

Abstract
The alpha-Proteobacterium Bartonella is a common parasite of voles and mice, giving rise to short-lived (4 weeks to 2 months) infections. Here, we report high sequence diversity in genes of the VirB/VirD type IV secretion system (T4SS), amongst Bartonella from natural rodent populations in NE Poland. The VirB5 protein is predicted to consist of three conserved alpha helices separated by loops of variable length which include numerous indels. The C-terminal domain includes repeat stretches of KEK residues, reflecting underlying homopolymeric stretches of adenine residues. A total of 16 variants of VirB5, associated with host identity, but not bacterial taxon, were identified from 22 Bartonella isolates. One was clearly a recombinant from two others, another included an insertion of two KEK repeats. The virB5 gene appears to evolve via both mutation and recombination, as well as slippage mediated insertion/deletion events. The recombinational units are thought to be relatively short, as there was no evidence of linkage disequilibrium between virB5 and the bepA locus only 5.5 kb distant. The diversity of virB5 is assumed to be related to immunological role of this protein in Bartonella infections; diversity of virB5 may assist persistence of Bartonella in the rodent population, despite the relatively short (3–4 weeks) duration of individual infections. It is clear from the distribution of virB5 and bepA alleles that recombination within and between clades is widespread, and frequently crosses the boundaries of conventionally recognised Bartonella species.
Electronic supplementary material
The online version of this article (doi:10.1007/s00248-012-0033-y) contains supplementary material, which is available to authorized users.
Introduction
The alpha-Proteobacterium Bartonella includes such significant human pathogens as Bartonella quintana, Bartonella henselae and Bartonella bacilliformis. However the greatest diversity is found amongst rodent-infecting forms; of more than 1,400 citrate synthase (gltA) sequences within GenBank, over 530 are from rodents. Rodents are infected at high prevalence (sometimes exceeding 60 %, see Ref. [1]), by co-existing Bartonella species [1–6] which undergo genetic exchange both within and between taxa [7–11], and these bacteria form a significant component of the parasite community infecting rodents, although their impact on rodents appears to be minimal [6, 12]. Most significantly, these rodent-infecting forms may represent an important reservoir for emergent strains of Bartonella which infect humans [13–17]. The host specificity of Bartonella is unclear. At one level, species appear to lack specificity [10], and opportunistic human infections with, e.g. Bartonella vinsonii or Bartonella grahamii suggest that many Bartonella taxa may have a wide host range. On the other hand, while species may lack host specificity, individual clades within species show close fidelity to particular genera of host rodents [11], suggesting that research on host specificity has focussed at an inappropriate taxonomic level.
Bartonella has a three-phase life cycle [18]. An initial proliferative phase infecting endothelial cells persists for several days, after which bacteria escape to invade erythrocytes, within which they remain until either the death of the cell or uptake in the blood meal of an insect. In laboratory rodents, lesions in liver and kidneys have also been observed [19], although their relevance to the epidemiology of natural Bartonella infections is unknown. The third phase of the life cycle is within the gut of an insect vector. Invasion of both endothelial and red blood cells depends upon type IV secretion systems (T4SS), encoded within pathogenicity islands in the Bartonella genome [20, 21]. The T4SS of Bartonella is similar to the type A T4SS of other gram-negative bacteria [22, 23], but levels of homology with other bacteria are low [24]. T4SS gene clusters have been duplicated and lost during genomic rearrangements in Bartonella, but typically two or three systems are present [20]; trw, responsible for red blood cell infection [24] and virB/virD and vbh, involved in endothelial cell infection [20, 25]. The trw system in Bartonella is unique in that several genes within the cluster are duplicated [24, 26]. Sequencing of the B. grahamii genome [21] revealed an astonishing diversity of pathogenicity genes, and, including those encoded by a plasmid, the sequenced isolate could access a total of 69 T4SS genes, compared to none in B. bacilliformis [20, 21].
Despite the evidence from whole genome sequencing, and experimental knowledge of T4SS function [20, 27], little is known of the diversity of T4SS genes in natural Bartonella infections. Here, we present evidence of the molecular diversity and mode of evolution of two T4SS genes, virB5 (encoding a putative adhesin involved in host cell invasion) and bepA, located 5.5 kb from virB5 (see Fig. 1) and encoding a crucial inoculated effector protein which prevents host cell apoptosis [28], in Bartonella isolates circulating in rodent populations in an old field and forest system in North East Poland.
Figure 1.
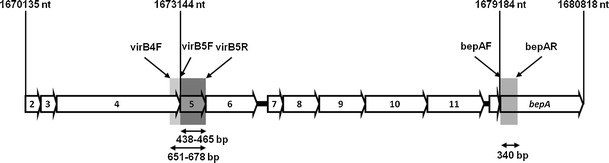
Schematic drawing of virB gene complex showing both virB5 and bepA genes, based on data available for B. grahamii (isolate as4up) genome (accession number: NC012846, see [21]); virB (2–11) and bepA genes marked, with primers used to amplify virB5 gene and bepA gene fragment, and the size of amplicons. Positions in genome (nt) based on isolate NC012846
Materials and Methods
The field study site represents a complex of forest and abandoned agricultural land in NE Poland, the focus of several previous studies of rodent-pathogen epidemiology [6, 11, 29–36]. This work follows [37], concentrating on a longitudinal field study of rodents in an old field habitat, where the voles Microtus oeconomus and Microtus arvalis and the mouse Apodemus flavicollis were collected, and a forest, where an area of 2 ha was sampled monthly for A. flavicollis and Myodes glareolus [6, 11, 36] from June 2007 until May 2009, with short breaks during winter (December–March). Detailed trapping, marking and collection protocols have been presented elsewhere [6, 11, 38]. After collection of blood and ectoparasites, the rodents were marked with an ear tag (World Precision Instruments Inc., USA) and released close to their original capture point. Animal handling and sampling methodologies conformed to permission granted by the Polish Local Ethical Committee (737/2007). Blood (50–100 μl) was taken by caudal vein bleeding into 1 mM EDTA and stored at 4°C prior to DNA extraction using a proprietary method (AxyPrep Miniprep Blood, AxyGen, USA). A 316 bp fragment of the gltA gene was amplified using primers Bh.CS782p and Bh.Cs1137n [39]. Further data were collected from cultures of Bartonella isolated from rodents euthanised with an ether overdose, and then bled by cardiac puncture. The blood was stored in 1 mM EDTA at 4°C until use, then plated on Columbia blood agar and incubated at 37°C in a 5 % CO2 atmosphere for up to 10 days [40]. Each isolate was subcultured three times before single colonies were selected. Cells were harvested and DNA isolated by boiling in sterile water for 10 min, and centrifuging at 12,000 rpm for a further 15 min [41]. The supernatant was stored frozen and used with the following primers to amplify genes of the T4SS-bepA (bepAF: 5′-TCATCATTAATTTTATCCGAACACCAC-3′, bepAR: 5′-TGAGATAAATTCTTCGCGCGTTAA-3′) and virb5. The first pair of primers (virB5F: 5′-TGGAATGAATGATGAGATCGCC-3′ and virB5R: 5′-TAAAGTCGGACATCAGAWTTCYCAAG-3′) was used on all isolates but failed to consistently amplify virB5. The forward primer (virB4F: 5′-TACAGTCAGAATTAAGCAGAG-3′) was therefore designed to complement the 3′ end of the virB4 gene (immediately 5′ to virB5; see Fig. 1) and used with the virB5 reverse primer (virB5R), to successfully amplify virB5 from a further 12 isolates. In the PCR reaction, after first denaturation in 94°C for 300 s, genes were amplified with 45 cycles of 94°C for 45 s, 51°C (using primer pair bepAF/bepAR) or 52°C (virB5F/virB5R and virB4F/virB5R) for 45 s, 72°C for 60s, and followed by a single 7-min extension step at 72°C. Sequencing used amplification primers on both strands of purified amplicons (AxyPrep™ Clean, AxyGen, USA or ExoSAP-IT®, Affymetrics, USA) with ABI technology (Genomed, Poland or ABI-lab, University of Oslo, Norway). New sequences obtained in this study were deposited in GenBank under accession numbers: HM450047–HM450063 and JN797608-JN797610 (virB5), and HM450064–HM450072 (bepA); see Electronic Supplementary Material.
Sequence outputs were checked visually for ambiguous peaks, which, where appropriate, were re-sequenced. Because of indels within the virB5 gene, it proved difficult to achieve an unambiguous alignment, and therefore three approaches to alignment were adopted. The first was to use standard and Expresso alignment algorithms within the T-Coffee package [42]. The second was to use the Prank algorithm via the WebPrank server (www.ebi.ac.uk/). The final approach was by homology modelling using the Escherichia coli TraC protein as a template [43, 44] via the SWISS-MODEL package (swissmodel.expasy.org [45–47]). The modelled protein structures were then used as a guide for alignment of the DNA sequences. Maximum Likelihood and Bayesian phylogenetic analysis, including analyses of selection on codon positions and of rates of evolution of different lineages, was carried out using PhyML and PAML [48, 49], after choosing the best DNA evolution model (jModelTest [50]). Nested clade analysis was performed as described [51–53], using log-linear models (SPSS) to test statistical hypotheses concerning the segregation of virB5 and bepA variation with clade identity or host identity. Analysis of recombination was carried out using the RDP-3 software package [54].
Results
A total of 37 variant gltA sequences were recovered from 154 Bartonella strains isolated over 2 years from four rodent species [11]. Most could be referred either to B. grahamii (five variants) or to Bartonella taylorii (26 variants), but one isolate of Bartonella birtlesii and three of Bartonella doshiae were also identified. Two variants had recombinant gltA genes [11]; one of these (Mo1) was B. doshiae-like at other housekeeping gene loci. The characterisation of virB5 and bepA genes was attempted only for the 42 Bartonella isolates cultured on blood agar. VirB5 was amplified from 30 isolates, bepA from 17; both sequences were obtained from 12 isolates.
Out of 30 isolates for which virB5 was amplified, 20 were B. taylorii clades, six were B. grahamii, three were B. doshiae (including one recombinant) and one B. birtlesii [11], according to gltA gene identity. In all, the inferred VirB5 protein product was between 145- and 173-amino acids (aa) in length; amplicons from B. taylorii and B. birtlesii isolates were clearly shorter than those from non-recombined B. doshiae and B. grahamii isolates (inferred length 145–152 and 148 aa vs. 159 and 173 aa, respectively). Because of apparent similarities of sequence, the Bartonella elizabethae sequence from GenBank (AF195504) was included in the alignment. The B. grahamii isolate from Uppsala (GenBank accession number NC012846) proved identical to B. grahamii isolates collected in the present work (Mg4, Af4). Homology modelling of Bartonella VirB5 molecules suggested that this protein is made up of three helices, a short helix at the carboxy terminus following the signal peptide, followed by two long helices separated by a loop. The most reliable structures generated were for B. elizabethae and B. taylorii (Table 1). The model for B. elizabethae (QMEAN4 = 0.7, Z-score = −1.39, see [55]), featured a long (24 aa) loop between helices 2 and 3. The poorest fit model was for B. grahamii, which could not be modelled using the automatic algorithm. However, a manual alignment, based on the alignment of B. elizabethae to TraC, and the Prank alignment of B. grahamii to B. elizabethae, allowed the generation of the same protein structure as in the case of the other Bartonella species (Table 1). All VirB variants sequenced during the present work could be modelled with the same structure, with variable length loops due to indels between helices 2 and 3, and at the end of helix 3 (Table 1). Helices 1, 2 and 3 represented the most highly conserved parts of the translated molecule.
Table 1.
Properties of VirB5 proteins as deduced from SWISS-MODEL homology modelling, using Escherichia coli Trac1 as template
| Strain | QMEAN4 score | Z-score | Length (aa) (position in the protein (aa)) | Length (aa) of the loop between helixes 2 and 3 | Indel(s) position(s) in the protein (aa) | Protein length (aa) | ||
|---|---|---|---|---|---|---|---|---|
| Helix 1 | Helix 2 | Helix 3 | ||||||
| Bartonella elizabethae (AF195504) | 0.7 | −1.4 | 11 (29–39) | 25 (45–69) | 31 (101–131) | 30 | 72–89 | 177 |
| 92–93 | ||||||||
| 97–103 | ||||||||
| 151–153 | ||||||||
| 157–160 | ||||||||
| 171–172 | ||||||||
| Bartonella grahamii (NC012846) | 0.4 | −4.1 | 11 (29–39) | 25 (45–69) | 31 (98–128) | 28 | 72–85 | 173 |
| 88–89 | ||||||||
| 93–99 | ||||||||
| 147–149 | ||||||||
| 153–156 | ||||||||
| 167–168 | ||||||||
| Bartonella doshiae (Ma16) | 0.5 | −2.6 | 11 (29–39) | 25 (45–69) | 35 (81–115) | 11 | 72–75 | 159 |
| 81–87 | ||||||||
| 149–155 | ||||||||
| Bartonella birtlesii (Af5) | 0.6 | −2.0 | 11 (29–39) | 25 (45–69) | 31 (81–111) | 11 | 72–78 | 148 |
| Bartonella taylorii (Af1) | 0.7 | −1.4 | 11 (29–39) | 25 (45–69) | 29 (80–108) | 10 | 72–75 | 145 |
| B. taylorii (Ma12) | 0.6 | −1.8 | 11 (29–39) | 25 (45–69) | 31 (81–111) | 11 | 72–78a | 148 |
| B. taylorii (Mo2) | 0.6 | −1.8 | 11 (29–39) | 25 (45–69) | 32 (80–111) | 10 | 72–78a | 154 |
| 140–145 | ||||||||
| B. taylorii (Af6) | 0.6 | −1.5 | 11 (29–39) | 25 (45–69) | 31 (81–111) | 11 | 72–78a | 148 |
| B. taylorii (Ma13) | 0.5 | −2.5 | 11 (29–39) | 25 (45–69) | 36 (80–115) | 10 | 72–78 | 152 |
| 84–87 | ||||||||
Indel positions marked following the alignment obtained using prank algorithm
aIdentical amino acid indels
Phylogenetic Reconstruction of virB5
Phylogenetic reconstructions using alignments generated using different algorithms did not differ in the shape of resultant trees generated using maximum likelihood (PhyML), although the alignment did affect the maximum likelihood estimates of the trees, with Prank with a GTR model (estimated using jModelTest) giving the best estimate (ML = −2,769). This alignment was therefore used in subsequent analyses. A phylogenetic reconstruction using this alignment divided the 16 basic variants into six clades (Fig. 2), corresponding to B. doshiae, B. grahamii, B. birtlesi, two clades of B. taylorii and one mixed clade of B. taylorii and B. grahamii. When included, B. elizabethae made a 7th clade, linked as sister group to the B. grahamii clade. The virB5 clades were structured according to host species infected. Thus, clade B in Fig. 2 infected mainly voles from the field system, although it also included one isolate from A. flavicollis from forest. Clade C consisted of four isolates from voles, three from M. arvalis and one from M. glareolus. Clade F was collected from three M. arvalis from the fallows, and from one representative of each of the forest rodents, M. glareolus and A. flavicollis. Two clades (A, eight isolates, and D, one isolate, see Fig. 2) were found only in A. flavicollis. This association of virB5 alleles with host identity was significant (χ 2 = 33.8, df = 15, P = 0.004).
Figure 2.
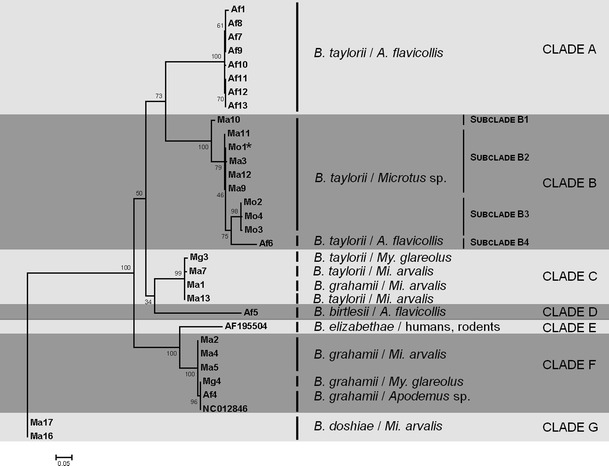
Phylogeny of obtained sequences of virB5 gene (465 bp), generated using PhyML with a GTR substiution model and the Prank amino acid alignment. Isolate identified as recombinant on basis of housekeeping genes (see Ref. [11]) marked with an asterisk
Differences between clades were much greater than those within clades, and were due to mutation, recombination and loss/gain of indels. Within clade A, isolates differed by up to two nucleotides; only Af1 (13–14 bases difference from the rest of clade A isolates) could not have been a result of clonal expansion. Clades C and F varied by, respectively, up to five and three bases while both clade G isolates were identical. Clade B was most diverse, with four subclades (Fig. 2). Subclade B1 consisted of only one recombinant isolate from M. arvalis (see below). Subclade B2, from both Microtus species (Ma3, Ma9, Ma11, Ma12 and Mo1), varied by up to three bases. Subclade B3 isolates from Mi. oeconomus differed by up to two bases, and were sister group to B2, with a characteristic insertion of 6-amino acids (aa 148–153, LKKLEK) in the carboxy terminus of the protein generated by an imperfect repetition of two sections of DNA (TTAAAA and GAAAAA). Subclade B3 alleles were not recently derived from subclade B2, as there were 16 other mutational changes between these alleles. The fourth subclade B4, consisted only of the isolate Af6. By other phylogenetic approaches (Maximum Parsimony) this isolate is basal to the entire tree, and it is very different in sequence to the other clade B isolates. Its position within this clade is therefore uncertain.
The isolate Ma10 from subclade B1 (Fig. 2), proved to be recombinant between a clade A allele and an allele from subclade B2 (infecting Apodemus and Microtus, respectively), with a breakpoint between positions 73 and 77 of the DNA sequence. Statistical support was high (P = 7.4 × 10−11, RDP; 2.9 × 10−30, GENECONV; 2.3 × 10−5, BootScan; 3.2 × 10−5, Chimera; 3.5 × 10−5, MaxChi; 3.2 × 10−29, SiScan; 3.3 × 10−16, 3 Seq). Visual inspection showed 17 base changes, shared between clade A and the Ma10 isolate but differing from subclade B2, up to position 73 and 73 changes from position 77 to the end of the gene, which were shared between Ma10 and subclade B2 but not with clade A. There were also indels of 18 bases between positions 223 and 240 and of 12 bases between positions 391 and 402, shared between Ma10 and subclade B2 but absent from clade A. Only a single base change (G436T) appeared to have occurred in Ma10 which was not shared with either clade A or subclade B2.
Differences between clades were due to both mutational change and to indels. Each clade had a characteristic pattern of indels, which could account for up to 100 bases difference between the shortest and the longest sequences (Table 1). Nevertheless, the clades also differed substantially due to mutational change, with more than 130 base changes (c. 30 % of the gene) between the most diverse isolates. Mutational change and indels were correlated, and phylogenies based on alignments in which indels were omitted had the same topology as those for which they were included.
BepA
Only a fragment of the bepA gene was amplified, between amino acids 26 and 138 of the complete protein, encompassing the Fic-1 domain as identified by Schulein et al. [28]. In the present work, bepA amplified successfully from 17 Bartonella isolates, of which five could be classified as B. taylorii, ten as B. grahamii and one as B. birtlesii [11]. The final isolate which amplified was Mo1, the B. doshiae isolate with a recombinant gltA. Compared with virB5, there was relatively little variation in bepA at either nucleotide or amino acid level, and the fragments proved easy to align and to belong to three clades of sequence variants (Fig. 3). No relationship was found between host identity or bacterial species identity. Clade A was amplified from eight isolates (predominantly B. grahamii with a single B. taylorii isolate) collected from M. arvalis (three isolates), M. glareolus (two isolates) and A. flavicollis (three isolates). The bepA from the fully sequenced Swedish B. grahamii isolate (NC012846), also belonged to this clade. Only two single nucleotide polymorphisms occurred within this group, one silent, the other substituting Q60P (numbering based on NC012846 [21]). Clade B was made up of six isolates from M. arvalis (two B. taylorii and four B. grahamii) and one from Mi. oeconomus (the B. doshiae with recombinant gltA). Six isolates within this clade had identical nucleotide sequences but one of the M. arvalis isolates (Ma1) differed by a single nucleotide causing an inferred amino acid change (L54M, numbering according to NC012846 [21]). Clades A and B were five nucleotides different, with a characteristic E121K substitution. The third bepA clade was however completely different, with 49 nucleotide changes relative to clades A and B, resulting in 19- and 18-amino acid substitutions, respectively. It was found in three isolates, two referrable to B. taylorii and the third to B. birtlesii, all from A. flavicollis. Within this group, there was a single silent nucleotide substitution (A309G in Af6; DNA numbering based on NC012846 [21]).
Figure 3.
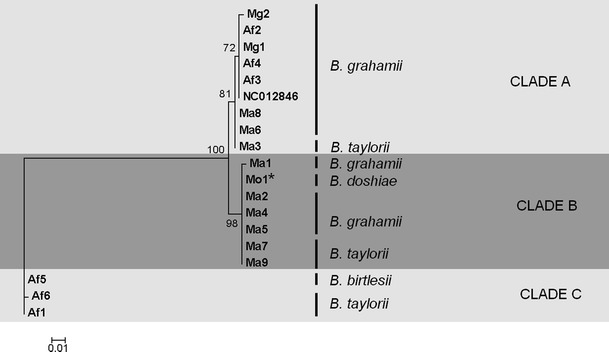
Phylogeny of obtained sequences of bepA gene fragment (340 bp), generated using PhyML with a GTR substition model and the Prank amino acid alignment. Recombinant isolate (see Ref. [11]) marked with an asterisk
For the 12 isolates where both virB5 and bepA were successfully amplified, there was no consistent pattern of linkage between alleles (Fig. 4). For example, Af1 (B. taylorii) and Af5 (B. birtlesii) shared a bepA allele but exhibited the most disparate virB5 alleles, suggesting that recombination between these loci is common, despite their close proximity (no more than 5.5 kb apart [21]). Alleles of both bepA and virB5 were distributed between all Bartonella spp. as established using housekeeping genes [11]. There was however no consistent phylogenetic pattern in the distribution of virB5 alleles (see Table 2). For example, identical virB5 alleles amplified from isolates Ma1 and Ma7, which were entirely different at all sequenced housekeeping gene loci except groEl. Conversely, isolate Mo1, identical with both B. doshiae isolates (Ma16 and Ma17) according to housekeeping genes except for a recombinant gltA [11], had an entirely distinct virB5 allele.
Figure 4.
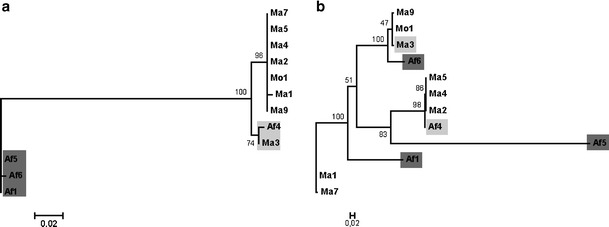
Phylogenetic trees of isolates, for which both a bepA and b virB gene sequences were obtained, generated using PhyML with a GTR substition model and the Prank amino acid alignment. Isolates of particular clades of bepA (a) marked with the same colors on virB5 tree (b)
Table 2.
Allele patterns (chosen housekeeping genes, rDNA and T4SS gene fragments) of Bartonella isolates from different virB5 clades
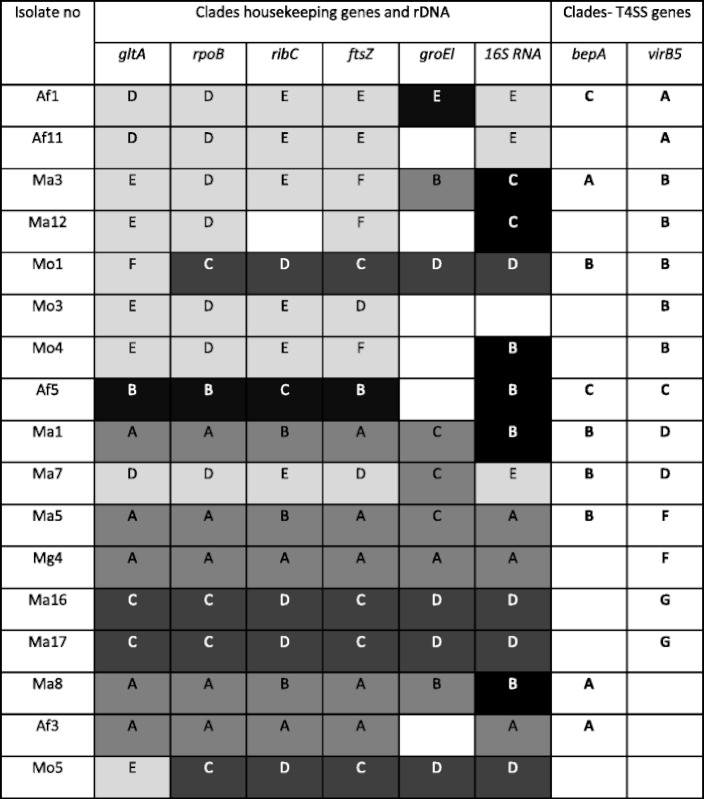
Bacteria species based on housekeeping genes and 16S rDNA; shade colour—from the brightest: Bartonella taylorii, Bartonella grahamii, Bartonella doshiae and Bartonella birtlesii
Distribution and Recombinant Pathways of bepA and virB5 Within Rodent Populations
Analysis of gltA demonstrated a complex of Bartonella species and clades which recombine with each other to varying degrees ([11]; Fig. 5). VirB5 variants (clade F on Fig. 2) from clades of B. grahamii diverged from each other by no more than three nucleotides, and were congruent with clonal expansion inferred from gltA. BepA variants from B. grahamii were also consistent with this hypothesis. VirB5 clade A variants were characteristic of B. taylorii gltA clade A (see Fig. 2), and were also sufficiently similar to indicate clonal expansion. However, a second clade of virB variants (Clade C in Fig. 2) also occurred within B. taylorii gltA clade A (Ma7 and Ma13); these two forms could not have resulted from clonal diversification alone. One virB clade C variant (Ma1) was also recovered from B. grahamii, indicating recombination of the whole virB5 gene between these species. VirB5 clade B had the most interesting distribution within the Bartonella network. Most isolates (Ma3, Ma9, Ma11 and Ma12) came from a single gltA variant within B. taylorii clade B (see Fig. 5), with recombinant links to B. doshiae (see [11]); Mo1 was recombinant between B. taylorii and B. doshiae [11]. The somewhat different virB5 subclade B3 from Mo2, Mo3 and Mo4 also circulated within B. taylorii gltA clade B (Fig. 5). The presence of identical bepA alleles suggested a recombinant pathway between B. taylorii gltA clade B, B. grahamii and the recombinant B.taylorii/B. doshiae isolate Mo1. Finally sharing of the basal bepA allele C between B. birtlesii and B. taylorii clade A suggests that these isolates may also recombine.
Figure 5.
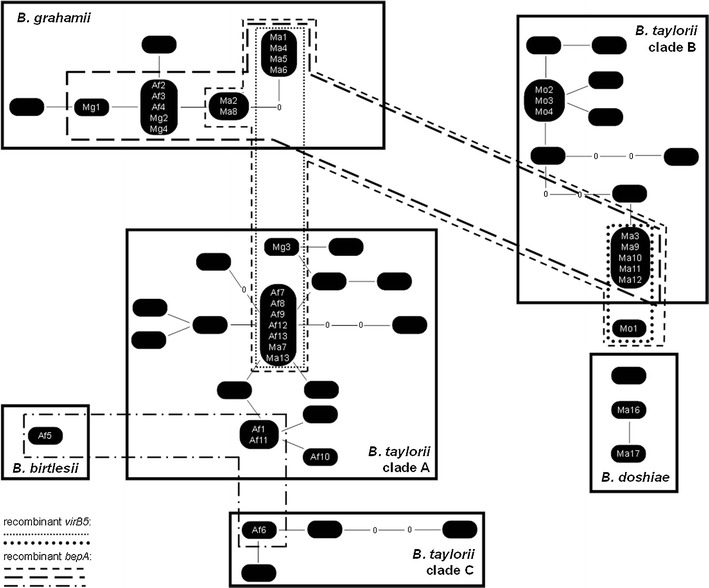
Cladogram of the Bartonella isolates collected from rodents at the study site (as described in Ref. [11]), and relevant haplotypes from Welc-Falęciak et al. [37], based on 292 bp fragment of gltA, showing recombinant events of virB5 and bepA. Black boxes indicate variants of gltA that have been found in the study, ‘missing’ variants (i.e. predicted but not collected) are indicated by 0. Isolates for which sequencing of virB5 and/or bepA were successfully marked on (Af A. flavicollis, Mg M. glareolus, Ma M. arvalis, Mo M. oeconomus). Clades sharing virB5 and bepA variants between ‘species’ and/or main clades of B. taylorii are marked. Main clades (B. grahamii, B. taylorii clades A, B and C, and B. doshiae) are based on 95 % probability that all isolates within them have arisen through mutation (calculated in TCS [56])
Discussion
The Bartonella genome is extensively reshaped by recombination, both at a genomic scale [10, 20, 21] and more locally at the level of individual genes [11]. Indeed, Nysted et al. [9] focussed on the virB5 analogue at the trw locus to demonstrate duplication and recombination within a single clade of B. grahamii, identifying potentially recombinant stretches derived interspecifically (B. henselae sequences within the B. grahamii and Bartonella tribocorum genes) or from an unknown donor. Here, we present the first account of virB5 evolution in a natural Bartonella population and show the relative importance of recombination and mutation in the evolution of this gene. Part of the pressure for recombination is thought to come from selection for optimal combinations of invasion genes, particularly through microevolution of adhesins such as BadA [10, 57] and of the T4SS cell invasion systems. We demonstrate that the sequence of virB5 (identified as an adhesin by [44]) is linked to host identity, that its structure is shaped by recombination, and that it is exchanged between Bartonella clades and species which are only distantly related. Although this represents the first demonstration of such extensive intragenic rearrangements within Bartonella, in fact this process is well known in the related alpha-Proteobacterium Helicobacter pylori. Within a relatively small number of isolates of virB5, we have identified one clear example of recombination between two clades, another where the subclades have been derived by acquisition or loss of an indel of 7-amino acids based on a partial DNA repeat, a third clade derived by loss of 3-amino acids at the C terminus, and other examples where recombination or loss/gain of indels can be suspected in the origin of the genes. At the same time, there is extensive mutational drift between virB5 clades showing different patterns of indels, although there is little mutational change within them. It should be stressed that we present here the minimum diversity of virB5 likely to exist within the Bartonella community analysed, as a further 12 isolates failed to sequence, suggesting the existence of further variants.
Although VirB5 in other alpha-Proteobacteria is a three-helix, 220-amino acid acidic protein [44] of which the E. coli TraC protein is a prototype [43], the inferred Bartonella VirB5 protein is much shorter (145–177 aa), consisting of one short and two long helices. The second long helix, and the C terminal domain, both contain numerous charged residues and are the most variable parts of the molecule, including numerous indels. Although Backert et al. [44] make the case for VirB5 being involved in docking with integrins on the host cell surface, this seems not to be the case for the Bartonella VirB5, as the fibronectin recognition domain of the three-helix VirB5 molecules is absent [44]. However, the large number of charged residues in the C terminal domain of the Bartonella VirB5 protein does suggest a role in intra-molecular interactions. This could be related to the binding of the VirB5 molecule into the pilus formed by the T4SS [18]; in B. henselae VirB5 binds strongly to VirB3 [58], and in Agrobacterium this protein is an integral part of the pilus structure [59]. Additionally, this protein is a strong and specific immunogen in Bartonella infections, being characterised as the 17 kDa antigen in clinical studies [60]. Naturally immune sera had highly specific recognition patterns, suggesting exposure of native VirB5 to the host immune system [60], and making it likely that VirB5 of Bartonella is exposed on the outer surface of the pilus, where it has a species-specific conformation. In its alternation of runs of charged lysine and glutamate residues, VirB5 in Bartonella shows interesting parallels with the immunologically relevant glutamate-rich merozoite surface proteins of Plasmodium (e.g. [61, 62]). It is also similar in this respect to the VirB10/CagY protein of H. pylori, which includes runs of lysine and glutamate doublets and may be implicated in immunological evasion [63].
Evolution of virB5 in B. taylorii has clearly taken place through homologous recombination, and at least one (subclade B1) of the seven virB5 clades identified in this work is unambigiously derived by recombination from two others (B2 and A). The double duplication of 3-amino acids in subclade B3 is presumably due to misalignment of the repetitive (CTGAGAAA) motif between partner chromosomes during a recombinant event. The longest VirB5 molecules, those of B. grahamii and B. elizabethae have also evolved by duplication of short segments, with an insertion of 9-amino acids built up partly from a (GAAAAA) repeat. A second, imperfect (GCTTCAA) repeat (n = 3) gives a further insertion of 12-amino acids. Imperfect copying of partial repeat regions within virB5 appears to have been an important mechanism for the microevolution of this gene. The virB5 gene has numerous poly-A tracts, up to a maximum of A8, at which slippage during replication can occur [64]. This provides considerable plasticity to gene structure, and the presence of large numbers of glutamate and lysine residues may be related both to an antigenic function of VirB5, and to the importance of homopolymeric tracts at which slippage can occur.
Apart from small-scale duplications and evidence of recombination within the gene, there was also clear evidence of recombinant exchange of the whole virB5 gene between B. taylorii and B. grahamii. This corresponded exactly with exchange of the housekeeping gene groEl between the same two clades of B. taylorii and B. grahamii [11], although it is not clear how much more of the genome was swapped between these isolates. Sharing of virB5 variants showed little or no correlation with the patterns of gltA alleles present in the Bartonella isolates, further suggesting widespread recombination between isolates. However, the distribution of virB5 variants correlated much more closely with host identity than did gltA. Recombination on a large scale in the Bartonella genome has been obvious since the comparisons of Frank et al. [65] and Saenz et al. [20], showing different genome sizes and gene arrangements across the genus, and a role for phage mediated recombination [21] was suggested following the observation of phage transcription in B. bacilliformis [66] and B. grahamii [21], packaging genomic fragments of c. 14 kb. The work of Nystedt et al. [9] also suggested recombination in a virB5 homologue (trw), in this case inferred from a phylogenetic context. There was little evidence in the present work for linkage disequilibrium between virB5 and the bepA gene, and phylogenies based on the two genes are quite different, although these genes are only 5.5 kb apart [21]. This further suggests that recombination involves relatively small stretches of DNA, certainly no larger than the 14-kb fragments packaged by the Bartonella gene transfer agent (GTA) [21]. Barbian and Minnick [66] indicate that not all Bartonella species possess this GTA, as evidenced by the presence or absence of a 14-kb extra-chromosomal band in whole genomic DNA preparations. B. grahamii does possess this GTA (confirmed by Berglund et al. [21]), but Bartonella claridgeiae, B. vinsonii and B. elizabethae do not; B. taylorii, B. birtlesii and B. doshiae have not been studied, and therefore we do not know whether all of the clades of Bartonella included in the present work had the GTA and were capable of recombination via this route.
The present work demonstrates a close parallel between Bartonella isolates in rodents and the epidemiological situation in Helicobacter, with a difference in the temporal scale of persistence of variants. In H. pylori, novel antigenic variants [63, 67, 68] appear within the same host, infected with H. pylori for perhaps 30 years. These variants allow the H. pylori infection to escape from host immunological surveillance [67], and take advantage of transient infection with co-colonising strains to undertake recombination [69]. In the context of Bartonella in rodents, individual infections are transient, lasting only 4–11 weeks [6, 70, 71], in rodents which live a maximum of 13–15 months [6]. However, genetic exchange and recombination within virB5, a molecule known to be antigenically relevant, probably allows numerous Bartonella isolates to circulate within the wider rodent population; temporal persistence in this case is replaced by spatial persistence. Clearly, the role of the arthropod vectors in generating and maintaining allelic diversity through recombination in Bartonella is important, and requires further elucidation. This is particularly important given the reputation that rodent Bartonella isolates are acquiring as emerging human pathogens [72].
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
(DOC 51 kb)
Acknowledgements
Supported by Polish Ministry of Science and Higher Education, through Faculty of Biology, University of Warsaw intramural grant 1791-47 and The Norwegian Research Council Yggdrasil grant 202556.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
References
- 1.Birtles RJ, Hazel SM, Bennett M, Bown K, Raoult D, Begon M. Longitudinal monitoring of the dynamics of infections due to Bartonella species in UK woodland rodents. Epidemiol Infect. 2001;126:323–329. doi: 10.1017/S095026880100526X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Birtles RJ, Harrison TG, Saunders NA, Molyneux DH. Proposals to unify the genera Grahamella and Bartonella, with descriptions of Bartonella talpae comb. nov., Bartonella peromysci comb. nov., and three new species, Bartonella grahamii sp.nov., Bartonella taylorii sp. nov., and Bartonella doshiae sp. nov.”. Int J Syst Evol Microbiol. 1995;45:1–8. doi: 10.1099/00207713-45-1-1. [DOI] [PubMed] [Google Scholar]
- 3.Bermond D, Heller R, Barrat F, Delacour G, Dehio C, Alliot A, et al. Bartonella birtlesii sp. nov., isolated from small mammals (Apodemus spp.) Int J Syst Evol Microbiol. 2000;50:1973–1979. doi: 10.1099/00207713-50-6-1973. [DOI] [PubMed] [Google Scholar]
- 4.Holmberg M, Mills JN, McGill S, Benjamin G, Ellis BA. Bartonella infection in sylvatic small mammals of central Sweden. Epidemiol Infect. 2003;130:149–157. doi: 10.1017/S0950268802008075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knap N, Duh D, Birtles R, Trilar T, Petrovec M, Avsic-Zupanc T. Molecular detection of Bartonella species infecting rodents in Slovenia. FEMS Immunol Med Microbiol. 2007;50:45–50. doi: 10.1111/j.1574-695X.2007.00226.x. [DOI] [PubMed] [Google Scholar]
- 6.Paziewska A, Harris PD, Zwolińska L, Bajer A, Siński E (2012) Differences in the ecology of Bartonella spp. infecting Apodemus flavicollis and Myodes glareolus in a boreal forest. Parasitology. doi:10.1017/S0031182012000170 [DOI] [PubMed]
- 7.Inoue K, Kabeya H, Kosoy MY, Bai Y, Smirnov G, McColl D, Artsob H, Maruyama S. Evolutional and geographical relationships of Bartonella grahamii isolates from wild rodents by multi-locus sequencing analysis. Microb Ecol. 2009;57:534–541. doi: 10.1007/s00248-009-9488-x. [DOI] [PubMed] [Google Scholar]
- 8.Inoue K, Maruyama S, Kabeya H, Yamada N, Ohashi N, Sato Y, Yukawa M, Masuzawa T, Kawamori F, Kadosaka T, Takada N, Fujita H, Kawabata H. Prevalence and genetic diversity of Bartonella species isolated from wild rodents in Japan. Appl Environ Microbiol. 2008;74:5086–5092. doi: 10.1128/AEM.00071-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nystedt B, Frank AC, Thollesson M, Andersson SG. Diversifying selection and concerted evolution of a type IV secretion system in Bartonella. Mol Biol Evol. 2008;25:287–300. doi: 10.1093/molbev/msm252. [DOI] [PubMed] [Google Scholar]
- 10.Berglund EC, Ehrenborg C, Pettersson OV, Granberg F, Näslund K, Holmberg M, Andersson SGE. Genome dynamics of Bartonella grahamii in micro-populations of woodland rodents. BMC Genomics. 2010;11:152. doi: 10.1186/1471-2164-11-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paziewska A, Harris PD, Zwolińska L, Bajer A, Siński E. Recombination within and between species of the alpha proteobacterium Bartonella infecting rodents. Microb Ecol. 2011;61:134–145. doi: 10.1007/s00248-010-9735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Telfer S, Lambin X, Birtles R, Beldomenico P, Burthe S, Paterson S, Begon M. Species interactions in a parasite community drive infection risk in a wildlife population. Science. 2010;330:243–246. doi: 10.1126/science.1190333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daly JS, Worthington MG, Brenner DJ, Moss CW, Hollis DG, Weyant RS, Steigerwalt AG, Weaver RE, Daneshvar MI, O’Connor SP. Rochalimaea elizabethae sp. nov. isolated from a patient with endocarditis. J Clin Microbiol. 1993;31:872–881. doi: 10.1128/jcm.31.4.872-881.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Welch DF, Carroll KC, Hofmeister EK, Persing DH, Robison DA, Steigerwalt AG, Brenner DJ. Isolation of a new subspecies, Bartonella vinsonii subsp. arupensis, from a cattle rancher: identity with isolates found in conjunction with Borrelia burgdorferi and Babesia microti among naturally infected mice. J Clin Microbiol. 1999;37:2598–2601. doi: 10.1128/jcm.37.8.2598-2601.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kosoy M, Murray M, Gilmore RD, Jr, Bai Y, Gage KL. Bartonella strains from ground squirrels are identical to Bartonella washoensis isolated from a human patient. J Clin Microbiol. 2003;41:645–650. doi: 10.1128/JCM.41.2.645-650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fenollar F, Sire S, Raoult D. Bartonella vinsonii subsp. arupensis as an agent of blood culture-negative endocarditis in a human. J Clin Microbiol. 2005;43:945–947. doi: 10.1128/JCM.43.2.945-947.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Probert W, Louie JK, Tucker JR, Longoria R, Hogue R, Moler S, Graves M, Palmer HJ, Cassady J, Fritz CL. Meningitis due to a “Bartonella washoensis”- like human pathogen. J Clin Microbiol. 2009;47:2332–2335. doi: 10.1128/JCM.00511-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dehio C. Infection-associated type IV secretion systems of Bartonella and their diverse roles in host cell interaction. Cell Microbiol. 2008;10:1591–1598. doi: 10.1111/j.1462-5822.2008.01171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colton L, Zeidner N, Kosoy MY. Experimental infection of Swiss Webster mice with four rat Bartonella strains: host specificity, bacteremia kinetics, dose dependent response, and histopathology. Comp Immunol Microbiol Infect Dis. 2011;34:465–473. doi: 10.1016/j.cimid.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 20.Saenz HL, Engel P, Stoeckli MC, Lanz C, Raddatz G, Vayssier-Taussat M, Birtles R, Schuster SC, Dehio C. Genomic analysis of Bartonella identifies type IV secretion systems as host adaptability factors. Nat Genet. 2007;39:1469–1475. doi: 10.1038/ng.2007.38. [DOI] [PubMed] [Google Scholar]
- 21.Berglund EC, Frank AC, Calteau A, Pettersson OV, Granberg F, Eriksson A-S, Näslund K, Holmberg M, Lindroos H, Andersson SGE. Run-off replication of host adaptability genes is associated with gene transfer agents in the genome of mouse-infecting Bartonella grahamii. PLoS Genet. 2009;5:e1000546. doi: 10.1371/journal.pgen.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmiederer M, Anderson B. Cloning, sequencing, and expression of three Bartonella henselae genes homologous to the Agrobacterium tumefaciens VirB region. DNA Cell Biol. 2000;19:141–147. doi: 10.1089/104454900314528. [DOI] [PubMed] [Google Scholar]
- 23.Christie PJ. Type IV secretion: the Agrobacterium VirB/D4 and related conjugation systems. Biochim Biophys Acta. 2004;1694:219–234. doi: 10.1016/j.bbamcr.2004.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seubert A, Hiestand R, de la Cruz F, Dehio C. A bacterial conjugation machinery recruited for pathogenesis. Mol Microbiol. 2003;49:1253–1266. doi: 10.1046/j.1365-2958.2003.03650.x. [DOI] [PubMed] [Google Scholar]
- 25.Scheidegger F, Ellner Y, Guye P, Rhomberg TA, Weber H, Augustin HG, Dehio C. Distinct activities of Bartonella henselae type IV secretion effector proteins modulate capillary sprout formation. Cell Microbiol. 2009;11:1088–1101. doi: 10.1111/j.1462-5822.2009.01313.x. [DOI] [PubMed] [Google Scholar]
- 26.Alsmark CM, Frank AC, Karlberg EO, Legault BA, Ardell DH, Canbäck B, Eriksson AS, Näslund AK, Handley SA, Huvet M, La Scola B, Holmberg M, Andersson SG. The louse-borne human pathogen Bartonella quintana is a genomic derivative of the zoonotic agent Bartonella henselae. Proc Natl Acad Sci U S A. 2004;101:9716–9721. doi: 10.1073/pnas.0305659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schulein R, Dehio C. The VirB/VirD4 type IV secretion system of Bartonella is essential for establishing intraerythrocytic infection. Mol Microbiol. 2002;46:1053–1067. doi: 10.1046/j.1365-2958.2002.03208.x. [DOI] [PubMed] [Google Scholar]
- 28.Schulein R, Guye P, Rhomberg TA, Schmid MC, Schröder G, Vergunst AC, Carena I, Dehio C. A bipartite signal mediates the transfer of type IV secretion substrates of Bartonella henselae into human cells. Proc Nat Acad Sci U S A. 2005;102:856–861. doi: 10.1073/pnas.0406796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bajer A, Pawełczyk A, Behnke JM, Gilbert FS, Siński E. Factors affecting the component community structure of haemoparasites in bank voles (Clethrionomys glareolus) from the Mazury Lake District region of Poland. Parasitology. 2001;122:43–54. doi: 10.1017/S0031182000007058. [DOI] [PubMed] [Google Scholar]
- 30.Behnke JM, Barnard CJ, Bajer A, Bray D, Dinmore J, Frake K, Osmond J, Race T, Siński E. Variation in the helminth community structure in bank voles (Clethrionomys glareolus) from three comparable localities in the Mazury Lake District region of Poland. Parasitology. 2001;123:401–414. doi: 10.1017/S0031182001008605. [DOI] [PubMed] [Google Scholar]
- 31.Bajer A, Bednarska M, Pawełczyk A, Behnke JM, Gilbert FS, Siński E. Prevalence and abundance of Cryptosporidium parvum and Giardia spp. in wild rural rodents from the Mazury Lake District region of Poland. Parasitology. 2002;125:21–34. doi: 10.1017/S0031182002001865. [DOI] [PubMed] [Google Scholar]
- 32.Bajer A, Behnke JM, Gilbert FS, Siński E. Factors affecting the component community structure of haemoparasites in common voles (Microtus arvalis) from the Mazury Lake District region of Poland. Parasitol Res. 2004;92:270–284. doi: 10.1007/s00436-003-1040-1. [DOI] [PubMed] [Google Scholar]
- 33.Pawełczyk A, Bajer A, Behnke JM, Gilbert FS, Sinski E. Factors affecting the component community structure of haemoparasites in common voles (Microtus arvalis) from the Mazury Lake District region of Poland. Parasitol Res. 2004;92:270–284. doi: 10.1007/s00436-003-1040-1. [DOI] [PubMed] [Google Scholar]
- 34.Bajer A, Behnke JM, Pawełczyk A, Kuliś K, Sereda MJ, Siński E. Medium-term temporal stability of the helminth component community structure in bank voles (Clethrionomys glareolus) from the Mazury Lake District region of Poland. Parasitology. 2005;130:213–228. doi: 10.1017/S0031182004006389. [DOI] [PubMed] [Google Scholar]
- 35.Siński E, Pawełczyk A, Bajer A, Behnke JM. Abundance of wild rodents, ticks and environmental risk of Lyme borreliosis: a longitudinal study in an area of Mazury Lakes district of Poland. Ann Agric Environ Med. 2006;13:295–300. [PubMed] [Google Scholar]
- 36.Paziewska A, Zwolińska L, Harris PD, Bajer A, Siński E. Utilisation of rodent species by larvae and nymphs of hard ticks (Ixodidae) in two habitats in NE Poland. Exp Appl Acarol. 2010;50:79–91. doi: 10.1007/s10493-009-9269-8. [DOI] [PubMed] [Google Scholar]
- 37.Welc-Falęciak R, Paziewska A, Bajer A, Behnke JM, Siński E. Bartonella spp. infection in rodents from different habitats in the Mazury Lake District, Northeast Poland. Vector Borne Zoonotic Dis. 2008;8:467–474. doi: 10.1089/vbz.2007.0217. [DOI] [PubMed] [Google Scholar]
- 38.Harris PD, Paziewska A, Zwolińska L, Siński E. Seasonality of the ectoparasite community of woodland rodents in a Mazurian Forest, Poland. Wiad Parazytol. 2009;55:377–388. [PubMed] [Google Scholar]
- 39.Norman AF, Regnery R, Jameson P, Greene C, Krause DC. Differentiation of Bartonella-like isolates at the species level by PCR-restriction fragment length polymorphism in the citrate synthase gene. J Clin Microbiol. 1995;33:1797–1803. doi: 10.1128/jcm.33.7.1797-1803.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chmielewski T, Podsiadły E, Tylewska-Wierzbanowska S. Presence of Bartonella spp. in various human populations. Pol J Microbiol. 2007;56:33–38. [PubMed] [Google Scholar]
- 41.Bown KJ, Bennett M, Begon M. Flea-borne Bartonella grahamii and Bartonella taylorii in bank voles. Emerg Infect Dis. 2004;10:684–687. doi: 10.3201/eid1004.030455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di Tommaso P, Moretti S, Xenarios I, Orobitg M, Montanyola A, Chang J-M, Taly J-F, Notredame C. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 2011;39:W13–W17. doi: 10.1093/nar/gkr245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yeo H-J, Yuan Q, Beck MR, Baron C, Waksman G. Structural and functional characterization of the VirB5 protein from the type IV secretion system encoded by the conjugative plasmid pKM101. Proc Nat Acad Sci U S A. 2003;100:15947–15952. doi: 10.1073/pnas.2535211100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Backert S, Fronzes R, Waksman G. VirB2 and VirB5 proteins: specialized adhesins in bacterial type-IV secretion systems? Trends Microbiol. 2008;16:409–413. doi: 10.1016/j.tim.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 45.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 46.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL Workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 48.Yang Z, Wong WSW, Nielsen R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- 49.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 50.Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- 51.Templeton AR, Boerwinkle E, Sing CF. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. I. Basic theory and an analysis of alcohol dehydrogenase activity in Drosophila. Genetics. 1987;117:343–351. doi: 10.1093/genetics/117.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Templeton AR, Crandall KA, Sing CF. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data: III Cladogram estimation. Genetics. 1992;132:619–633. doi: 10.1093/genetics/132.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Templeton AR, Sing CF. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. IV. Nested analyses with cladogram uncertainty and recombination. Genetics. 1993;134:659–669. doi: 10.1093/genetics/134.2.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heath L, van der Walt E, Varsani A, Martin DP. Recombinant patterns in aphthoviruses mirror those found in other picornaviruses. J Virol. 2006;80:11827–11832. doi: 10.1128/JVI.01100-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benkert P, Biasini M, Schwede T. Towards the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27:343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9:1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- 57.Kaiser PO, Riess T, Wagner CL, Linke D, Lupas AN, Schwarz H, Raddatz G, Shäfer A, Kempf VA. The head of Bartonella adhesin A is crucial for host cell interaction of Bartonella henselae. Cell Microbiol. 2008;10:2223–2234. doi: 10.1111/j.1462-5822.2008.01201.x. [DOI] [PubMed] [Google Scholar]
- 58.Shamaei-Tousi A, Cahill R, Frankel G. Interaction between protein subunits of the type IV secretion system of Bartonella henselae. J Bacteriol. 2004;186:4796–4801. doi: 10.1128/JB.186.14.4796-4801.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yuan Q, Carle A, Gao C, Sivanesan D, Khaled Ahmed A, Höppner C, Krall L, Domke N, Baron C. Identification of the VirB4-VirB8-VirB5-VirB2 pilus assembly sequence of type IV secretion systems. J Biol Chem. 2005;280:26349–26359. doi: 10.1074/jbc.M502347200. [DOI] [PubMed] [Google Scholar]
- 60.Sweger D, Resto-Ruiz S, Johnson DP, Schmiederer M, Hawke N, Anderson B. Conservation of the 17-Kilodalton antigen gene within the genus Bartonella. Clin Diagn Lab Immunol. 2000;7:251–257. doi: 10.1128/cdli.7.2.251-257.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huber W, Felger I, Matile H, Lipps HJ, Steiger S, Beck HP. Limited sequence polymorphism in the Plasmodium falciparum merozoite surface protein 3. Mol Biochem Parasitol. 1997;87:231–234. doi: 10.1016/S0166-6851(97)00067-4. [DOI] [PubMed] [Google Scholar]
- 62.Singh S, Soe S, Weisman S, Barnwell JW, Pérignon JL, Druilhe P. A conserved multi-gene family induces cross-reactive antibodies effective in defense against Plasmodium falciparum. PLoS One. 2009;4:e5410. doi: 10.1371/journal.pone.0005410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aras RA, Fischer W, Perez-Perez GI, Crosatti M-L, Ando T, Haas R, Blaser MJ. Plasticity of repetitive DNA sequences within a bacterial (type IV) secretion system component. J Exp Med. 2003;198:1349–1359. doi: 10.1084/jem.20030381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Criss AK, Bonney KM, Chang RA, Duffin PM, LeCuyer BE, Seifert HS. Mismatch correction modulates mutation frequency and pilus phase and antigenic variation in Neisseria gonorrhoeae. J Bacteriol. 2010;192:316–325. doi: 10.1128/JB.01228-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Frank AC, Alsmark CM, Thollesson M, Andersson SGE. Functional divergence and horizontal transfer of type IV secretion systems. Mol Biol Evol. 2005;22:1325–1336. doi: 10.1093/molbev/msi124. [DOI] [PubMed] [Google Scholar]
- 66.Barbian KD, Minnick MF. A bacteriophage-like particle from Bartonella bacilliformis. Microbiology. 2000;146:599–609. doi: 10.1099/00221287-146-3-599. [DOI] [PubMed] [Google Scholar]
- 67.Kraft C, Suerbaum S. Mutation and recombination in Helicobacter pylori: mechanisms and role in generating strain diversity. Int J Med Microbiol. 2005;295:299–305. doi: 10.1016/j.ijmm.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 68.Schak JR, Dick JJ, Meinersmann RJ, Perez-Perez GI, Blaser MJ. Repeat associated plasticity in the Helicobacter pylori RD gene family. J Bacteriol. 2009;191:6900–6910. doi: 10.1128/JB.00706-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Falush D, Kraft C, Correa P, Taylor NS, Fox JG, Achtman M, Suerbaum S. Recombination and mutation during long-term gastric colonisation by Helicobacter pylori: estimates of clock rates, recombination size and minimal age. Proc Nat Acad Sci U S A. 2001;98:15056–15061. doi: 10.1073/pnas.251396098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koesling J, Aebischer T, Falch C, Schulein R, Dehio C. Cutting edge: antibody-mediated cessation of hemotropic infection by the intraerythrocytic mouse pathogen Bartonella grahamii. J Immunol. 2001;167:11–14. doi: 10.4049/jimmunol.167.1.11. [DOI] [PubMed] [Google Scholar]
- 71.Schulein R, Seubert A, Gille C, Lanz C, Hansmann Y, Piémont Y, Dehio C. Invasion and persistent intracellular colonization of erythrocytes. A unique parasitic strategy of the emerging pathogen Bartonella. J Exp Med. 2001;193:1077–1086. doi: 10.1084/jem.193.9.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vorou RM, Papavassiliou VG, Tsiodras S. Emerging zoonoses and vector-borne infections affecting humans in Europe. Epidemiol Infect. 2007;135:1231–1247. doi: 10.1017/S0950268807008527. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 51 kb)