent translation and subsequent trafficking of GluA1 subunits to the plasma membrane mediated via CaMKK/AKT/mTOR signaling.
To confirm the involvement of TRPC channels and identify which subunits are involved, we selectively knocked down TRPC subunits using siRNA. While previous studies have shown that TRPC3 and TRPC6 are required for BDNF-mediated spine formation (Amaral and Pozzo-Miller, 2007a; Zhou et al., 2008), we also chose to examine TRPC5 since we previously reported these channels to be upstream of CaMKK in neurons (Davare et al., 2009). Neurons were cotransfected on DIV 7 with HA-AKT alone or with siRNA directed toward TRPC3, TRPC5, or TRPC6. After 72 h, neurons were subsequently treated with BDNF for 10 min and then subjected to immunoprecipitation to isolate HA-AKT before immunoblotting for AKTT308 phosphorylation. We found that small interfering TRPC5 (siTRPC5) and siTRPC6, but not siTRPC3, were able to largely suppress AKTT308 phosphorylation induced by BDNF, indicating that TRPC channels composed of these subunits are required for the activation of CaMKK/AKT pathway (Fig. 5G,H). Together these data strongly suggest that TRPC channels are the major mediators of BDNF-induced activation of AKT by CaMKK in hippocampal neurons.
BDNF induces the surface expression of newly translated GluA1
Although several studies have shown that pharmacological inhibition of the translation machinery prevents GluA1 membrane insertion, this approach does not directly address the question of whether the surface GluA1 itself was indeed newly translated or preexisting in some other subcellular compartment. Alternatively, it is conceivable that BDNF could simply induce the translation of accessory proteins necessary for surface trafficking of existing GluA1 and/or membrane retainment. In such a scenario, GluA1 subunits, although not translated, would be under the control of newly translated adaptor/trafficking proteins responsible for their membrane delivery or retention (Jourdi et al., 2003).
To determine whether BDNF induced the membrane insertion of newly translated GluA1 subunits, we took advantage of the recently developed surface sensing of translation method (Schmidt et al., 2009). Puromycin, a structural analog of aminoacyl tRNAs, is incorporated into nascent polypeptide chains. When used at low concentrations, puromycin incorporation into newly synthesized proteins reflects directly the rate of mRNA translation. Using this approach, we were able to directly assess whether surface GluA1 subunits contained puromycin, indicating they were acutely translated in response to BDNF. Monoclonal antibodies to puromycin demonstrated, as shown in Figure 6A, that BDNF with puromycin, compared with puromycin pulse alone, induced a robust increase in puromycin incorporation into total cellular proteins. Importantly, the incorporation of puromycin was prevented by the translational inhibitor anisomycin, confirming that puromycin was incorporated into nascent proteins. To determine whether newly translated GluA1 was being incorporated into the plasma membrane, we pulsed neuronal cultures with 10 ng/ml puromycin for 10 min, immediately followed by a 30 min stimulation with BDNF in the absence or presence of anisomycin. Surface GluA1 was detected by subjecting neuronal cultures to surface biotinylation followed by immunoprecipitation using a GluA1 antibody before immunodetection of streptavidin and puromycin. The increase in surface GluA1 in response to BDNF correlated with increased incorporation of puromycin, and both were blocked by anisomycin treatment (10 μm for 30 min) (Fig. 6B,C). These data strongly suggest that BDNF induces the translation of GluA1 and this newly translated pool of GluA1 is subsequently incorporated into the plasma membrane.
Figure 6.

BDNF induces surface delivery of newly translated GluA1. A, Immunoblot of hippocampal total cell lysates following a 10 min puromycin (10 ng/ml) pulse followed by BDNF stimulation (40 min) in the absence or presence of anisomycin (Aniso, 10 μm) and probed with puromycin or β-tubulin antibodies. B, Western blot showing levels of surface biotinylated GluA1 (streptavidin), and puromycin incorporation (puromycin) from hippocampal lysates immunoprecipitated with an antibody to GluA1 following BDNF stimulation in the absence or presence of anisomycin. β-Tubulin was probed from accompanying total cell lysates. C, Quantification of the ratio of surface biotinylated GluA1 and puromycin incorporation to β-tubulin for indicated conditions (n = 5 from 5 independent experiments). Error bars indicate SEM. *p < 0.05 by Student's t test.
Synaptic incorporation of newly translated GluA1 by BDNF
Is the increase in GluA1 protein consequential for functional AMPARs at synapses? It has been previously reported that BDNF also induces the upregulation of several PDZ proteins, including SAP97, GRIP1, and PICK1 (Jourdi et al., 2003). Given that the interactions between AMPA receptor subunits with specific PDZ proteins are important for the delivery of subunits into synapses, we wanted to determine whether this newly synthesized surface pool of receptors was incorporated into synapses. To test this possibility, we performed whole-cell patch-clamp recordings from cultured hippocampal neurons treated with and without BDNF. Synaptic currents were monitored by isolating miniature EPSCs by addition of TTX (0.5 μm) and GABAzine (10 μm) to the bath perfusate to block voltage-dependent Na+ channels and GABAA receptors, respectively. Additionally, d-APV (25 μm) was added to the bath perfusate to block the contribution of NMDA receptors, allowing the isolation of AMPA receptor-mediated mEPSCs. Perfusion of BDNF for 15 min led to a significant increase in the amplitude of mEPSC recorded from primary hippocampal cultures compared with controls, which received heat-inactivated BDNF (Fig. 7A). It should be noted that the frequency (control, 2.0 ± 0.2 Hz; BDNF, 2.1 ± 0.1 Hz), rise time (control, 1.06 ± 0.06 ms; BDNF, 1.07 ± 0.05 ms) and decay times (control, 6.98 ± 0.10 ms; BDNF, 7.57 ± 0.07 ms) of mEPSCs were not altered by the addition of BDNF. Importantly, the increase in mEPSC amplitude was sensitive to the coapplication of anisomycin to the bath perfusate (Fig. 7A), suggesting that the majority of the increase in mEPSC amplitude required de novo protein synthesis.
Figure 7.

BDNF-induced increase in synaptic strength requires protein synthesis of GluA1 subunits coupled to expression of CP-AMPARs. A, Left, Example traces of mEPSCs recorded at room temperature at a holding potential of −70 mV for each condition indicated. Traces were an average of 100–150 consecutive events recorded during each treatment condition. In the indicated experiments, neurons were pretreated with anisomycin for 30 min before the addition of BDNF to impair translation. Right, Group data for mean mEPSC amplitude for control (n = 12), BDNF (n = 6; *p < 0.05 by Student's t test) and BDNF in the presence of anisomycin (Aniso; n = 6). B, Representative Western blot for GluA1 and GluA2 subunits expressed in HEK cells in the absence or presence of 0.4 or 4.0 nm siRNA for GluA1 or GluA2. β-Tubulin was used as a loading control. C, Quantification of GluA1 and GluA2 knockdown for conditions indicated in B for four independent experiments. D, E, Left, Average mEPSCs recorded during baseline and following treatment with BDNF with inclusion of siGluA1 (D) or siGluA2 (E) in the patch pipette. Right, Individual (gray) and mean (black) mEPSC amplitudes for siGluA1 (D; n = 7) and siGluA2 (E; n = 5). *p < 0.05 by Student's paired t test. F, Group data illustrating the mean reduction in mEPSC amplitude as a percentage of baseline responses following IEM treatment under baseline conditions (left; n = 12) and following BDNF stimulation (right; n = 7). Error bars indicate SEM. *p < 0.05 by Student's paired t test.
To directly test whether GluA1 subunits were specifically being translated and incorporated into synapses, we infused siRNA directed toward either GluA1 (siGluA1) or GluA2 (siGluA2) through the whole-cell patch pipette. We tested the specificity of both siRNAs in HEK cells. HEK cells were grown to 60% confluence and transfected with either wtGluA1 or wtGluA2 for 24 h with or without cotreatment of 0.4 or 4 nm siGluA1 or siGluA2. We found that siGluA1 significantly reduced the expression of GluA1 but did not alter the expression of GluA2, while siGluA2 robustly reduced GluA2 protein and only modestly reduced GluA1 expression, thus demonstrating the subunit specificity of these siRNAs (Fig. 7B,C). Hippocampal pyramidal neurons were then patched with or without infusion of either siGluA1 (2.0 nm) or siGluA2 (2 nm). Following the establishment of whole-cell recording, mEPSCs were recorded for 10 min to establish baseline and then perfused with BDNF for 15 min. Infusion of siGluA1 (Fig. 7D) but not siGluA2 (Fig. 7E) attenuated the increase in mEPSC amplitude mediated by BDNF toward baseline responses. This strongly suggests that the increase in mEPSC amplitude induced by BDNF was due to synaptic insertion of AMPARs containing newly translated GluA1 but not GluA2 subunits. To control for the possibility that GluA1 subunits may run down in neurons infused with siGluA1 due to potential “off target” effects, we compared the properties of basal AMPA receptor-mediated mEPSCs before and 15 min after infusion of siGluA1. We found that infusion of 2 nm siGluA1 alone had no effect on basal mEPSC amplitude (0–3 min, 11.0 ± 1.12 pA; 15–20 min, 9.9 ± 1.06 pA) or decay time (0–3 min, 3.1 ± 0.5 ms; 15–20 min, 3.9 ± 0.7 ms), suggesting that the failure of BDNF to increase mEPSC amplitude in cells loaded with siGluA1 likely stems not from “off target” effects, but rather inhibition of nascent GluA1 synthesis.
The above observations implied that the increase in mEPSC amplitude induced by BDNF could be caused by the increased presence of GluA2-lacking CP-AMPARs. To detect the presence of CP-AMPARs, we performed whole-cell voltage-clamp recordings of AMPA receptor-mediated mEPSCs in the presence of IEM-1460, a selective antagonist of GluA2-lacking (i.e., calcium-permeable) AMPA receptors. IEM-1460 has been shown to have greater selectivity than philanthotoxin-433 for CP-AMPARs versus GluA2-containing AMPARs as well as NMDARs (Magazanik et al., 1997; Buldakova et al., 1999; Samoilova et al., 1999; Buldakova et al., 2007). Given that some synaptic CP-AMPARs are present during this early stage (DIV 7–9) of development (Kumar et al., 2002; Eybalin et al., 2004; Ho et al., 2007), we first determined their basal contribution to recorded spontaneous mEPSCs. The mean mEPSC amplitude was attenuated by 40 ± 3.2% of baseline responses following perfusion of IEM-1460 (30 μm) for 10 min. More importantly, the increase in mEPSC amplitude induced by BDNF was completely abolished by IEM-1460 (Fig. 7F). These results are consistent with the synaptic incorporation of CP-AMPARs containing newly translated GluA1 subunits.
Discussion
It is well known that BDNF plays an important role in the development of synapses (Poo, 2001; Kuipers and Bramham, 2006). Previous studies have also demonstrated that BDNF acutely regulates the expression and membrane trafficking of GluA1 subunits during the period of increased synaptogenesis (Caldeira et al., 2007; Nakata and Nakamura, 2007). Consistent with this, many laboratories have also shown that in various brain regions, including hippocampus, excitatory glutamatergic synapses initially express GluA2-lacking CP-AMPARs (Caldeira et al., 2007; Nakata and Nakamura, 2007). However, the cellular and molecular mechanisms induced by BDNF that mediate changes in GluA1 surface expression remain poorly understood. Using multiple experimental approaches, we now identify a role for CaMKK (Fig. 8) that in young hippocampal neurons BDNF acutely induces the direct translation and subsequent synaptic insertion of GluA1-containing AMPARs, which leads to the increased synaptic expression of CP-AMPARs.
Figure 8.
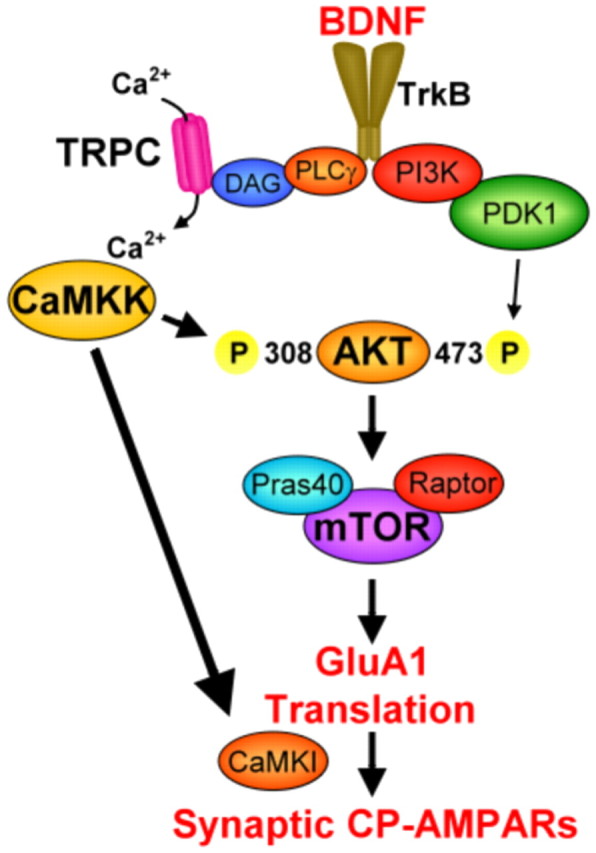
Schematic of signaling pathway involving CaMKK regulation of GluA1 synthesis and synaptic incorporation as CP-AMPARs. Signaling shown in bold font is documented in the current report whereas signaling in normal font is taken from the literature as cited in the text.
The trafficking of AMPARs to the synaptic membrane is thought to play an important role during synaptogenesis and synapse maturation, as well as during synaptic plasticity (Malinow and Malenka, 2002; Song and Huganir, 2002; Derkach et al., 2007). Many of these processes are also dependent on protein synthesis because impairments in the translational machinery result in abnormal neuronal morphology and physiology, which have been linked to various neurological disorders (Wang et al., 2010). We have provided evidence demonstrating that BDNF directly induces the synthesis of GluA1 subunits. As BDNF has been shown to alter the protein expression of over a hundred synaptic proteins (Liao et al., 2007), it is possible that BDNF also alters the expression of additional auxiliary proteins that may be necessary for GluA1 trafficking to or retention in the synapse. Indeed, BDNF has been shown to increase the expression of SAP97, a key PDZ protein associated with GluA1 stability at the postsynaptic density (Jourdi et al., 2003). Although, we have strong evidence to suggest that newly synthesized GluA1 subunits traffic to the synaptic membrane, expression of auxiliary proteins, such as TARPS (transmembrane AMPAR regulatory proteins) or SAP97, may produce similar results.
One particular finding is that newly translated GluA1-containing CP-AMPARs appear to be required for the increased synaptic strength induced by BDNF. Interestingly, immature hippocampal neurons do basally express a significant fraction of extrasynaptic and synaptic CP-AMPARs, but these receptors are not used for enhancing synaptic strength. This is unexpected and suggests these new GluA1-containing AMPARs might serve a separate function. For example, GluA1 may have a different composition of associated proteins or different downstream signaling, beyond their high Ca2+ permeability, which might be important for synaptogenesis and synapse maturation.
We found that BDNF induced the synthesis, trafficking, and synaptic incorporation of GluA1 subunits within 15 min, resulting in increased synaptic strength. While this study does not address whether synthesis of GluA1 occurs locally in the dendrite, growing evidence has made it clear that local dendritic protein synthesis does occur (Sutton and Schuman, 2006) and that GluA1 subunits can indeed be translated within the dendritic compartment (Ju et al., 2004; Smith et al., 2005; Sutton et al., 2006). It should also be noted that whereas several laboratories have observed increases in mEPSC frequency without accompanying changes in mEPSC amplitude in cultured hippocampal neurons following BDNF treatment (Lessmann et al., 1994; Lessmann and Heumann, 1998; Li et al., 1998; Schinder et al., 2000; Madara and Levine, 2008), we only detected changes in mEPSC amplitude. The origin of this discrepancy is unclear, but it may be due to developmental differences in BDNF signaling. For example, BDNF fails to trigger increases in surface GluA1 in neurons cultured to 14 DIV (Caldeira et al., 2007). In addition, the increase in mEPSC amplitude we observe after 15 min of BDNF treatment may be accounted for by insertion of a very small number of CP-AMPARs that exhibit enhanced unity conductance (see Guire et al., 2008, Fig. 6). This small number of additional CP-AMPARs at existing synapses would probably not be sufficient to give a detectable increase in mEPSC frequency.
We have also identified a signaling pathway induced by BDNF which enhances GluA1 synthesis and synaptic strength. Activation of TrkB receptors upon BDNF binding has been shown to engage the Ras-Raf MAPK/ERK cascade, the PI3 kinase cascade, and the phospholipase C-γ-IP3-TRPC cascade (Segal and Greenberg, 1996). Our data suggest that TRPC channels can activate mTOR-dependent translation of GluA1 subunits by activating the CaMKK/AKT signaling pathway. Although, both recombinant BDNF (Li et al., 1999; Nakata and Nakamura, 2007) and endogenous BDNF (Li et al., 2010) have been shown to activate TRPC channels in hippocampal neurons, this is the first report linking these channels to mTOR-dependent translation. Previous studies have linked BDNF to the activation of TRPC3 (Li et al., 1999, 2010; Amaral and Pozzo-Miller, 2007a) and/or TRPC6 (Zhou et al., 2008). The current study demonstrates that BDNF also activates TRPC5. It should be noted that TRPC5 has been shown to be stimulated by receptor tyrosine kinases (Schaefer et al., 2000). Our data specifically links activation of TRPC5 and TRPC6 to BDNF-induced phosphorylation of AKT at T308, whereas knockdown of TRPC3 had no effect. These data demonstrate that channels containing TRPC5 and TRPC6 are likely to contribute to the Ca2+ flux responsible for the activation of CaMKK required for BDNF-induced mTOR-dependent translation of GluA1.
The present study also implicates AKT as a downstream substrate of CaMKK which mediates the effects of BDNF in hippocampal neurons. In addition to phosphoinositide binding at the plasma membrane, AKT requires the phosphorylation of T308 within its kinase domain as well as phosphorylation of S473 within its C-terminal domain (Alessi et al., 1996). While phosphorylation of T308 is important for its catalytic activity, S473 is thought to aid in the stabilization of its substrates (Yang et al., 2002a,b). In most biological systems T308 as well as S473 are thought to be phosphorylated by phosphoinositide-dependent kinase-1 (PDK1) (Alessi et al., 1997; Stephens et al., 1998; Toker and Newton, 2000). However, our group has previously shown that CaMKK can phosphorylate recombinant AKT in vitro (Yano et al., 1998). Induction of theta-burst LTP via NMDARs results in rapid CaMKK-mediated phosphorylation of CaMKI (maximal at 5 min) but very slow CaMKK (i.e., blocked by STO-609) phosphorylation of AKT-T308 that peaked at 1 h (Schmitt et al., 2005). Here we show TRPC-mediated activation of CaMKK phosphorylates AKT-T308 within 5 min. Specificity of CaMKK phosphorylation was also observed as only the critical T-loop residue (T308) within AKT, but not S473, was phosphorylated by CaMKK. Thus, it appears that the source of Ca2+-influx (e.g., NMDAR vs TRPC) dictates the temporal substrate-specific targets downstream of CaMKK, as has been observed for other Ca2+-responsive systems (Bading et al., 1993). It is assumed that S473 is subsequently phosphorylated by PDK1 as in other biological systems. We also provide evidence that CaMKK phosphorylation of AKT at T308 is required for its kinase activity as YB1, a direct target of AKT, was unable to be phosphorylated upon CaMKK inhibition. Thus, our finding extends the list of neuronal CaMKK substrates to include AKT.
Previous work from our laboratory has established multiple roles of the CaMKK/CaMKI pathway in numerous aspects of neuronal development and plasticity (Wayman et al., 2008; Fortin et al., 2011). The current work highlights a novel role for CaMKK in mediating Ca2+-dependent downstream signaling events associated with regulation of protein synthesis, an area of growing interest in synaptic plasticity and neurological diseases (Sutton and Schuman, 2006). The translation of GluA1 and subsequent synaptic expression of GluA1-containing CP-AMPARs mediated by BDNF provides a mechanism to either increase postsynaptic Ca2+ concentrations or other signaling that may be important for synapse maturation. Having previously linked CP-AMPARs to the actin cytoskeleton via the Rac/PAK/LIMK pathway following NMDAR-dependent LTP, we suggest CP-AMPARs may play a similar role earlier in development. Alternatively, CP-AMPARs may serve to unsilence developing synapses, which lack NMDAR currents, by the stepwise acquisition of GluA1-containing CP-AMPARs (Isaac, 2003; Poncer, 2003). Our findings suggest that BDNF acting through CaMKK may facilitate the incorporation of CP-AMPARs during this critical time in synapse development (Fig. 8). Endogenous BDNF likely contributes to this process as treatment of hippocampal slices with the BDNF scavenger TrkB-IgG reduces dendritic spine density of CA1 pyramidal neurons (Tyler and Pozzo-Miller, 2001). BDNF activation of the CaMKK and PDK1 pathways can account for the AKT/mTOR-dependent synthesis of GluA1, but what triggers the synaptic expression of the nascent GluA1 as CP-AMPARs? As mentioned above, several possibilities, such as scaffold proteins (e.g., SAP97), may be involved. Another likely candidate is CaMKI, which is also activated by BDNF (Fig. 1D). Indeed, infusion of activated CaMKI via a patch pipette into hippocampal neurons results in rapid enhancement of synaptic CP-AMPAR currents (Guire et al., 2008). This effect presumably involves some type of trafficking event(s) since it was blocked by inhibition of actin polymerization with latrunculin A. Further experiments need to explore BDNF-mediated mechanisms underlying synaptic trafficking of newly synthesized GluA1. Another critical area of future investigation is the identification of targets specifically downstream of CP-AMPARs.
Footnotes
This work was supported by National Institutes of Health Grant NS027037 (T.R.S.).
References
- Aizenman CD, Muñoz-Elías G, Cline HT. Visually driven modulation of glutamatergic synaptic transmission is mediated by the regulation of intracellular polyamines. Neuron. 2002;34:623–634. doi: 10.1016/s0896-6273(02)00674-8. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Alsina B, Vu T, Cohen-Cory S. Visualizing synapse formation in arborizing optic axons in vivo: dynamics and modulation by BDNF. Nat Neurosci. 2001;4:1093–1101. doi: 10.1038/nn735. [DOI] [PubMed] [Google Scholar]
- Amaral MD, Pozzo-Miller L. TRPC3 channels are necessary for brain-derived neurotrophic factor to activate a nonselective cationic current and to induce dendritic spine formation. J Neurosci. 2007a;27:5179–5189. doi: 10.1523/JNEUROSCI.5499-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral MD, Pozzo-Miller L. BDNF induces calcium elevations associated with IBDNF, a nonselective cationic current mediated by TRPC channels. J Neurophysiol. 2007b;98:2476–2482. doi: 10.1152/jn.00797.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Buldakova SL, Vorobjev VS, Sharonova IN, Samoilova MV, Magazanik LG. Characterization of AMPA receptor populations in rat brain cells by the use of subunit-specific open channel blocking drug, IEM-1460. Brain Res. 1999;846:52–58. doi: 10.1016/s0006-8993(99)01970-8. [DOI] [PubMed] [Google Scholar]
- Buldakova SL, Kim KK, Tikhonov DB, Magazanik LG. Selective blockade of Ca2+ permeable AMPA receptors in CA1 area of rat hippocampus. Neuroscience. 2007;144:88–99. doi: 10.1016/j.neuroscience.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Caldeira MV, Melo CV, Pereira DB, Carvalho R, Correia SS, Backos DS, Carvalho AL, Esteban JA, Duarte CB. Brain-derived neurotrophic factor regulates the expression and synaptic delivery of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J Biol Chem. 2007;282:12619–12628. doi: 10.1074/jbc.M700607200. [DOI] [PubMed] [Google Scholar]
- Davare MA, Saneyoshi T, Guire ES, Nygaard SC, Soderling TR. Inhibition of calcium/calmodulin-dependent protein kinase kinase by protein 14-3-3. J Biol Chem. 2004;279:52191–52199. doi: 10.1074/jbc.M409873200. [DOI] [PubMed] [Google Scholar]
- Davare MA, Fortin DA, Saneyoshi T, Nygaard S, Kaech S, Banker G, Soderling TR, Wayman GA. Transient receptor potential canonical 5 channels activate Ca2+/calmodulin kinase Iγ to promote axon formation in hippocampal neurons. J Neurosci. 2009;29:9794–9808. doi: 10.1523/JNEUROSCI.1544-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Evdokimova V, Ovchinnikov LP, Sorensen PH. Y-box binding protein 1: providing a new angle on translational regulation. Cell Cycle. 2006;5:1143–1147. doi: 10.4161/cc.5.11.2784. [DOI] [PubMed] [Google Scholar]
- Eybalin M, Caicedo A, Renard N, Ruel J, Puel JL. Transient Ca2+-permeable AMPA receptors in postnatal rat primary auditory neurons. Eur J Neurosci. 2004;20:2981–2989. doi: 10.1111/j.1460-9568.2004.03772.x. [DOI] [PubMed] [Google Scholar]
- Fortin DA, Davare MA, Srivastava T, Brady JD, Nygaard S, Derkach VA, Soderling TR. Long-term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by CaM-kinase I. J Neurosci. 2010;30:11565–11575. doi: 10.1523/JNEUROSCI.1746-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin DA, Srivastava T, Soderling TR. Structural modulation of dendritic spines during synaptic plasticity. Neuroscientist. 2011 doi: 10.1177/1073858411407206. Advanced online publication. Retrieved December 4, 2011. PMID: 21670426. [DOI] [PubMed] [Google Scholar]
- Goold CP, Nicoll RA. Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron. 2010;68:512–528. doi: 10.1016/j.neuron.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guire ES, Oh MC, Soderling TR, Derkach VA. Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I. J Neurosci. 2008;28:6000–6009. doi: 10.1523/JNEUROSCI.0384-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Ho MT, Pelkey KA, Topolnik L, Petralia RS, Takamiya K, Xia J, Huganir RL, Lacaille JC, McBain CJ. Developmental expression of Ca2+-permeable AMPA receptors underlies depolarization-induced long-term depression at mossy fiber CA3 pyramid synapses. J Neurosci. 2007;27:11651–11662. doi: 10.1523/JNEUROSCI.2671-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA–gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- Huang S, Bjornsti MA, Houghton PJ. Rapamycins: mechanism of action and cellular resistance. Cancer Biol Ther. 2003;2:222–232. doi: 10.4161/cbt.2.3.360. [DOI] [PubMed] [Google Scholar]
- Isaac JT. Postsynaptic silent synapses: evidence and mechanisms. Neuropharmacology. 2003;45:450–460. doi: 10.1016/s0028-3908(03)00229-6. [DOI] [PubMed] [Google Scholar]
- Jourdi H, Iwakura Y, Narisawa-Saito M, Ibaraki K, Xiong H, Watanabe M, Hayashi Y, Takei N, Nawa H. Brain-derived neurotrophic factor signal enhances and maintains the expression of AMPA receptor-associated PDZ proteins in developing cortical neurons. Dev Biol. 2003;263:216–230. doi: 10.1016/j.ydbio.2003.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju W, Morishita W, Tsui J, Gaietta G, Deerinck TJ, Adams SR, Garner CC, Tsien RY, Ellisman MH, Malenka RC. Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat Neurosci. 2004;7:244–253. doi: 10.1038/nn1189. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Kuipers SD, Bramham CR. Brain-derived neurotrophic factor mechanisms and function in adult synaptic plasticity: new insights and implications for therapy. Curr Opin Drug Discov Devel. 2006;9:580–586. [PubMed] [Google Scholar]
- Kumar SS, Bacci A, Kharazia V, Huguenard JR. A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J Neurosci. 2002;22:3005–3015. doi: 10.1523/JNEUROSCI.22-08-03005.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie NR, Biondi RM, Alessi DR. Phosphoinositide-regulated kinases and phosphoinositide phosphatases. Chem Rev. 2001;101:2365–2380. doi: 10.1021/cr000091i. [DOI] [PubMed] [Google Scholar]
- Lessmann V, Heumann R. Modulation of unitary glutamatergic synapses by neurotrophin-4/5 or brain-derived neurotrophic factor in hippocampal microcultures: presynaptic enhancement depends on pre-established paired-pulse facilitation. Neuroscience. 1998;86:399–413. doi: 10.1016/s0306-4522(98)00035-9. [DOI] [PubMed] [Google Scholar]
- Lessmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. Neuroreport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-D-aspartic acid receptor activity. Proc Natl Acad Sci U S A. 1998;95:10235–10239. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HS, Xu XZ, Montell C. Activation of a TRPC3-dependent cation current through the neurotrophin BDNF. Neuron. 1999;24:261–273. doi: 10.1016/s0896-6273(00)80838-7. [DOI] [PubMed] [Google Scholar]
- Liao L, Pilotte J, Xu T, Wong CC, Edelman GM, Vanderklish P, Yates JR., 3rd BDNF induces widespread changes in synaptic protein content and up-regulates components of the translation machinery: an analysis using high-throughput proteomics. J Proteome Res. 2007;6:1059–1071. doi: 10.1021/pr060358f. [DOI] [PubMed] [Google Scholar]
- Li YX, Zhang Y, Lester HA, Schuman EM, Davidson N. Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J Neurosci. 1998;18:10231–10240. doi: 10.1523/JNEUROSCI.18-24-10231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Calfa G, Inoue T, Amaral MD, Pozzo-Miller L. Activity-dependent release of endogenous BDNF from mossy fibers evokes a TRPC3 current and Ca2+ elevations in CA3 pyramidal neurons. J Neurophysiol. 2010;103:2846–2856. doi: 10.1152/jn.01140.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SQ, Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405:454–458. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, Hell JW. Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 2007;26:4879–4890. doi: 10.1038/sj.emboj.7601884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madara JC, Levine ES. Presynaptic and postsynaptic NMDA receptors mediate distinct effects of brain-derived neurotrophic factor on synaptic transmission. J Neurophysiol. 2008;100:3175–3184. doi: 10.1152/jn.90880.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magazanik LG, Buldakova SL, Samoilova MV, Gmiro VE, Mellor IR, Usherwood PN. Block of open channels of recombinant AMPA receptors and native AMPA/kainate receptors by adamantane derivatives. J Physiol. 1997;505(Pt 3):655–663. doi: 10.1111/j.1469-7793.1997.655ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Memmott RM, Dennis PA. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal. 2009;21:656–664. doi: 10.1016/j.cellsig.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata H, Nakamura S. Brain-derived neurotrophic factor regulates AMPA receptor trafficking to post-synaptic densities via IP3R and TRPC calcium signaling. FEBS Lett. 2007;581:2047–2054. doi: 10.1016/j.febslet.2007.04.041. [DOI] [PubMed] [Google Scholar]
- Narisawa-Saito M, Carnahan J, Araki K, Yamaguchi T, Nawa H. Brain-derived neurotrophic factor regulates the expression of AMPA receptor proteins in neocortical neurons. Neuroscience. 1999;88:1009–1014. doi: 10.1016/s0306-4522(98)00496-5. [DOI] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci. 2006;9:602–604. doi: 10.1038/nn1678. [DOI] [PubMed] [Google Scholar]
- Poncer JC. Hippocampal long term potentiation: silent synapses and beyond. J Physiol Paris. 2003;97:415–422. doi: 10.1016/j.jphysparis.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Rosner M, Siegel N, Valli A, Fuchs C, Hengstschläger M. mTOR phosphorylated at S2448 binds to raptor and rictor. Amino Acids. 2010;38:223–228. doi: 10.1007/s00726-008-0230-7. [DOI] [PubMed] [Google Scholar]
- Rozov A, Burnashev N. Polyamine-dependent facilitation of postsynaptic AMPA receptors counteracts paired-pulse depression. Nature. 1999;401:594–598. doi: 10.1038/44151. [DOI] [PubMed] [Google Scholar]
- Samoilova MV, Buldakova SL, Vorobjev VS, Sharonova IN, Magazanik LG. The open channel blocking drug, IEM-1460, reveals functionally distinct alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors in rat brain neurons. Neuroscience. 1999;94:261–268. doi: 10.1016/s0306-4522(99)00326-7. [DOI] [PubMed] [Google Scholar]
- Saneyoshi T, Wayman G, Fortin D, Davare M, Hoshi N, Nozaki N, Natsume T, Soderling TR. Activity-dependent synaptogenesis: regulation by a CaM-kinase kinase/CaM-kinase I/betaPIX signaling complex. Neuron. 2008;57:94–107. doi: 10.1016/j.neuron.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer M, Plant TD, Obukhov AG, Hofmann T, Gudermann T, Schultz G. Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J Biol Chem. 2000;275:17517–17526. doi: 10.1074/jbc.275.23.17517. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Berninger B, Poo M. Postsynaptic target specificity of neurotrophin-induced presynaptic potentiation. Neuron. 2000;25:151–163. doi: 10.1016/s0896-6273(00)80879-x. [DOI] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6:275–277. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- Schmitt JM, Guire ES, Saneyoshi T, Soderling TR. Calmodulin-dependent kinase kinase/calmodulin kinase I activity gates extracellular-regulated kinase-dependent long-term potentiation. J Neurosci. 2005;25:1281–1290. doi: 10.1523/JNEUROSCI.4086-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schratt GM, Nigh EA, Chen WG, Hu L, Greenberg ME. BDNF regulates the translation of a select group of mRNAs by a mammalian target of rapamycin-phosphatidylinositol 3-kinase-dependent pathway during neuronal development. J Neurosci. 2004;24:7366–7377. doi: 10.1523/JNEUROSCI.1739-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg PH, Higuchi M, Sprengel R. RNA editing of brain glutamate receptor channels: mechanism and physiology. Brain Res Brain Res Rev. 1998;26:217–229. doi: 10.1016/s0165-0173(97)00062-3. [DOI] [PubMed] [Google Scholar]
- Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- Slipczuk L, Bekinschtein P, Katche C, Cammarota M, Izquierdo I, Medina JH. BDNF activates mTOR to regulate GluR1 expression required for memory formation. PLoS One. 2009;4:e6007. doi: 10.1371/journal.pone.0006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WB, Starck SR, Roberts RW, Schuman EM. Dopaminergic stimulation of local protein synthesis enhances surface expression of GluR1 and synaptic transmission in hippocampal neurons. Neuron. 2005;45:765–779. doi: 10.1016/j.neuron.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Sommer B, Köhler M, Sprengel R, Seeburg PH. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67:11–19. doi: 10.1016/0092-8674(91)90568-j. [DOI] [PubMed] [Google Scholar]
- Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002;25:578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P, Coadwell J, Hawkins PT. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science. 1998;279:710–714. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM. Dendritic protein synthesis, synaptic plasticity, and memory. Cell. 2006;127:49–58. doi: 10.1016/j.cell.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Takei N, Kawamura M, Hara K, Yonezawa K, Nawa H. Brain-derived neurotrophic factor enhances neuronal translation by activating multiple initiation processes: comparison with the effects of insulin. J Biol Chem. 2001;276:42818–42825. doi: 10.1074/jbc.M103237200. [DOI] [PubMed] [Google Scholar]
- Toker A, Newton AC. Cellular signaling: pivoting around PDK-1. Cell. 2000;103:185–188. doi: 10.1016/s0092-8674(00)00110-0. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Enslen H, Soderling TR. Characterization of a Ca2+/calmodulin-dependent protein kinase cascade. Molecular cloning and expression of calcium/calmodulin-dependent protein kinase kinase. J Biol Chem. 1995;270:19320–19324. doi: 10.1074/jbc.270.33.19320. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Inuzuka H, Ishikawa Y, Ikeda M, Saji I, Kobayashi R. STO-609, a specific inhibitor of the Ca(2+)/calmodulin-dependent protein kinase kinase. J Biol Chem. 2002;277:15813–15818. doi: 10.1074/jbc.M201075200. [DOI] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci. 2001;21:4249–4258. doi: 10.1523/JNEUROSCI.21-12-04249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdoorn TA, Burnashev N, Monyer H, Seeburg PH, Sakmann B. Structural determinants of ion flow through recombinant glutamate receptor channels. Science. 1991;252:1715–1718. doi: 10.1126/science.1710829. [DOI] [PubMed] [Google Scholar]
- Wang DO, Martin KC, Zukin RS. Spatially restricting gene expression by local translation at synapses. Trends Neurosci. 2010;33:173–182. doi: 10.1016/j.tins.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Kaech S, Grant WF, Davare M, Impey S, Tokumitsu H, Nozaki N, Banker G, Soderling TR. Regulation of axonal extension and growth cone motility by calmodulin-dependent protein kinase I. J Neurosci. 2004;24:3786–3794. doi: 10.1523/JNEUROSCI.3294-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, Soderling TR. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron. 2006;50:897–909. doi: 10.1016/j.neuron.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Wayman GA, Lee YS, Tokumitsu H, Silva A, Soderling TR. Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron. 2008;59:914–931. doi: 10.1016/j.neuron.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Cron P, Thompson V, Good VM, Hess D, Hemmings BA, Barford D. Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol Cell. 2002a;9:1227–1240. doi: 10.1016/s1097-2765(02)00550-6. [DOI] [PubMed] [Google Scholar]
- Yang J, Cron P, Good VM, Thompson V, Hemmings BA, Barford D. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat Struct Biol. 2002b;9:940–944. doi: 10.1038/nsb870. [DOI] [PubMed] [Google Scholar]
- Yano S, Tokumitsu H, Soderling TR. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature. 1998;396:584–587. doi: 10.1038/25147. [DOI] [PubMed] [Google Scholar]
- Zhou J, Du W, Zhou K, Tai Y, Yao H, Jia Y, Ding Y, Wang Y. Critical role of TRPC6 channels in the formation of excitatory synapses. Nat Neurosci. 2008;11:741–743. doi: 10.1038/nn.2127. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Esteban JA, Hayashi Y, Malinow R. Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat Neurosci. 2000;3:1098–1106. doi: 10.1038/80614. [DOI] [PubMed] [Google Scholar]