

Background: Mice lacking the neurosecretory protein Chromogranin A are obese, presumably because of resistance to catecholamines and leptin.
Results: Catestatin (CST) reduces adiposity, an effect likely mediated by restoring leptin sensitivity and modulating adrenergic signaling.
Conclusion: CST promotes lipolysis by blocking α-AR signaling and stimulating fatty acid oxidation.
Significance: We propose CST as a candidate antiobesity agent.
Keywords: Adipose Tissue Metabolism, Adrenergic Receptor, Catecholamines, Leptin Receptor, Lipids
Abstract
Chromogranin A knock-out (Chga-KO) mice display increased adiposity despite high levels of circulating catecholamines and leptin. Consistent with diet-induced obese mice, desensitization of leptin receptors caused by hyperleptinemia is believed to contribute to the obese phenotype of these KO mice. In contrast, obesity in ob/ob mice is caused by leptin deficiency. To characterize the metabolic phenotype, Chga-KO mice were treated with the CHGA-derived peptide catestatin (CST) that is deficient in these mice. CST treatment reduced fat depot size and increased lipolysis and fatty acid oxidation. In liver, CST enhanced oxidation of fatty acids as well as their assimilation into lipids, effects that are attributable to the up-regulation of genes promoting fatty acid oxidation (Cpt1α, Pparα, Acox, and Ucp2) and incorporation into lipids (Gpat and CD36). CST did not affect basal or isoproterenol-stimulated cAMP production in adipocytes but inhibited phospholipase C activation by the α-adrenergic receptor (AR) agonist phenylephrine, suggesting inhibition of α-AR signaling by CST. Indeed, CST mimicked the lipolytic effect of the α-AR blocker phentolamine on adipocytes. Moreover, CST reversed the hyperleptinemia of Chga-KO mice and improved leptin signaling as determined by phosphorylation of AMPK and Stat3. CST also improved peripheral leptin sensitivity in diet-induced obese mice. In ob/ob mice, CST enhanced leptin-induced signaling in adipose tissue. In conclusion, our results implicate CST in a novel pathway that promotes lipolysis and fatty acid oxidation by blocking α-AR signaling as well as by enhancing leptin receptor signaling.
Introduction
Chromogranin A (CHGA in humans, Chga in mice), a 48-kDa acidic secretory proprotein (1–3), gives rise to several peptides of biological importance, including the dysglycemic hormone pancreastatin (CHGA250–301) (4, 5), the vasodilator vasostatin (CHGA1–76) (6), and the antihypertensive peptide catestatin (CST; CHGA352–372),3 which inhibits nicotine-induced catecholamine release (7–9). Initially identified as a physiological brake in catecholamine secretion (7), CST has been established as a pleiotropic hormone having effects on promoting angiogenesis (10), lowering of blood pressure (8, 11, 12), and cardiac contractility (13–15), as well as enhancing baroreflex sensitivity (16, 17) and heart rate variability (18).
In addition to the above cardiovascular functions, CST has an antimicrobial activity (19, 20) and also regulates mast cell migration, cytokine production and release (21), smooth muscle cell proliferation (22), and monocyte migration (23). CST can act both extracellularly and intracellularly because the peptide can cross the cell membrane (24, 25).
Fat cell functions are regulated by catecholamines through four types of adrenergic receptors (AR): β1, β2, β3, and α2 (26, 27). Activation of the three β-ARs is positively coupled to adenylyl cyclase by stimulatory GTP-sensitive proteins, resulting in enhanced production of cyclic AMP. Cyclic AMP activates protein kinase A, which in turn phosphorylates hormone-sensitive lipase, leading to hydrolysis of triglycerides (lipolysis). In contrast, α2-AR activation has the opposite effects on lipolysis because it is coupled to inhibitory GTP-sensitive proteins (28–31). Therefore, the net action of catecholamines depends on the balance between β- and α-ARs (27). Sustained activation of sympathetic nervous system or increased plasma catecholamines is often associated with desensitization of β-AR (32). In vivo studies have shown that the lipolytic action of catecholamines is blunted in obese subjects (33, 34). Catecholamine-induced regulation of lipolysis through β-AR desensitization has also been demonstrated in vitro (32, 35). Repeated treatment with epinephrine results in the suppression of basal and epinephrine-stimulated lipolysis in lean and obese subjects (36). Even the in vivo lipolytic response to epinephrine is desensitized by prior exposure to epinephrine (37). In view of the above, we hypothesize that the increased fat mass in hyperadrenergic Chga-KO mice (38) reflects β-AR desensitization by increased plasma catecholamines (8). Because catecholamines are known to inhibit leptin secretion (39–41), β-AR desensitization may prevent such inhibition and lead to increased leptin level along with the increased adipose mass as found in Chga-KO mice and other obese models. Chronic hyperleptinemia in turn may desensitize leptin receptor and perpetuate the obese phenotype. Therefore, we reasoned that CST could break this cycle and reduce obesity by restoring AR and leptin receptor sensitivity through normalization of catecholamine and leptin levels. Indeed, we found that chronic CST administration to obese Chga-KO mice resulted in a dramatic lean phenotype. CST treatment also reduced body weight and adipose mass in DIO mice without reducing food intake. Interestingly, CST could enhance leptin effects on adipose tissue metabolism and signaling in both DIO and leptin-deficient ob/ob mice. Our findings suggest that the reduction in fat mass after chronic CST treatment is due to increased lipolysis and lipid mobilization through CST action on α2-AR and leptin receptor. In line with this, CST promoted fatty acid oxidation and leptin signaling.
EXPERIMENTAL PROCEDURES
Animals
Adult male (7 months old) WT (31.8 ± 1.2 g) and Chga-KO (39.2 ± 1.5 g) mice in the mixed genetic background (129SvJ × C57BL/6) were studied. Both genotypes were generated from the original founder carrying mixed genotype (50% 129SvJ, 50% C57BL/6) and were maintained by sibling mating. The animals were kept in a 12-h dark/light cycle and fed standard chow ad libitum. Male C57BL/6 mice, 8 weeks old, were fed a 60% high fat diet (D12492; Research Diets, Inc., New Brunswick, NJ) for 16 weeks before using for experiments. Male leptin-deficient (C57BL/6J-ob/ob) mice from the Jackson Laboratory were maintained on a standard chow diet. The institutional animal care and utilization committee approved all procedures. Chga-KO, DIO, and ob/ob mice were treated daily with saline or CST (5 μg/g of body weight intraperitoneally, for 12 days for Chga-KO mice and 16 days for DIO and ob/ob mice).
Measurement of Glycerol, Adipokine, Lipid, and CST Levels in Blood and in Conditioned Media
Mice were fasted for 12 h prior to blood draw. Triglycerides and nonesterified fatty acids (NEFA) were assayed using kits from Wako Diagnostics (Richmond, VA). Glycerol was assayed using a kit from Sigma. Media from the explant cultures and mouse serum were analyzed for glycerol and NEFA as a measure of lipolysis. ELISA kits were used to determine plasma levels of leptin, adiponectin (Millipore, Billerica, MA), and CST (Bachem, Torrance, CA). For CST assay, plasma samples and reference standards were passed through mini C18 columns, and the flow-through fractions were assayed. The same kits were used for measurements in culture media.
Treatment of Fat Pad Explants with CST and Leptin
Adipose tissue explants were prepared as described (42). Epididymal fat pads from 12-h fasted WT, Chga-KO, DIO, and ob/ob mice with or without CST treatment were collected in Krebs-Ringer phosphate buffer containing 10 mm Hepes and 0.5% BSA. The tissues were minced to 1–2 mm size and treated with 100 nm CST, 1 μm leptin, or saline for 30 min (for signaling analysis) or 3 h (for lipolysis and β-oxidation assays). Explants were also treated acutely with CST, leptin, a combination, or saline. At the end, incubation media were saved for analysis of glycerol and fatty acids. The explants were washed prior to homogenization for immunoblotting and analysis of fatty acid oxidation.
Preparation of Primary Adipocytes
Primary adipocytes were isolated from epididymal fat pads essentially as described (43). Adipose tissues from WT and Chga-KO mice were minced in Krebs-Ringer bicarbonate-Hepes buffer, pH 7.4, containing 10 mm bicarbonate, 30 mm Hepes, 200 nm adenosine, 2.5 mm glucose, and 1% fatty acid-free BSA, and digested for 30–40 min with Type I collagenase (10 mg/g tissue; Invitrogen) with gentle swirling in a 37 °C incubator. The digestion mixture was then filtered through a nylon strainer and centrifuged at 400 × g for 1 min. The oily layer (released from broken cells) above floating fat cells was skimmed off, and fat cells were recovered from the top and washed three times with warm Krebs-Ringer bicarbonate-Hepes.
Immunoblotting of Signaling Molecules
Adipose tissue explants after treatments ex vivo and tissues from mice treated in vivo were homogenized in a buffer containing phosphatase and protease inhibitors (20 mm Tris/HCl, pH 7.5, 250 mm sucrose, 2 mm EDTA, 2 mm EGTA, 2 mm Na3VO4, 10 mm NaF, 2 mm Na4P2O7, 1 mm PMSF, 20 μg/ml leupeptin, 10 μg/ml aprotinin, and 1 μm microcystin-LR) as described (38, 44). Homogenates were subjected to SDS-PAGE and immunoblotted. Primary antibodies for AMPK and Stat3 were from Cell Signaling Technology (Beverly, MA). The chemiluminescence kit was from Pierce.
Incorporation and Oxidation of Fatty Acid in Vivo
The mice were injected with saline or CST (5 μg/g of body weight intraperitoneally, twice daily) for 12 days. One hr after the final injection, [U-14C]palmitate (5 μCi, 100 μl, 0.2 mm) was injected intraperitoneally, and the mice were sacrificed 3 h later. Liver and adipose tissues (∼100 mg) were homogenized in 0.8 ml of 3.5 n perchloric acid and extracted by vortexing in 3 ml of a mixture of methanol and chloroform (2:1, v/v). To the final homogenate, 1.2 ml of 3.5 n perchloric acid was added. The mixture was vortexed and centrifuged. The lower (chloroform) layer contained all the lipids derived from the incorporation of [14C]palmitate, whereas the upper acidic layer contained partially oxidized acid-soluble metabolites of [14C]palmitate. The lower layer was further fractionated by thin layer chromatography on silica gel plates using a hexane:diethyl ether:acetic acid (79:20:1, v/v/v) mixture as the developing solvent. Lipogenesis from palmitate was determined based on the radioactivity of the free palmitic acid band compared with other lipids (phospholipids, triglycerides, diacylglycerol, etc.) on the TLC plate. Complete oxidation of [14C]palmitate was measured in cultured cells but not in mice.
Fatty Acid Oxidation in Explants and Cultured Cells
Oxidation of radiolabeled palmitate in the homogenates of adipose tissue explants as described previously (45). For oxidation studies of cultured cells, HepG2 (hepatocytes) and 3T3-L1 preadipocytes were obtained from ATCC and cultured following supplier's protocol. Confluent 3T3-L1 cultures were differentiated into adipocytes by treating with a mixture of dexamethasone (100 nm), isobutylmethylxanthine (1 μm) and insulin (100 nm) for 10 days. The media were then switched to serum-free DMEM with 1% BSA. Hepatocytes were assayed for lipogenesis and fat oxidation in response to CST treatment. Serum-free cultures were treated with CST (100 nm) for 24 h followed by a 2-h incubation with 0.2 mm [U-14C]palmitate (0.5 μCi/ml). Fatty acid oxidation in cells was determined by modifying a published method (46). Specifically, the culture media in 24-well plates were acidified with 10% HClO4 after incubation with the labeled fatty acid and immediately covered with a thick filter paper sheet (cut to size) soaked in 2 n NaOH and placed underneath the plastic lid. The whole plate was sealed with parafilm. After further incubation for 2 h, the filter paper sheet was marked as circles around the rim of the wells, and then the circles were excised. The filter discs were counted to determine the amount of 14CO2 absorbed in the papers. The cells in the culture wells were lysed in 1 n NaOH, and protein content was assayed using Folin's reagent (Bio-Rad).
Real Time PCR
RNA was extracted using a kit (RNeasy Plus; Qiagen) according to the manufacturer's specifications. After DNase digestion, 100 ng of RNA was transcribed into cDNA in a 20-μl reaction using a high capacity cDNA kit (Invitrogen), analyzed, and amplified. PCR was performed in a 25-μl reaction containing 5 μl of cDNA (one-fifth diluted), 2× SYBR Green PCR Master Mix, and the primers were described in Table 1 (400 nm each). Differences in cycle threshold values (ΔCt) between target and the housekeeping gene GAPDH were used to calculate the levels of expression.
TABLE 1.
Primers for quantitative RT-PCR
| Acox1 | Forward | GTC GAC CTT GTT CGC CA |
| Reverse | GGT TCC TCA GCA CGG CTT | |
| CD36 | Forward | TCC AGC CAA TGC CTT TGC |
| Reverse | TGG AGA ATT ACT TTT TCA GTG CAG AA | |
| Cpt1α | Forward | CAG GAT TTT GCT GTC AAC CTC |
| Reverse | GAG CAT CTC CAT GGC GTA G | |
| Gapdh | Forward | TAT GTC GTG GAG TCT ACT GGT GT |
| Reverse | GTC ATC ATA CTT GGC AGG TTT CT | |
| Gpat4 | Forward | TGT CTG GTT TGA GCG TTC TG |
| Reverse | TTC TGG GAA GAT GAG GAT GG | |
| Pparα | Forward | GGG CTC TCC CAC ATC CTT |
| Reverse | CCC ATT TCG GTA GCA GGT AGT C | |
| Pparγ1 | Forward | GAG TGT GAC GAC AAG ATT TG |
| Reverse | GGT GGG CCA GAA TGG CAT CT | |
| Srebp-1c | Forward | GGA GCC ATG GAT TGC ACA TT |
| Reverse | GCT TCC AGA GAG GAG GCC AG | |
| Ucp2 | Forward | CAG CCA GCG CCC AGT ACC |
| Reverse | CAA TGC GGA CGG AGG CAA AGC |
Statistics
The data are expressed as the means ± S.E. Curve fitting was accomplished using the program Kaleidagraph (Synergy Software, Reading, PA). Statistical analyses were done by Student's t test or one-way analysis of variance followed by Bonferroni's post hoc test. Statistical significance was defined as p < 0.05.
RESULTS
Effects of CST on Adiposity, Plasma Lipid, and Leptin Levels in Overweight Chga-KO Mice
Plasma CST concentration was 3.8 ng/ml in WT mice fed with a normal chow diet (Fig. 1A). Administration of CST (5 μg/g of body weight intraperitoneally/day for 12 days) to these mice raised CST concentration to 7.0 ng/ml (Fig. 1A), a level maintained for at least 4 h (data not shown). High fat diet (60% fat, for 16 weeks) decreased CST levels to 2.8 ng/ml, which increased to 5.8 ng/ml upon CST administration. CST administration to CST-deficient Chga-KO mice achieved a lower CST level (2.3 ng/ml) than WT mice. Chronic CST administration (5 μg/g of body weight intraperitoneally/day for 12 days) to Chga-KO mice reduced epididymal fat pad size to WT level (∼25% reduction with respect to body weight of Chga-KO mice) without affecting body weight (Fig. 1, B and C) or liver weight (saline: 1.20 ± 0.08 g versus CST: 1.27 ± 0.07 g). Interestingly, CST decreased plasma triglyceride levels in Chga-KO mice (Fig. 1D). This decrease in overall lipid content may be caused in part by increased lipolysis as shown by increased glycerol and NEFA levels in plasma (Fig. 1, E and F). We found that Chga-KO mice have higher leptin levels than WT (Fig. 1G), consistent with the established consequence of pancreastatin deficiency in the Chga-KO mice (38). Interestingly, CST treatment of Chga-KO mice lowered plasma leptin to a level below WT (Fig. 1G), suggesting that leptin at subphysiological levels is sufficient to maintain the Chga-KO mice in a lean state. CST also inhibited leptin production in cultured 3T3-L1 adipocytes (Fig. 1H), suggesting a direct effect on leptin secretion independent of other circulating factors. Although leptin is known to facilitate fat oxidation and decrease food intake (47, 48), sustained hyperleptinemia may desensitize its receptor and lead to obesity as seen in DIO models (49, 50). We therefore reasoned that CST restored leptin action in Chga-KO mice by reversing the desensitization effect of chronic leptin excess. CST did not increase food intake of Chga-KO mice (data not shown), suggesting that CST, despite lowering leptin levels, preserved leptin signaling in the brain. Leptin-deficient ob/ob mice with sensitive leptin receptors responded to short term CST treatment by reducing food intake, whereas DIO mice, with peripheral leptin resistance and with a barrier against circulating leptin for hypothalamic action, failed to respond (as discussed later in Fig. 6, A and B).
FIGURE 1.

Plasma parameters of WT and Chga-KO mice treated with saline or CST. A, plasma CST from saline-treated (Sal) or CST-treated (5 μg/g of body weight intraperitoneally/day) WT mice (28 week old) on normal chow (NCD) and high fat diet (HFD, 60% fat for 16 weeks), as well as from Chga-KO mice (28 week old) on normal chow diet. B–G, body weights (B) and epididymal fat pad size normalized to body weight (C) in 28-week-old WT and Chga-KO mice treated with saline or CST for 12 days. Plasma triglyceride (D), glycerol (E), NEFA (F), and leptin (G) in 28-week-old WT and Chga-KO mice after treatment with saline or CST for 12 days. H, leptin in adipocyte culture media after treatment with saline or CST.
FIGURE 6.
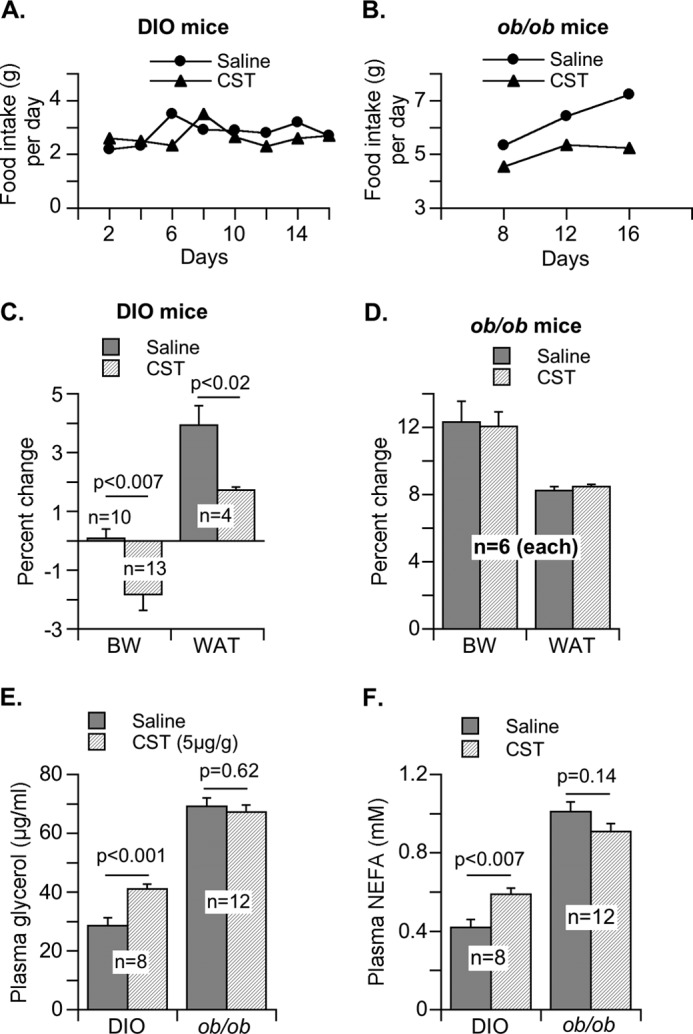
Effects of CST administration to DIO and ob/ob mice on food intake, body weight, adipose tissue weight, and lipolysis. CST (5 μg/g of body weight intraperitoneally/day) or saline was administered to DIO and ob/ob mice for 16 days. A and B, food intake for DIO mice (A) and ob/ob mice (B) was measured every other day. C and D, percentages of change in final body weight (BW) and white adipose tissue (WAT) mass (as % of body weight) were determined for DIO (C) and ob/ob (D) mice. E and F, plasma glycerol (E) and NEFA (F) were quantified for DIO and ob/ob mice at the end of CST treatment.
Effects of CST on Lipogenesis, Fatty Acid Oxidation, and Gene Expression in Chga-KO Mice
In Chga-KO mice treated with CST, we found tissue-specific effects on [14C]palmitate incorporation into lipids. The incorporation was decreased by CST in adipose tissue but enhanced in liver (Fig. 2, A and B). In contrast, CST stimulated palmitate oxidation into acid-soluble metabolites in both adipose tissue and liver (Fig. 2, C and D). The effect of CST on [14C]palmitate oxidation in cultured hepatocytes (HepG2) and adipocytes (3T3-L1) was measured based on 14CO2 formation (Fig. 2E). Given that adipose tissue in CST-treated mice showed increased palmitate oxidation but decreased incorporation into lipids, we conclude that CST inhibits the expansion of adipose tissue and also promotes fatty acid uptake in liver for oxidation. Liver mRNA analyses revealed that CST augmented the expression of carnitine palmitoyltransferase 1α (Cpt1α), peroxisome proliferator-activated receptor-α (Pparα), acyl-CoA oxidase 1 (Acox1), and uncoupling protein 2 (Ucp2) genes involved in fatty acid oxidation (Fig. 3, A–D). In contrast, CST had no effect on the expression of lipogenic genes such as sterol regulatory element-binding protein 1 (Srebp-1) and peroxisome proliferator-activated receptor-γ (Pparγ) (Fig. 3E). Interestingly, CST stimulated the expression of cluster of differentiation 36 (CD36), a transporter mediating cellular uptake of long chain fatty acids, as well as the lipogenic gene glycerol-3-phosphate acyltransferase (Gpat4) (Fig. 3E). This indicates that CST stimulates fatty acid incorporation into triglycerides but not de novo lipogenesis. Overall, CST appears to promote lipid flux from adipose tissue toward liver for catabolism.
FIGURE 2.
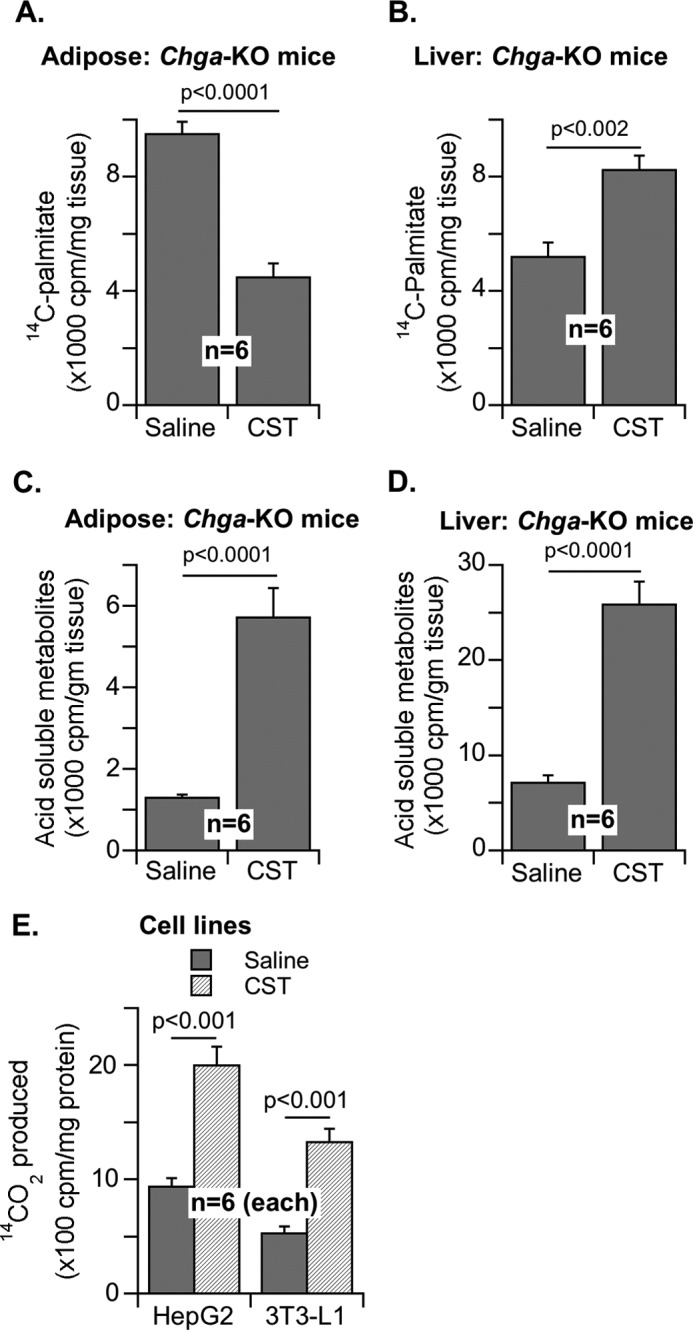
Effects of CST on lipogenesis from fatty acid and fatty acid oxidation in hepatic and adipose tissues. A and B, incorporation of [14C]palmitate into lipids in adipose (A) and liver (B) in Chga-KO mice after saline or CST treatment (5 μg/g of body weight intraperitoneally/day) for 12 days. C and D, partial oxidation to acid-soluble metabolites in adipose tissue (C) and liver (D). E, complete oxidation to 14CO2 in hepatocytes (HepG2) and adipocytes (3T3-L1) after saline or CST treatment.
FIGURE 3.
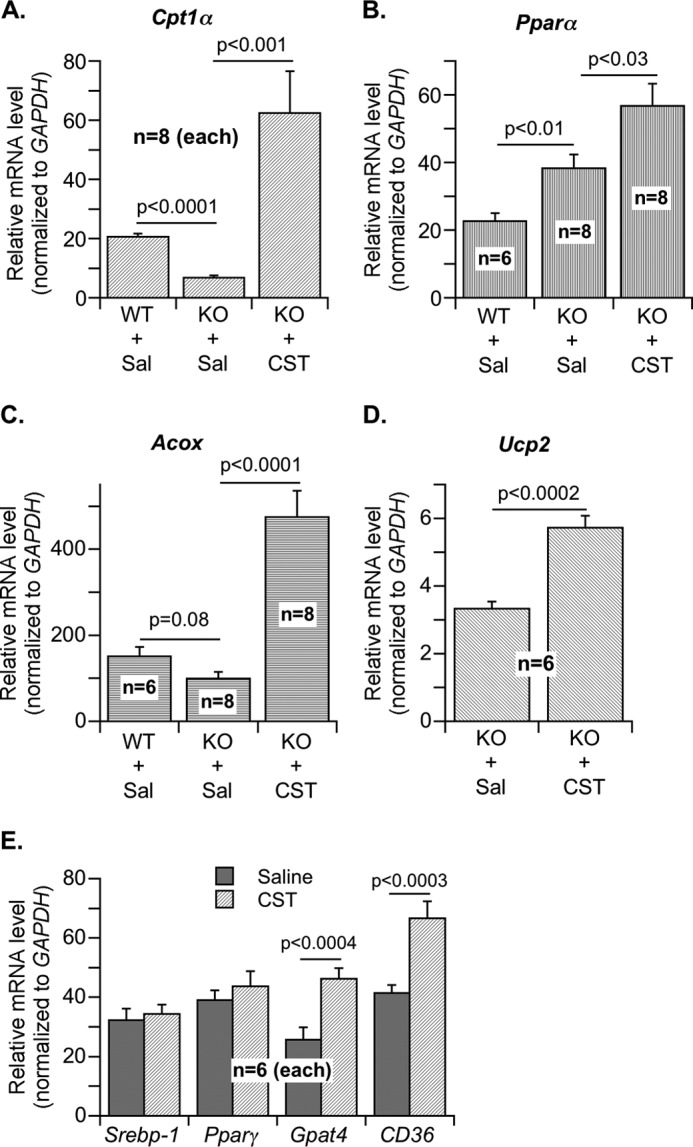
Effects of CST on hepatic expression of genes involved in lipogenesis and fatty acid oxidation. A–D, relative mRNA expression of genes involved in fatty acid oxidation in liver of WT and Chga-KO mice after treatment with saline (Sal) or CST (5 μg/g of body weight intraperitoneally/day) for 12 days. A, carnitine palmitoyltransferase 1α (Cpt1α). B, peroxisome proliferator-activated receptor-α (Pparα). C, acyl-CoA oxidase 1 (Acox1). D, uncoupling protein 2 (Ucp2). E, relative expression of genes involved in lipogenesis in liver of Chga-KO mice after treatment with saline or CST (5 μg/g of body weight, intraperitoneally/day) for 12 days: sterol regulatory element-binding protein 1 (Srebp-1), peroxisome proliferator-activated receptor-γ (Pparγ), glycerol-3-phosphate acyltransferase (Gpat4), and cluster of differentiation 36 (CD36).
Modulation of Adrenergic Receptor-mediated Lipolysis by CST in Adipocytes
We examined the direct effects of CST in cultured cells. In both 3T3-L1 adipocytes and PC-12 neuroendocrine cells, cAMP production was stimulated by isoproterenol but not by CST (Fig. 4A), indicating that CST did not stimulate β-AR signaling. In contrast, CST attenuated phospholipase C (PLC) activation by both phenylephrine (an α-AR agonist) and epinephrine in 3T3-L1 adipocytes (Fig. 4B). Because epinephrine activates both α- and β-AR, the inhibition of its effect on PLC by CST might represent selective inhibition of α-AR. Of note, CST itself mildly stimulated PLC but inhibited the stimulatory effect of α-AR agonists (Fig. 4B), suggesting that PLC activation by CST itself is α-AR-independent.
FIGURE 4.
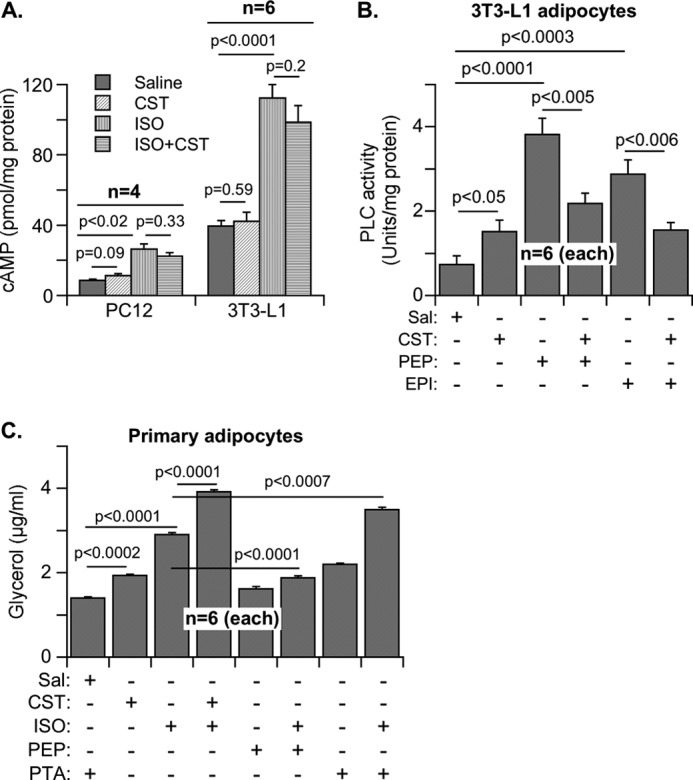
Regulation of adrenergic signaling by CST as measured by cAMP production, phospholipase C activity, and lipolytic glycerol release. A, effects of CST (100 nm, 10 min) on cAMP production in PC-12 and 3T3-L1 cells pretreated with saline (Sal) or isoproterenol (ISO) (10 μm) for 10 min. B, effects of CST on phospholipase C activity in 3T3-L1 adipocytes pretreated with phenylephrine or epinephrine (10 μm for 10 min). C, effects of CST on glycerol release from adipocytes pretreated with ISO, phenylephrine (PEP), and phentolamine (PTA) (10 μm for 10 min).
As in Chga-KO mice, CST also inhibited leptin release from 3T3-L1 adipocytes (Fig. 1H) and stimulated glycerol release from primary adipocytes (Fig. 4C). Consistent with the literature (30), we found that in adipocytes, the α-AR antagonist phentolamine stimulated lipolysis and potentiated the lipolytic effects of the β-AR agonist isoproterenol (Fig. 4C). In contrast, the α-AR agonist phenylephrine dampened the lipolytic effect of isoproterenol (Fig. 4C). Both the α-antagonist phentolamine and CST potentiated the effects of isoproterenol (Fig. 4C). These findings suggest that CST recapitulates the lipolytic effect of the α-AR antagonist phentolamine. This commonality of CST with phentolamine, coupled with its ability to inhibit phenylephrine action (Fig. 4B), suggests that CST acts by suppressing α-AR signaling.
CST Resensitizes Chga-KO Mice to Leptin
Leptin signals through AMPK and MAPK pathways and activates the transcription factor Stat3 (51–53). Chronic elevation of plasma leptin level causes desensitization of its receptor, leading to attenuation of Stat3 phosphorylation (49, 50). Acute CST treatment of adipose tissue explants from Chga-KO mice stimulated AMPK phosphorylation (Fig. 5A), an effect likely independent of leptin action because similar stimulation was also seen in cultured hepatocytes. Leptin signaling appeared to be subdued in Chga-KO mice as evidenced by the decreased phosphorylation of AMPK and Stat3 compared with WT following acute leptin treatment (Fig. 5, B and D). CST treatment restored the ability of leptin to stimulate the phosphorylation of AMPK (Fig. 5C) as well as Stat3 (Fig. 5D), suggesting that CST-induced lowering of plasma leptin in Chga-KO mice might have resensitized leptin receptor.
FIGURE 5.
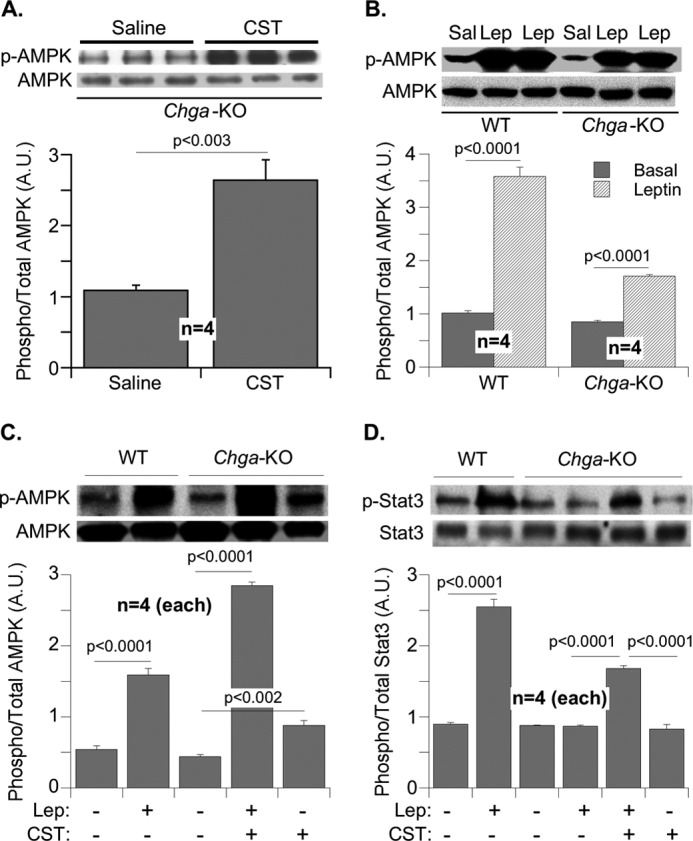
Regulation of AMPK and Stat3 signaling by CST in adipose tissue explants. A, explants from Chga-KO mice were treated with saline (Sal) or CST (100 nm for 30 min) and immunoblotted for pAMPK and AMPK. B, adipose explants from WT and Chga-KO mice were immunoblotted for pAMPK and AMPK after treatment with saline or leptin (Lep, 1 μm) for 30 min. C and D, pAMPK and AMPK (C) and pStat3 and Stat3 (D) signaling in adipose explants from WT and Chga-KO mice after treatment for 30 min with saline, CST (100 nm), leptin (1 μm), or leptin plus CST.
Modulation of Peripheral Leptin Action by CST in Leptin-resistant DIO Mice and Leptin-deficient ob/ob Mice with Sensitive Leptin Receptors
In contrast to insulin-sensitive Chga-KO mice, DIO and ob/ob mice are insulin-resistant and obese. However, DIO mice exhibit peripheral leptin resistance (54–56), whereas ob/ob mice maintain functional leptin receptors and full responsiveness to exogenous leptin (57–60). Because our focus in this work has been to study the regulation of lipid metabolism and leptin action by CST in adipose tissue, not in hypothalamus, we examined their effects ex vivo in adipose tissue explants. Adipose tissues from these two models of obesity, DIO and ob/ob mice, offer the opportunity to further clarify our observations in Chga-KO mice. When CST was administered to DIO and ob/ob mice for 16 days, food intake in DIO mice did not change, but there was a distinct indication that food intake in ob/ob mice started to level off (Fig. 6, A and B). Interestingly, decreased food intake by ob/ob mice was not reflected in any decrease in body weight or adipose tissue mass, whereas CST treatment for 16 days decreased body weight and adipose mass in DIO mice without a change in food intake (Fig. 6, C and D). Similarly, lipolysis as measured by the plasma concentrations of glycerol and NEFA was not affected in ob/ob mice but was increased by CST treatment in DIO mice (Fig. 6, E and F). From this experiment it appears that in terms of food intake during the treatment period, CST might have produced a central effect in leptin-sensitive ob/ob mice but not in leptin-resistant DIO mice. It is likely that a longer treatment with CST will be necessary to manifest CST effect in ob/ob mice and to translate the observed decrease in food intake into changes in body weight, tissue size, and overall metabolism. However, increased lipolysis and decreased body weight and adipose tissue mass indicated a significant peripheral effect in DIO mice. We observed that an average of 1.1 g (2%) decrease in body weight was accompanied with an approximately 2-g decrease in adipose tissue mass.
CST and Leptin Effects on Adipose Explants of DIO and ob/ob Mice with or without Prior CST Treatment in Vivo
Although leptin-deficient ob/ob mice possess functional leptin receptors and maintain leptin sensitivity (57–60), DIO mice develop peripheral leptin resistance but maintain partial sensitivity to centrally administered leptin (54–56). To examine the direct effects of leptin on adipose tissue lipolysis and fatty acid oxidation, and the influence of CST on leptin action, we treated DIO and ob/ob mice with CST or saline and exposed adipose explants to leptin for 30 min (for AMPK and Stat3 signaling) or 3 h (for lipolysis and fatty acid oxidation analysis). After incubation with leptin, an analysis of glycerol and NEFA released into the media demonstrated that although the explants from CST-treated DIO mice (exposed to both CST and leptin in vivo) can release some glycerol in the media, the greatest lipolytic response was produced when leptin was added to the cultures of CST-treated explants (Fig. 7A). CST effects on NEFA release by the explants from CST-treated DIO mice was not significant. It is possible that the released NEFA might have undergone further metabolism during the 3-h incubation. Nevertheless, the addition of leptin to the cultures of CST-treated explants resulted in augmented release of NEFA (Fig. 7B). It should be noted that leptin treatment did not stimulate lipolysis of explants from saline-treated mice (Fig. 7, A and B). These findings suggest that leptin resistance exists in adipose tissues of DIO mice and that prior CST treatment in vivo might have improved leptin receptor functions. In contrast to DIO mice, 16 days of CST administration alone to ob/ob mice did not influence lipolysis in the explants, whereas the addition of leptin to the cultures of explant from saline-treated ob/ob mice (CST naive) stimulated lipolysis (Fig. 7, C and D), suggesting that (i) functional leptin receptors were present in the adipose tissue explants from ob/ob mice and (ii) CST did not directly influence leptin receptor functions. However, adding leptin to the incubation with CST-treated explants from ob/ob mice produced the highest lipolytic response (Fig. 7, C and D). The regulation of palmitate oxidation by the explants in response to CST and leptin followed a pattern similar to lipolysis. Specifically, neither CST nor leptin alone stimulated oxidation in explants from DIO mice, but the combination had a stimulatory effect (Fig. 7E), and the treatment with leptin alone (not CST alone) was stimulatory for oxidation in ob/ob explants, but the combination showed highest oxidation (Fig. 7F).
FIGURE 7.
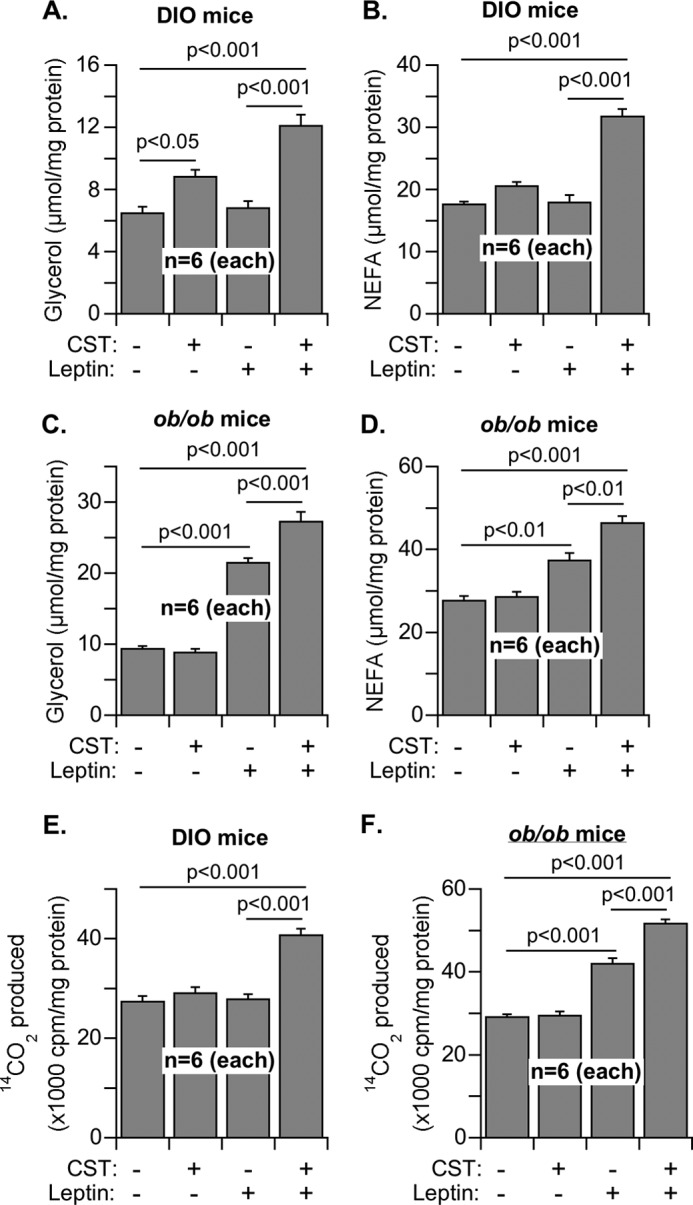
Lipolysis and fatty acid oxidation in the adipose tissue explants of DIO and ob/ob mice after treatment with saline or CST for 16 days. A–D, explants were incubated with saline or leptin (1 μm) for 3 h, and the concentrations of glycerol (A and C) and NEFA (B and D) released into the media from DIO (A and B) and ob/ob (C and D) explants were determined as a measure of lipolysis. E and F, homogenates of the explants from DIO (E) and ob/ob (F) were used to determine their capacity for oxidation of [U-14C]palmitate in response to the treatment with saline, CST, leptin, and CST + leptin. The 14CO2 released was captured and counted as the measure of fatty acid oxidation.
Leptin treatment alone did not stimulate Stat3 or AMPK phosphorylation in DIO explants treated with saline (Fig. 8, A and B). The treatment of DIO explants with CST in vivo stimulated the phosphorylation of Stat3 but not AMPK. It appears that CST may have a direct, leptin-independent effect on Stat3 phosphorylation. Sequential treatment with CST (in vivo) and leptin (ex vivo) resulted in the highest phosphorylation of both Stat3 and AMPK (Fig. 8, A and B). In ob/ob explants, the treatments with leptin alone stimulated phosphorylation of both Stat3 and AMPK (Fig. 8, C and D). Again, the treatment with a combination of leptin and CST showed the highest response. The CST stimulation of Stat3 phosphorylation in insulin-resistant models (DIO and ob/ob mice, Fig. 8) should be contrasted with the CST effects in insulin-sensitive Chga-KO mice (Fig. 5). The lack of CST stimulation of Stat3 phosphorylation in the explants from Chga-KO mice (Fig. 5D) may represent the effects of other missing Chga peptides in the Chga-KO mice that gave rise to the increased insulin sensitivity in those mice. Conversely, CST alone significantly stimulated pAMPK signaling in Chga-KO mice (Fig. 5A). It suggests that other Chga-derived peptides could have a suppressive effect on AMPK signaling stimulated by CST.
FIGURE 8.
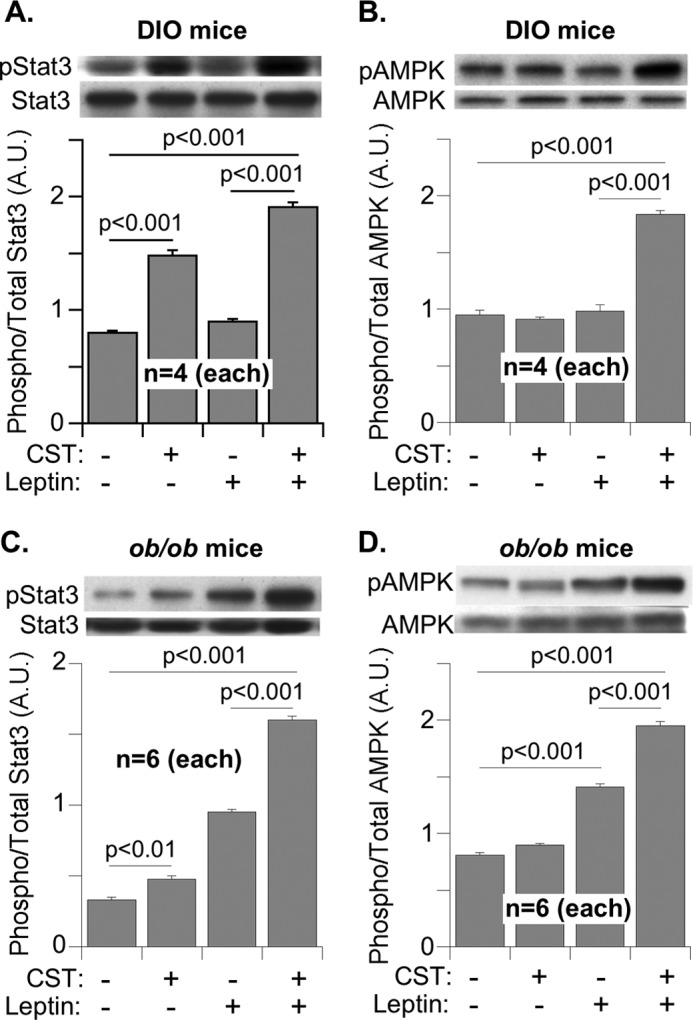
Stat3 and AMPK signaling in the adipose tissue explants from DIO and ob/ob mice treated with saline, CST (in vivo), and leptin (ex vivo). Explants from DIO mice (A and B) and ob/ob mice (C and D) were incubated with saline or leptin (1 μm) for 30 min, homogenized, and immunoblotted for phospho-Stat3 and total Stat3 (A and C) as well as phospho-AMPK and total AMPK signals (B and D). The results are presented as the ratio of signal strength of phosphorylated over total.
DISCUSSION
In this study, we found a novel CST function that reduces adiposity and mobilizes lipids from fat depot. These CST effects were examined in Chga-KO mice, where the lack of endogenous CST provided an ideal background for demonstrating the effects of exogenous CST. Another advantage of these mice over WT is their expanded adiposity on a regular chow diet, obviating the need for diet-induced obesity. Moreover, their circulating catecholamine levels are higher than WT (38) and desensitize adipose tissue to catecholamine-induced lipolysis. Likewise, Chga-KO mice also possess higher than WT plasma levels of leptin, adiponectin, and ketone bodies (38), yet their adiposity was not reduced despite increased lipid oxidation. These observations suggested that Chga-KO mice might be resistant to hormones such as catecholamines and adipokines and that alleviation of the resistance could potentially explain the metabolic effect of exogenous CST. In fact, CST treatment in Chga-KO mice not only lowered high levels of circulating catecholamines and leptin (38) but also reduced adipose tissue size by ∼25% (Fig. 1C).
It appears that CST promotes lipolysis in adipose tissue (Fig. 1E) as well as fatty acid uptake and oxidation in liver (Fig. 2). In other words, CST treatment created an environment where lipolytic products (glycerol and fatty acids) were not re-esterified for storage. Therefore, its net metabolic effect is to favor lipid disposal. Of note, the lipid disposal promoted by CST was not mediated by increased catecholamine or leptin release. On the contrary, CST inhibits catecholamine release (7, 61, 62) and leptin production (Fig. 1G).
Because CST did not modulate basal or isoproterenol-induced cAMP levels in PC-12 cells (Fig. 4A), its lipolytic effect is probably not mediated by β-AR signaling. Instead, inhibition of α-AR signaling may underlie the lipolytic effect of CST given its ability to prevent phenylephrine from activating PLC. Existing literature indicates that α-AR signaling inhibits lipolysis, whereas α-AR blockade potentiates the lipolytic effect of β-AR signaling (29, 30, 63, 64). Acting like an α-AR antagonist, CST enhanced the lipolytic effect of β-AR agonists (Fig. 4C). Under physiological conditions, α-AR dominates over β-AR, leading to overall lipogenesis (28). Therefore, α-AR inhibition by CST might have shifted the balance toward lipolysis.
In obese states, increased circulating levels of leptin cause desensitization of its receptors, resulting in failure of leptin to reduce food intake and promote lipid oxidation (49, 50). In this context, the ability of CST to decrease leptin production and minimize chronic overexposure might have restored leptin sensitivity in brain and peripheral tissues. Therefore, the net result was increased oxidation of lipolytic product (NEFA). Leptin signaling through AMPK and Stat3 in Chga-KO mice following acute leptin treatment was subdued compared with WT. CST treatment restored leptin action in Chga-KO mice, suggesting resensitization of the leptin receptor.
To further clarify the interactions between CST and leptin pathways and to establish CST as an antiobesity factor, we examined the effects of CST in leptin-resistant DIO mice and leptin-deficient ob/ob mice. DIO mice are known to exhibit peripheral leptin resistance (54–56). We observed leptin resistance in adipose tissue explants from DIO mice where acute leptin treatment did not stimulate lipolysis, β-oxidation, or phosphorylation of Stat3 and AMPK. However, prior CST treatment of DIO mice for 16 days led to the sensitization of all acute leptin effects in the adipose tissue explants, suggesting sensitization of leptin receptor-like functions in adipose tissue. CST administration to DIO mice did not reduce food intake but actually caused a modest reduction in body weight proportional to the loss of adipose mass. As a result, the products of lipolysis, glycerol and NEFA, were increased in serum.
Unlike DIO mice, leptin-deficient ob/ob mice maintain leptin sensitivity (57–60). As a result, acute treatment of adipose explants with leptin stimulated lipolysis, fatty acid oxidation, and phosphorylation of Stat3 and AMPK. The leptin effects were enhanced in the explants from CST treated ob/ob mice, whereas CST treatment alone did not show significant peripheral effects. It should be noted that the treatment of ob/ob mice with CST for 16 days started to reduce food intake (by 25%) resembling a leptin-like effect. This is in contrast to DIO mice, where CST treatment did not reduce food intake. One of the reasons for the failure of leptin to reduce food intake by activating hypothalamic leptin receptors in DIO mice is limited delivery of circulating leptin across the blood-brain barrier. When administered through the intracerebroventricular route, leptin could activate hypothalamic receptor signaling in DIO mice (54–56). It is therefore highly possible that like leptin, intracerebroventricular administration of CST might reduce food intake in DIO mice and enhance hypothalamic leptin response. Administration of CST to DIO mice by the intraperitoneal route, on the other hand, could generate only peripheral response and improve peripheral leptin sensitivity. It is also possible that CST acted centrally in ob/ob mice to reduce food intake, but longer treatment is required (6–8 weeks instead of 16 days) to initiate a reduction of body weight and adipose tissue mass. We will address these possibilities in a future work. Restoration of leptin-mediated AMPK signaling and fatty acid oxidation in DIO mice by CST with concommitant reduction in body weight and fat mass suggests a crucial physiological role of CST in fine-tuning lipid metabolism to prevent obesity. We have also seen that CST stimulates lipolysis by antagonizing α-AR functions. Is there an inverse relationship between α-AR and leptin receptor functions where CST could be a key regulator? We will explore these possibilities in future. In conclusion, our data support an essential role of the endogenous bioactive peptide CST in restoring homeostasis during metabolic disorders by controlling catecholamine release and lipid disposal via modulation of adrenergic and leptin signaling.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 DA011311 (to S. K. M.). This work was also supported by grants from the Department of Veterans Affairs (to S. K. M., D. T. O., and N.-W. C.).
- CST
- catestatin
- DIO
- diet-induced obese
- AR
- adrenergic receptor
- NEFA
- nonesterified fatty acid(s)
- PLC
- phospholipase C
- AMPK
- 5′-adenosine-monophosphate-activated protein kinase
- Stat3
- signal transducer and activator of transcription 3.
REFERENCES
- 1. Winkler H., Fischer-Colbrie R. (1992) The chromogranins A and B. The first 25 years and future perspectives. Neuroscience 49, 497–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Taupenot L., Harper K. L., O'Connor D. T. (2003) The chromogranin-secretogranin family. New Engl. J. Med. 348, 1134–1149 [DOI] [PubMed] [Google Scholar]
- 3. Montero-Hadjadje M., Vaingankar S., Elias S., Tostivint H., Mahata S. K., Anouar Y. (2008) Chromogranins A and B and secretogranin II. Evolutionary and functional aspects. Acta Physiol. (Oxf.) 192, 309–324 [DOI] [PubMed] [Google Scholar]
- 4. Tatemoto K., Efendić S., Mutt V., Makk G., Feistner G. J., Barchas J. D. (1986) Pancreastatin, a novel pancreatic peptide that inhibits insulin secretion. Nature 324, 476–478 [DOI] [PubMed] [Google Scholar]
- 5. Sánchez-Margalet V., González-Yanes C., Najib S., Santos-Alvarez J. (2010) Metabolic effects and mechanism of action of the chromogranin A-derived peptide pancreastatin. Regul. Pept. 161, 8–14 [DOI] [PubMed] [Google Scholar]
- 6. Aardal S., Helle K. B., Elsayed S., Reed R. K., Serck-Hanssen G. (1993) Vasostatins, comprising the N-terminal domain of chromogranin A, suppress tension in isolated human blood vessel segments. J. Neuroendocrinol. 5, 405–412 [DOI] [PubMed] [Google Scholar]
- 7. Mahata S. K., O'Connor D. T., Mahata M., Yoo S. H., Taupenot L., Wu H., Gill B. M., Parmer R. J. (1997) Novel autocrine feedback control of catecholamine release. A discrete chromogranin a fragment is a noncompetitive nicotinic cholinergic antagonist. J. Clin. Invest. 100, 1623–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mahapatra N. R., O'Connor D. T., Vaingankar S. M., Hikim A. P., Mahata M., Ray S., Staite E., Wu H., Gu Y., Dalton N., Kennedy B. P., Ziegler M. G., Ross J., Mahata S. K. (2005) Hypertension from targeted ablation of chromogranin A can be rescued by the human ortholog. J. Clin. Invest. 115, 1942–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mahata S. K., Mahata M., Fung M. M., O'Connor D. T. (2010) Catestatin. A multifunctional peptide from chromogranin A. Regul. Pept. 162, 33–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Theurl M., Schgoer W., Albrecht K., Jeschke J., Egger M., Beer A. G., Vasiljevic D., Rong S., Wolf A. M., Bahlmann F. H., Patsch J. R., Wolf D., Schratzberger P., Mahata S. K., Kirchmair R. (2010) The neuropeptide catestatin acts as a novel angiogenic cytokine via a basic fibroblast growth factor-dependent mechanism. Circ. Res. 107, 1326–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fung M. M., Salem R. M., Mehtani P., Thomas B., Lu C. F., Perez B., Rao F., Stridsberg M., Ziegler M. G., Mahata S. K., O'Connor D. T. (2010) Direct vasoactive effects of the chromogranin A (CHGA) peptide catestatin in humans in vivo. Clin. Exp. Hypertens. 32, 278–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gaede A. H., Pilowsky P. M. (2012) Catestatin, a chromogranin A-derived peptide, is sympathoinhibitory and attenuates sympathetic barosensitivity and the chemoreflex in rat CVLM. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302, R365–R372 [DOI] [PubMed] [Google Scholar]
- 13. Angelone T., Quintieri A. M., Brar B. K., Limchaiyawat P. T., Tota B., Mahata S. K., Cerra M. C. (2008) The antihypertensive chromogranin a peptide catestatin acts as a novel endocrine/paracrine modulator of cardiac inotropism and lusitropism. Endocrinology 149, 4780–4793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mazza R., Gattuso A., Mannarino C., Brar B. K., Barbieri S. F., Tota B., Mahata S. K. (2008) Catestatin (chromogranin A344–364) is a novel cardiosuppressive agent. Inhibition of isoproterenol and endothelin signaling in the frog heart. Am. J. Physiol. Heart Circ. Physiol. 295, H113–H122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Imbrogno S., Garofalo F., Cerra M. C., Mahata S. K., Tota B. (2010) The catecholamine release-inhibitory peptide catestatin (chromogranin A344–363) modulates myocardial function in fish. J. Exp. Biol. 213, 3636–3643 [DOI] [PubMed] [Google Scholar]
- 16. Gayen J. R., Gu Y., O'Connor D. T., Mahata S. K. (2009) Global disturbances in autonomic function yield cardiovascular instability and hypertension in the chromogranin a null mouse. Endocrinology 150, 5027–5035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gaede A. H., Pilowsky P. M. (2010) Catestatin in rat RVLM is sympathoexcitatory, increases barosensitivity, and attenuates chemosensitivity and the somatosympathetic reflex. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299, R1538–R1545 [DOI] [PubMed] [Google Scholar]
- 18. Dev N. B., Gayen J. R., O'Connor D. T., Mahata S. K. (2010) Chromogranin a and the autonomic system. Decomposition of heart rate variability and rescue by its catestatin fragment. Endocrinology 151, 2760–2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Briolat J., Wu S. D., Mahata S. K., Gonthier B., Bagnard D., Chasserot-Golaz S., Helle K. B., Aunis D., Metz-Boutigue M. H. (2005) New antimicrobial activity for the catecholamine release-inhibitory peptide from chromogranin A. Cell Mol. Life Sci. 62, 377–385 [DOI] [PubMed] [Google Scholar]
- 20. Radek K. A., Lopez-Garcia B., Hupe M., Niesman I. R., Elias P. M., Taupenot L., Mahata S. K., O'Connor D. T., Gallo R. L. (2008) The neuroendocrine peptide catestatin is a cutaneous antimicrobial and induced in the skin after injury. J. Invest. Dermatol. 128, 1525–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aung G., Niyonsaba F., Ushio H., Kajiwara N., Saito H., Ikeda S., Ogawa H., Okumura K. (2011) Catestatin, a neuroendocrine antimicrobial peptide, induces human mast cell migration, degranulation and production of cytokines and chemokines. Immunology 132, 527–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guo X., Zhou C., Sun N. (2011) The neuropeptide catestatin promotes vascular smooth muscle cell proliferation through the Ca2+-calcineurin-NFAT signaling pathway. Biochem. Biophys. Res. Commun. 407, 807–812 [DOI] [PubMed] [Google Scholar]
- 23. Egger M., Beer A. G., Theurl M., Schgoer W., Hotter B., Tatarczyk T., Vasiljevic D., Frauscher S., Marksteiner J., Patsch J. R., Schratzberger P., Djanani A. M., Mahata S. K., Kirchmair R. (2008) Monocyte migration. A novel effect and signaling pathways of catestatin. Eur. J. Pharmacol. 598, 104–111 [DOI] [PubMed] [Google Scholar]
- 24. Sugawara M., Resende J. M., Moraes C. M., Marquette A., Chich J. F., Metz-Boutigue M. H., Bechinger B. (2010) Membrane structure and interactions of human catestatin by multidimensional solution and solid-state NMR spectroscopy. FASEB J. 24, 1737–1746 [DOI] [PubMed] [Google Scholar]
- 25. Helle K. B. (2010) The chromogranin A-derived peptides vasostatin-I and catestatin as regulatory peptides for cardiovascular functions. Cardiovasc. Res. 85, 9–16 [DOI] [PubMed] [Google Scholar]
- 26. Arner P. (1999) Catecholamine-induced lipolysis in obesity. Int. J. Obes. Relat. Metab. Disord. 23, (Suppl. 1) 10–13 [DOI] [PubMed] [Google Scholar]
- 27. Arner P. (2005) Human fat cell lipolysis. Biochemistry, regulation and clinical role. Best Pract. Res. Clin. Endocrinol. Metab. 19, 471–482 [DOI] [PubMed] [Google Scholar]
- 28. Lafontan M., Barbe P., Galitzky J., Tavernier G., Langin D., Carpene C., Bousquet-Melou A., Berlan M. (1997) Adrenergic regulation of adipocyte metabolism. Hum. Reprod. 12, (Suppl. 1) 6–20 [DOI] [PubMed] [Google Scholar]
- 29. Stich V., de Glisezinski I., Crampes F., Suljkovicova H., Galitzky J., Riviere D., Hejnova J., Lafontan M., Berlan M. (1999) Activation of antilipolytic α2-adrenergic receptors by epinephrine during exercise in human adipose tissue. Am. J. Physiol. 277, R1076–1083 [DOI] [PubMed] [Google Scholar]
- 30. Stich V., Pelikanova T., Wohl P., Sengenès C., Zakaroff-Girard A., Lafontan M., Berlan M. (2003) Activation of α2-adrenergic receptors blunts epinephrine-induced lipolysis in subcutaneous adipose tissue during a hyperinsulinemic euglycemic clamp in men. Am. J. Physiol. Endocrinol. Metab. 285, E599–E607 [DOI] [PubMed] [Google Scholar]
- 31. Lafontan M., Langin D. (2009) Lipolysis and lipid mobilization in human adipose tissue. Prog. Lipid Res. 48, 275–297 [DOI] [PubMed] [Google Scholar]
- 32. Mori S., Nojiri H., Yoshizuka N., Takema Y. (2007) Rapid desensitization of lipolysis in the visceral and subcutaneous adipocytes of rats. Lipids 42, 307–314 [DOI] [PubMed] [Google Scholar]
- 33. Jensen M. D. (1997) Lipolysis. Contribution from regional fat. Annu. Rev. Nutr. 17, 127–139 [DOI] [PubMed] [Google Scholar]
- 34. Bougnères P., Stunff C. L., Pecqueur C., Pinglier E., Adnot P., Ricquier D. (1997) In vivo resistance of lipolysis to epinephrine. A new feature of childhood onset obesity. J. Clin. Invest. 99, 2568–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lafontan M., Berlan M. (1993) Fat cell adrenergic receptors and the control of white and brown fat cell function. J. Lipid Res. 34, 1057–1091 [PubMed] [Google Scholar]
- 36. Townsend R. R., Klein S., Wolfe R. R. (1994) Changes in lipolytic sensitivity following repeated epinephrine infusion in humans. Am. J. Physiol. 266, E155–E160 [DOI] [PubMed] [Google Scholar]
- 37. Stallknecht B., Bülow J., Frandsen E., Galbo H. (1997) Desensitization of human adipose tissue to adrenaline stimulation studied by microdialysis. J. Physiol. 500, 271–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gayen J. R., Saberi M., Schenk S., Biswas N., Vaingankar S. M., Cheung W. W., Najjar S. M., O'Connor D. T., Bandyopadhyay G., Mahata S. K. (2009) A novel pathway of insulin sensitivity in chromogranin A null mice. A crucial role for pancreastatin in glucose homeostasis. J. Biol. Chem. 284, 28498–28509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fritsche A., Wahl H. G., Metzinger E., Renn W., Kellerer M., Häring H., Stumvoll M. (1998) Evidence for inhibition of leptin secretion by catecholamines in man. Exp. Clin. Endocrinol. Diabetes 106, 415–418 [DOI] [PubMed] [Google Scholar]
- 40. Scriba D., Aprath-Husmann I., Blum W. F., Hauner H. (2000) Catecholamines suppress leptin release from in vitro differentiated subcutaneous human adipocytes in primary culture via β1- and β2-adrenergic receptors. Eur. J. Endocrinol. 143, 439–445 [DOI] [PubMed] [Google Scholar]
- 41. Couillard C., Mauriège P., Prud'homme D., Nadeau A., Tremblay A., Bouchard C., Després J. P. (2002) Plasma leptin response to an epinephrine infusion in lean and obese women. Obes. Res. 10, 6–13 [DOI] [PubMed] [Google Scholar]
- 42. Thalmann S., Juge-Aubry C. E., Meier C. A. (2008) Explant cultures of white adipose tissue. Methods Mol. Biol. 456, 195–199 [DOI] [PubMed] [Google Scholar]
- 43. Karnieli E., Zarnowski M. J., Hissin P. J., Simpson I. A., Salans L. B., Cushman S. W. (1981) Insulin-stimulated translocation of glucose transport systems in the isolated rat adipose cell. Time course, reversal, insulin concentration dependency, and relationship to glucose transport activity. J. Biol. Chem. 256, 4772–4777 [PubMed] [Google Scholar]
- 44. Bandyopadhyay G. K., Yu J. G., Ofrecio J., Olefsky J. M. (2005) Increased p85/55/50 expression and decreased phosphotidylinositol 3-kinase activity in insulin-resistant human skeletal muscle. Diabetes 54, 2351–2359 [DOI] [PubMed] [Google Scholar]
- 45. Bandyopadhyay G. K., Yu J. G., Ofrecio J., Olefsky J. M. (2006) Increased malonyl-CoA levels in muscle from obese and type 2 diabetic subjects lead to decreased fatty acid oxidation and increased lipogenesis; thiazolidinedione treatment reverses these defects. Diabetes 55, 2277–2285 [DOI] [PubMed] [Google Scholar]
- 46. Mao X., Kikani C. K., Riojas R. A., Langlais P., Wang L., Ramos F. J., Fang Q., Christ-Roberts C. Y., Hong J. Y., Kim R. Y., Liu F., Dong L. Q. (2006) APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat. Cell Biol. 8, 516–523 [DOI] [PubMed] [Google Scholar]
- 47. Seeley R. J., van Dijk G., Campfield L. A., Smith F. J., Burn P., Nelligan J. A., Bell S. M., Baskin D. G., Woods S. C., Schwartz M. W. (1996) Intraventricular leptin reduces food intake and body weight of lean rats but not obese Zucker rats. Horm. Metab. Res. 28, 664–668 [DOI] [PubMed] [Google Scholar]
- 48. Minokoshi Y., Kim Y. B., Peroni O. D., Fryer L. G., Müller C., Carling D., Kahn B. B. (2002) Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415, 339–343 [DOI] [PubMed] [Google Scholar]
- 49. Wang M. Y., Orci L., Ravazzola M., Unger R. H. (2005) Fat storage in adipocytes requires inactivation of leptin's paracrine activity. Implications for treatment of human obesity. Proc. Natl. Acad. Sci. U.S.A. 102, 18011–18016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Knight Z. A., Hannan K. S., Greenberg M. L., Friedman J. M. (2010) Hyperleptinemia is required for the development of leptin resistance. PLoS One 5, e11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim Y. B., Uotani S., Pierroz D. D., Flier J. S., Kahn B. B. (2000) In vivo administration of leptin activates signal transduction directly in insulin-sensitive tissues. Overlapping but distinct pathways from insulin. Endocrinology 141, 2328–2339 [DOI] [PubMed] [Google Scholar]
- 52. Morris D. L., Rui L. (2009) Recent advances in understanding leptin signaling and leptin resistance. Am. J. Physiol. Endocrinol. Metab. 297, E1247–E1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vaisse C., Halaas J. L., Horvath C. M., Darnell J. E., Jr., Stoffel M., Friedman J. M. (1996) Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat. Genet. 14, 95–97 [DOI] [PubMed] [Google Scholar]
- 54. El-Haschimi K., Pierroz D. D., Hileman S. M., Bjørbaek C., Flier J. S. (2000) Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J. Clin. Invest. 105, 1827–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lin S., Thomas T. C., Storlien L. H., Huang X. F. (2000) Development of high fat diet-induced obesity and leptin resistance in C57Bl/6J mice. Int. J. Obes. Relat. Metab. Disord. 24, 639–646 [DOI] [PubMed] [Google Scholar]
- 56. Van Heek M., Compton D. S., France C. F., Tedesco R. P., Fawzi A. B., Graziano M. P., Sybertz E. J., Strader C. D., Davis H. R., Jr. (1997) Diet-induced obese mice develop peripheral, but not central, resistance to leptin. J. Clin. Invest. 99, 385–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pelleymounter M. A., Cullen M. J., Baker M. B., Hecht R., Winters D., Boone T., Collins F. (1995) Effects of the obese gene product on body weight regulation in ob/ob mice. Science 269, 540–543 [DOI] [PubMed] [Google Scholar]
- 58. Koch C., Augustine R. A., Steger J., Ganjam G. K., Benzler J., Pracht C., Lowe C., Schwartz M. W., Shepherd P. R., Anderson G. M., Grattan D. R., Tups A. (2010) Leptin rapidly improves glucose homeostasis in obese mice by increasing hypothalamic insulin sensitivity. J. Neurosci. 30, 16180–16187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Frühbeck G., Aguado M., Martínez J. A. (1997) In vitro lipolytic effect of leptin on mouse adipocytes. Evidence for a possible autocrine/paracrine role of leptin. Biochem. Biophys. Res. Commun. 240, 590–594 [DOI] [PubMed] [Google Scholar]
- 60. Frühbeck G., Aguado M., Gómez-Ambrosi J., Martínez J. A. (1998) Lipolytic effect of in vivo leptin administration on adipocytes of lean and ob/ob mice, but not db/db mice. Biochem. Biophys. Res. Commun. 250, 99–102 [DOI] [PubMed] [Google Scholar]
- 61. Mahata S. K., Mahata M., Wakade A. R., O'Connor D. T. (2000) Primary structure and function of the catecholamine release inhibitory peptide catestatin (chromogranin A344–364). Identification of amino acid residues crucial for activity. Mol. Endocrinol. 14, 1525–1535 [DOI] [PubMed] [Google Scholar]
- 62. Mahata S. K., Mahata M., Wen G., Wong W. B., Mahapatra N. R., Hamilton B. A., O'Connor D. T. (2004) The catecholamine release-inhibitory “catestatin” fragment of chromogranin A. Naturally occurring human variants with different potencies for multiple chromaffin cell nicotinic cholinergic responses. Mol. Pharmacol. 66, 1180–1191 [DOI] [PubMed] [Google Scholar]
- 63. Gómez-Ambrosi J., Frühbeck G., Aguado M., Milagro F. I., Margareto J., Martínez A. J. (2001) Divergent effects of an α2-adrenergic antagonist on lipolysis and thermogenesis. Interactions with a β3-adrenergic agonist in rats. Int. J. Mol. Med. 8, 103–109 [PubMed] [Google Scholar]
- 64. Polak J., Moro C., Bessière D., Hejnova J., Marquès M. A., Bajzova M., Lafontan M., Crampes F., Berlan M., Stich V. (2007) Acute exposure to long-chain fatty acids impairs α2-adrenergic receptor-mediated antilipolysis in human adipose tissue. J. Lipid Res. 48, 2236–2246 [DOI] [PubMed] [Google Scholar]