

Abstract
This study explored the effects of the antioxidant astaxanthin on paraoxonase and thioredoxin reductase activities as well as on other oxidative stress parameters and on the lipid profile in hypercholesterolemic rabbits. Rabbits were fed a standard or a hypercholesterolemic diet alone or supplemented with 50, 100 and 500 mg/100 g of astaxanthin for 60 days. Antioxidant enzymes activities, lipid profile and oxidative stress markers were evaluated in the serum. The hypercholesterolemic diet increased lipids, including unsaturated fatty acids level, whereas it decreased saturated fatty acids level. These changes were accompanied by increased levels of oxidized low-density lipoprotein and oxidized low-density lipoprotein antibodies, as well as lipid and protein oxidation. Astaxanthin (100 and 500 mg/100 g) prevented hypercholesterolemia-induced protein oxidation, whereas 500 mg/100 g of astaxanthin decreased protein oxidation per se. The activities of superoxide dismutase and thioredoxin reductase were enhanced, whereas paraoxonase activity was inhibited in hypercholesterolemic rabbits. All astaxanthin doses prevented changes in thioredoxin reductase and paraoxonase activities. This effect was not related to a direct effect of astaxanthin on these enzymes, because in vitro astaxanthin enhanced thioredoxin reductase and had no effect on paraoxonase activity. Astaxanthin could be helpful in cardiovascular diseases by restoring thioredoxin reductase and paraoxonase activities.
Keywords: fatty acids, atherosclerosis, oxidative stress
Introduction
Hypercholesterolemia is among the major risk factors for atherosclerosis.(1) Low-density lipoproteins (LDL), the major cholesterol carrier in the circulation, can undergo oxidative modification by vascular cells and the cellular uptake of oxidized LDL (LDLox) also leads to the generation of reactive oxygen species (ROS).(2) Accordingly, increased levels of LDLox and oxidized LDL antibodies (LDLoxAB) have been found in the serum of hypercholesterolemic (HC) subjects.(3)
Oxidative stress plays an important role in hypercholesterolemia and atherosclerosis.(4) LDL oxidation affects its lipid and protein components and oxidative modifications in these molecules are involved in the mechanisms leading to endothelial dysfunction during HC atherosclerosis.(4) Oxidative modifications in lipids and proteins can be assessed by measuring thiobarbituric acid reactive substances (TBARS) and protein carbonyl contents.(4) In addition, LDL size and oxidizability seem to be related to its fatty acid (FA) composition.(5) However, the relationships between LDL oxidizability, FA composition and oxidant/antioxidant status in hypercholesterolemia are still not clear.
Since LDL oxidation can initiate atherogenesis, antioxidant status should have a major impact not only on the rate of LDL oxidation but also on the development of atherosclerosis.(4) The antioxidant enzyme thioredoxin reductase-1 (TrxR-1) is a selenoflavoprotein involved in many processes that modulate intracellular ROS levels.(6) Since TrxR-1 has a broad range of substrates, such as thioredoxin and selenium compounds, and acts in the redox regulation, it could play an important protective role against LDL oxidation and atherosclerosis progression. Accordingly, an up-regulation of TrxR-1 in human macrophages has been described as a cellular defense against LDLox.(7) However, only few studies were found on the behavior of TrxR-1 in the extracellular environment during hypercholesterolemia and cardiovascular diseases.(8,9) Another line of defense against oxidative stress is paraoxonase (PON1), a circulating enzyme attached to high-density lipoprotein (HDL). It protects plasma lipoproteins from oxidative modification by hydrolyzing and reducing bioactive lipid hydroperoxides.(10) In humans, low PON1 activity is an independent predictor of acute coronary events.(11)
Supplementation with antioxidant-containing foodstuffs, such as carotenoid-rich fruits, decreases LDL oxidation, playing a beneficial role against cardiovascular diseases.(12) Astaxanthin (ASX) is a carotenoid synthesized by algae and distributed in marine seafood.(13) It effectively scavenges free radicals, thereby protecting FA and biological membranes from oxidative damage.(13) However, the possible protective role of ASX against FA changes induced by hypercholesterolemia has not been investigated. ASX has been suggested as a novel potential treatment for oxidative stress and inflammation in cardiovascular diseases,(14) but there are still many controversies about the protective potential of ASX in cardiovascular events. ASX had no protective effect against LDL oxidation and atherosclerotic lesions in Watanabe heritable hyperlipidemic rabbits(15) or against aortic damage in atherosclerotic New Zealand rabbits.(16) However, ASX prevented the oxidation of human LDL in vitro(17) and stabilised atherosclerotic plaques in rabbits.(18)
We have recently evaluated the effects of ASX in the aortic tissue of atherosclerotic rabbits.(16) Since ASX is carried in serum and hypercholesterolemia is an important event in atherosclerosis, the knowledge of ASX effects on the lipids and oxidative stress in the serum will complement previous data on the aortic tissue. Furthermore, the possible protective role of ASX on PON1 activity during experimental hypercholesterolemia has not been investigated.
The objective of the present study was to investigate the possible protective role of ASX in experimental hypercholesterolemia by evaluating PON1 and TrxR-1 activities as well as other oxidative stress parameters, lipid levels and FA composition in the serum.
Materials and Methods
Ethics
This study was approved by the Ethics and Animal Welfare Committee of the Federal University of Santa Maria (23081.019182/2007-10) and is in agreement with Brazilian guidelines for animal usage in research.
Serum ASX
To evaluate the resulting concentration of ASX in blood after dietary intake, eight-weeks-old male New Zealand white rabbits (1.7 to 2.0 kg of body weight) were fed natural ASX (2% powder, Fuji Chemical Industry Co. Ltd., Toyama, Japan) at 50, 100 and 500 mg/100 g diet in standard chow. The standard chow (SUPRA, São Leopoldo, Brazil) had (g/kg): 120 moisture, 170 crude protein, 20 ether extract, 140 ash, 130 crude fibre, 12 phosphorus and 6 calcium. ASX was dissolved in diethyl ether and sprayed on the standard chow to yield the ASX-supplemented diets. The ASX-supplemented chows were left to allow the ether to evaporate before storage, protected from light and under refrigeration. The ASX doses were chosen based on a previous study.(15) Three animals per experimental group were used in this experiment. Rabbits were previously fasted for twelve hours and then blood samples were taken from the marginal ear vein before (baseline) and 3, 6, 9, 12 and 24 h after receiving a single meal (100 g of chow). Blood samples were centrifuged to yield serum samples. Serum ASX concentrations were analyzed by HPLC. Samples were prepared as previously reported.(19) Briefly, 600 µl of acetone containing 2 µg/ml of β-apo-8'-carotenal, as an internal standard, was added to 100 µl of a serum sample. After vortex-mixing for 1 min, the mixture was extracted with 600 µl of hexane. The organic layer was collected and dried under a gentle stream of N2 gas at 25°C. A 100 µl aliquot of the mobile phase was added to reconstitute the residue and 20 µl was directly injected onto a C18 reversed-phase Shim-pack CLC ODS (M) column (250 × 4.6 mm, 5 µm) with a guard column (Shim-pack GODS, 10 × 4 mm) (Shimadzu, Tokyo, Japan). The mobile phase consisted of methanol, t-butylmethylether and 1% phosphoric acid aqueous solution at a flow-rate of 1.0 ml/min. A gradient was employed, from 15:81:4 to 80:16:4 in 19 min, returning to the initial proportion until the end of the run (at 25 min). The column eluent was monitored using a photodiode array detector at 474 nm. The retention times for ASX and the internal standard were approximately 5.0 and 12.0 min, respectively. The detection limit of ASX in serum samples was 0.05 µg/ml.
Hypercholesterolemia experiment
Eight-weeks-old male New Zealand white rabbits were housed in individual cages, under a 12:12 h light:dark cycle with access to100 g of chow per day and water ad libitum. After 1 week of adaptation, the rabbits weighing 1.83 ± 0.33 kg were randomly divided into eight experimental groups, each one with six animals. The control group was fed a standard diet chow (SUPRA, São Leopoldo, Brazil); the HC groups were fed a diet enriched with 1% cholesterol (Vetec, Brazil); and the supplemented groups were fed natural ASX (2% powder, Fuji Chemical Industry Co. Ltd., Toyama, Japan) at 50, 100 and 500 mg/100 g diet in standard or HC diets. The standard diet chow had (g/kg): 120 moisture, 170 crude protein, 20 ether extract, 140 ash, 130 crude fibre, 12 phosphorus and 6 calcium. Cholesterol and ASX were dissolved in diethyl ether and sprayed on the standard chow to yield the cholesterol enriched and ASX-supplemented diets. The standard chow of the control group was sprayed with diethyl ether (vehicle). The standard and supplemented chows were left to allow the ether to evaporate before storage, protected from light and under refrigeration. Chows were prepared once a week. All animals received a daily portion of 100 g chow for 60 days, and their body weight was checked once a week. The cholesterol dose was chosen based on a previous study.(20) Blood samples were collected from rabbits fasted for twelve hours by cardiac puncture before (baseline) and after 60 days of treatment. At the end of the experimental period, animals were sacrificed with an intravenous overdose of thiopental. Blood samples were collected into tubes without additives and centrifuged at 3,000 × g for 10 min. Serum was removed immediately and stored at −20°C until biochemical analyses.
Serum lipids
Total cholesterol (TC), triglycerides (TG) and HDL levels were determined by enzymatic methods, using commercial kit reagents (Doles, Goiania, GO, Brazil). Non-HDL cholesterol levels were calculated as the difference between TC and HDL and represent the sum of VLDL (very low-density lipoprotein), LDL and IDL (intermediate-density lipoprotein) fractions.
Lipid peroxidation (LPO)
After the addition of 7.2 mM butylated hydroxytoluene to prevent further oxidation, LPO was estimated by the measurement of TBARS in serum samples using a standard curve of 1,1,3,3-tetraethoxypropane.(21)
Protein oxidation
Protein oxidation was determined based on the reaction of the carbonyl groups with 2,4-dinitrophenylhydrazine (DNPH) to form 2,4-dinitrophenylhydrazone.(22)
LDLox and LDLoxAB
LDLox was determined by a capture ELISA kit according to the manufacturer’s instructions (Mercodia AB, Uppsala, Sweden).(23) LDLoxAB was determined using ELISA.(24)
PON1 activity
PON1 activity was assessed by measuring the rate of paraoxon hydrolysis to yield para-nitrophenol, at 412 nm and 25°C.(25)
Antioxidant enzymes
TrxR-1 activity was determined using 5,5'-dithiobis (2-nitrobenzoic acid) (DTNB) and reduced adenine dinucleotide phosphate (NADPH). This method is based on the reduction of DTNB, which is indicated by an increase in absorbance at 412 nm.(26) Superoxide dismutase (SOD) activity was determined based on its ability to inhibit the autoxidation of epinephrine to adrenochrome at an alkaline pH.(27)
FA composition
FA composition in serum was determined by a one-step reaction(28) with some modification. Serum (50 µl) was mixed with 1 ml methanol/isooctane (4:1, v/v) and 100 µl acetyl chloride and was incubated at 100°C for 60 min; then 60 g/l aqueous potassium carbonate containing 100 g/l sodium chloride was added. The mixture was shaken for 10 min at room temperature and centrifuged at 1,800 × g for 5 min. The isooctane phase, containing the FA methyl esters, was injected (1 µl, split ratio 50:1) in an Agilent Technologies gas chromatograph (HP 6890) fitted with a capillary column DB-23 (50% cyanopropylmethylpolysiloxane, 60 m × 0.25 mm × 0.25 µm) and flame ionization detection. Standard FA methyl esters (37-component FAME Mix, Sigma, St. Louis, MO) were used to identify the fatty acids.
Protein quantification
Protein was measured using bovine serum albumin as the standard.(29)
In vitro cell-free assays
The direct effect of ASX on TrxR-1 and PON1 activities was assessed in vitro in cell-free assays using the serum from four normocholesterolemic non-treated rabbits as the source of enzymes. The serum was preincubated at 37°C for 1 h in the presence of ASX (0, 3, 10, 30 or 100 µg/ml). After the preincubation, the enzymes activities were determined as described before for TrxR-1(26) and PON1.(25) ASX concentrations for these in vitro experiments were chosen based on the blood levels of ASX after oral intake, which were found in the present study and in a previous one.(30)
Statistical analysis
The data from the in vitro assays were analyzed using one-way ANOVA, whereas the data from the in vivo experiments were analyzed using two-way ANOVA. ANOVA tests were followed by post hoc Duncan’s multiple range test when necessary. Results were expressed as the mean ± standard error of the mean and differences were considered statistically significant when p<0.05.
Results
ASX was not detected in the serum of rabbits prior to ASX supplementation (baseline value). Serum ASX concentrations increased in a dose-dependent manner after a single meal containing ASX (50–500 mg/100 g) (p<0.05, data not shown). Peak serum concentrations of ASX were observed around 12 h after a single meal and these concentrations ranged from 0.67 to 6.96 µg/ml in rabbits fed 50 to 500 mg/100 g of ASX, respectively (p<0.05).
In the hypercholesterolemia study, no differences were found among the experimental groups with respect to weight gain during the experimental period (data not shown). The initial average body weight of the animals was 1.83 ± 0.33 kg, and the animals grew to a final body weight of 2.68 ± 0.22 kg. In addition, the baseline values (before the experimental period) of all parameters were not different among the experimental groups (data not shown).
The profile of serum FA is presented in Table 1. Feeding rabbits a HC diet for 60 days decreased the serum levels of total saturated FA (SFA), whereas the total unsaturated FA (UFA), monounsaturated FA (MUFA) and UFA/SFA ratio were increased when compared to the no-ASX control group (p<0.05). These changes were due to a decrease in the levels of palmitic and estearic acid and an increase in the levels of oleic acid (data not shown). HC rabbits also had decreased levels of arachidonic acid (data not shown). ASX had no effect on changes in the FA composition induced by a HC diet at any of the evaluated doses (p>0.05). In addition, neither cholesterol nor ASX supplementation changed the levels of total polyunsaturated FA (PUFA) in the serum of rabbits (p>0.05).
Table 1.
Fatty acid composition (% of total fatty acids) of serum from rabbits fed a cholesterol and/or ASX enriched diet
| Fatty acids | Control |
Cholesterol |
||||||
|---|---|---|---|---|---|---|---|---|
| ASX0 | ASX50 | ASX100 | ASX500 | ASX0 | ASX50 | ASX100 | ASX500 | |
| Σ SFA | 43.4 ± 1.9 | 45.6 ± 0.8 | 45.1 ± 1.7 | 45.3 ± 1.2 | 33.7 ± 1.4* | 33.0 ± 2.5* | 34.2 ± 2.4* | 33.7 ± 1.7* |
| Σ UFA | 56.5 ± 2.5 | 54.3 ± 0.5 | 54.8 ± 1.7 | 54.6 ± 1.2 | 66.2 ± 1.5* | 66.9 ± 2.5* | 65.6 ± 2.4* | 66.1 ± 1.7* |
| Σ MUFA | 21.4 ± 2.0 | 18.1 ± 1.0 | 18.5 ± 0.8 | 20.2 ± 0.5 | 35.1 ± 0.7* | 35.4 ± 0.7* | 32.4 ± 1.5* | 33.8 ± 0.9* |
| Σ PUFA | 35.1 ± 1.6 | 36.7 ± 1.8 | 36.2 ± 1.4 | 33.3 ± 2.2 | 31.3 ± 1.0 | 30.6 ± 2.5 | 33.3 ± 1.2 | 32.4 ± 1.8 |
| UFA/SFA ratio | 1.4 ± 0.1 | 1.3 ± 0.0 | 1.3 ± 0.1 | 1.3 ± 0.1 | 2.0 ± 0.1* | 2.1 ± 0.2* | 2.0 ± 0.2* | 2.0 ± 0.1* |
Results are means ± SEM (n = 6). ASX, astaxanthin; Σ SFA, sum of SFA; Σ UFA, sum of UFA; Σ MUFA, sum of MUFA; Σ PUFA, sum of PUFA. *Different from control-ASX0 group (p<0.05).
The cholesterol enriched diet increased TC, TG, HDL and non-HDL cholesterol serum levels when compared to control groups (Fig. 1, p<0.05), which confirms that the cholesterol enriched diet induced hyperlipidemia. Dietary supplementation with ASX did not prevent the increase in serum lipids at any of the doses evaluated (Fig. 1, p>0.05).
Fig. 1.

Total cholesterol (A), triglycerides (B), HDL (C) and non-HDL cholesterol (D) levels of rabbits fed with a cholesterol and/or ASX enriched diet (means ± SEM, n = 6). *Different from control groups (p<0.05).
The HC diet increased TBARS and protein carbonyl groups content, as well as LDLox and LDLoxAB levels when compared to the control group (Fig. 2, p<0.05). Although the increased levels of TBARS, LDLox and LDLoxAB were not attenuated by any dose of ASX, 100 and 500 mg/100 g of ASX did prevent the increase in protein carbonyl content (p<0.05). In addition, 500 mg/100 g of ASX decreased protein carbonyl content per se (p<0.05).
Fig. 2.

TBARS levels (A), protein carbonyl content (B), LDLox (C) and LDLoxAB (D) levels of rabbits fed with a cholesterol and/or ASX enriched diet (means ± SEM, n = 6). *Different from control groups (p<0.05). #Different from ASX 0-Chol group (p<0.05). &Different from ASX 0-control group (p<0.05). MDA = malondialdehyde.
The HC diet inhibited PON1 activity, but increased SOD and TrxR-1 activities when compared to the control group (p<0.05; Fig. 3). Although no ASX doses prevented the increase in SOD activity, changes in TrxR-1 and PON1 activities were attenuated by all ASX doses evaluated.
Fig. 3.
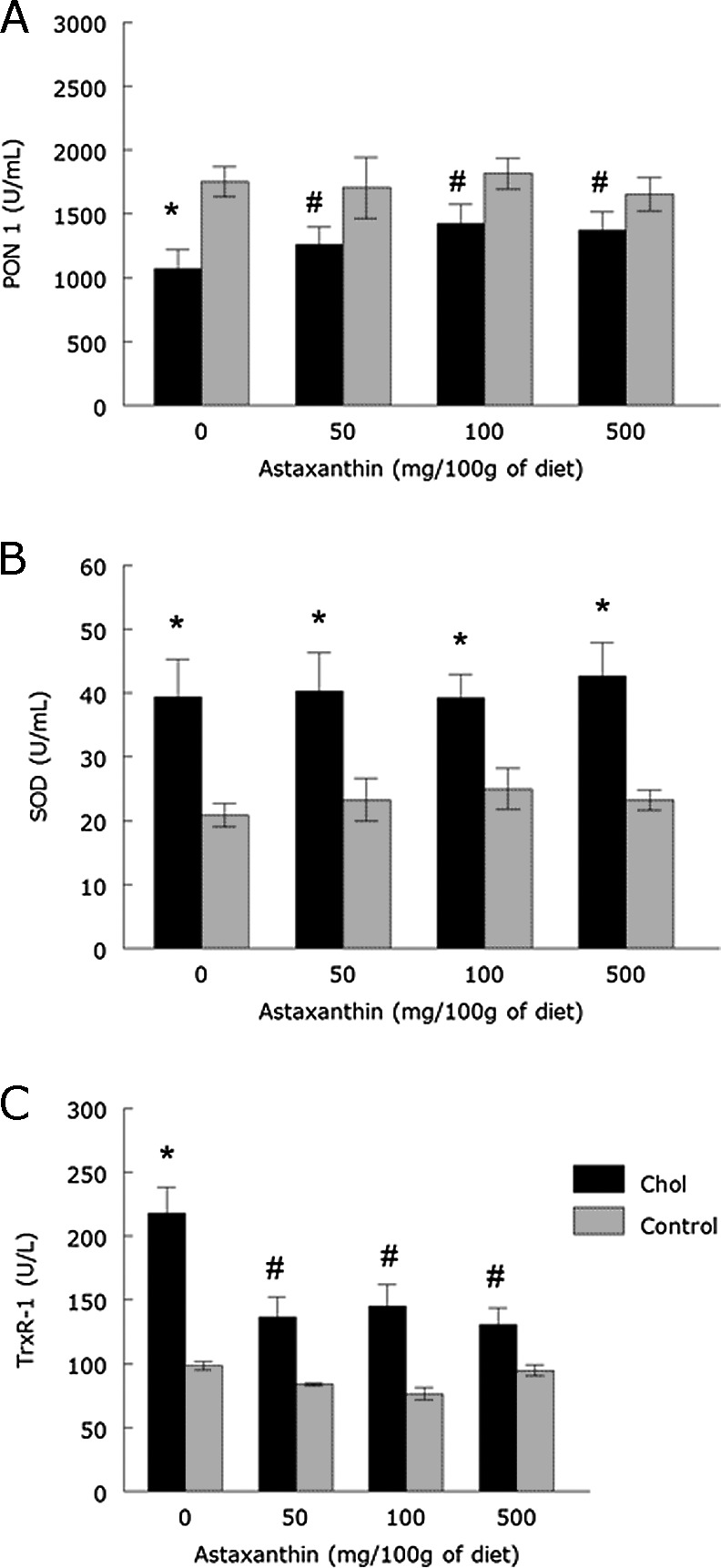
PON1 (A), SOD (B) and TrxR-1 (C) activities of rabbits fed with a cholesterol and/or ASX enriched diet (means ± SEM, n = 6). *Different from control groups (p<0.05). #Different from ASX 0-Chol group (p<0.05).
Cell-free in vitro assays were used to determine whether ASX had a direct effect on the activity of TrxR-1 or PON1. The serum of untreated normocholesterolemic rabbits was used as the source of enzymes in these assays. The incubation with ASX (30 and 100 µg/ml) caused a significant increase in TrxR-1 activity when compared to control (p<0.05; Fig. 4A). No ASX concentration (0–100 µg/ml) affected PON1 activity in vitro (p>0.05; Fig. 4B).
Fig. 4.
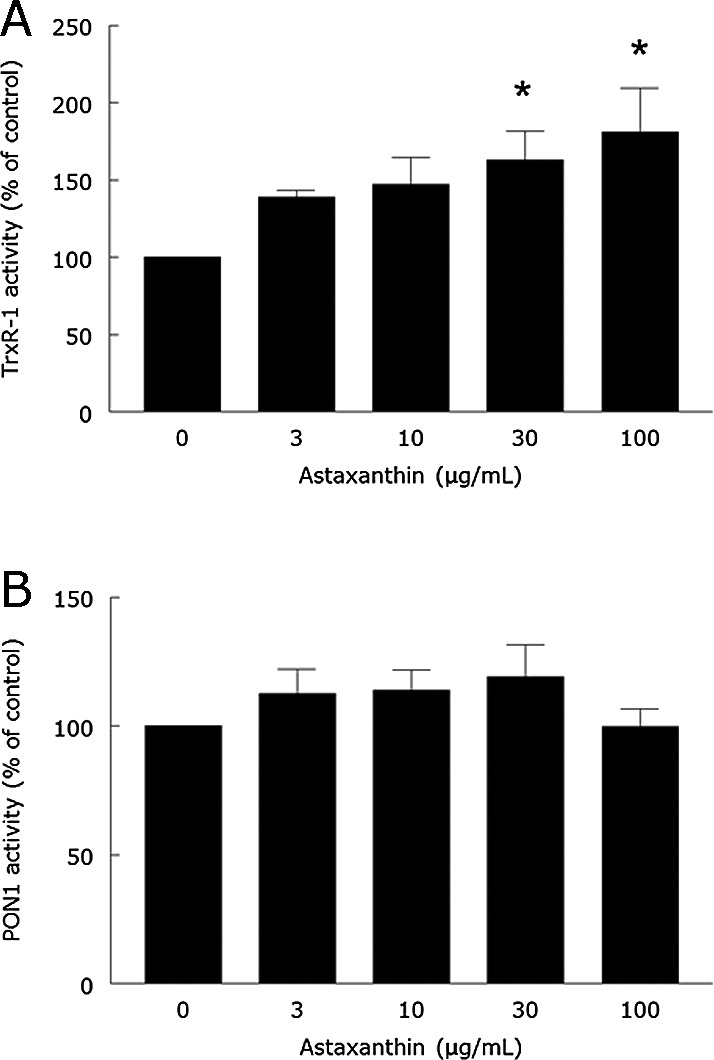
In vitro effect of ASX on serum TrxR-1 (A) and PON1 (B) activities from rabbits. Serum was preincubated for 1 h in the presence of 0, 3, 10, 30 or 100 µg/ml of ASX. Then, the activities of TrxR-1 and PON1 were determined as described in Materials and Methods section. Data are means ± SEM (n = 4). *Different from control (0 µg ASX/ml) at p<0.05.
Discussion
The present study clearly indicates that ASX is well absorbed into the systemic blood circulation and maximum levels of ASX in the serum of rabbits were reached 12 h after a single meal containing 50, 100 or 500 mg/100 g of ASX. These findings are consistent with a previous study in humans,(31) which showed that the maximum plasma level was reached 11.5 h after the oral ASX administration (doses of 10 and 100 mg).
Cholesterol-enriched diets induce MUFA synthesis(32) in order to provide free FA for the synthesis of cholesteryl esters, which is the major form of circulating cholesterol in the plasma of cholesterol-fed rabbits.(33) Accordingly, the contribution of MUFA to the total levels of serum FA increased after feeding rabbits with a HC diet for 60 days. The decreased levels of total SFA in cholesterol-fed rabbits may be explained because palmitic and estearic acid are consumed to synthesize MUFA.(32) As reported previously, the HC diet increased the levels of serum TC, TG, HDL and non-HDL cholesterol(20) along with changes in the FA profile. In fact, non-high-density lipoproteins, such as VLDL and LDL are the major carriers of cholesterol in circulation.(34)
Hypercholesterolemia increases the synthesis of prostaglandins and leukotrienes from arachidonic acid generating ROS.(2) Therefore, hypercholesterolemia is associated with the increased susceptibility of LDL to oxidation.(2) Accordingly, in the present study LDL oxidation in HC rabbits was accompanied by reduced arachidonic acid levels. LDL oxidation is dependent on the presence of oxidizable lipids, such as PUFA.(35) However, PUFA levels of HC rabbits remained unchanged, probably due to a balance between PUFA synthesis and degradation. In fact, PUFA may be lost by oxidation in salmon, but oxidative events induce a compensatory overproduction of PUFA in order to replace this loss.(36)
ASX has strong antioxidant properties, protecting FA and biological membranes from oxidative damage.(13) Additionally, ASX deficiency increases the conversion of FA to their long-chain highly unsaturated products in salmons in order to replace those FA lost due to the oxidative process.(36) However, in our study, ASX supplementation did not prevent changes in the FA composition in HC rabbits. This could be linked to the fact that xanthophylls, such as ASX, are found in chylomicrons and in the serum of humans in a non-esterified free form,(37) not requiring FA for its transport. The lack of hypolipidemic effect of ASX in HC rabbits in this study has already been reported,(15) but this work is the first to report the effects of ASX on serum FA composition during experimental hypercholesterolemia.
LDL oxidation in vivo affects its lipid and protein components at different stages.(4) In the first stage, lipids are oxidized with minor protein oxidation. In the second stage, both lipids and proteins are attacked by free radicals.(4) In agreement, HC rabbits showed increased serum lipid and protein oxidation along with high levels of LDLox. LDLox may damage membrane proteins and phospholipids, which can impair the endothelial function.(38) Oxidative modification of LDL induces the formation of LDLoxAB, which can be detected in serum.(3) Moreover, LDLoxAB have been shown to block the uptake of LDLox by macrophages, suggesting a possible role in preventing the formation of foam cells.(38) Accordingly, we observed increased LDLoxAB levels in the serum of HC rabbits, which supports the idea that antibodies were secreted in order to combat LDLox toxicity.
ASX did not prevent the increase in LPO, LDLox or LDLoxAB levels induced by hypercholesterolemia. However, ASX attenuated both basal and cholesterol-induced serum protein oxidation. In fact, ASX is a potent scavenger of oxygen radicals(13) and inhibits both the basal and heavy metal-induced protein oxidation.(39) Due to its polarity, ASX is assumed to be attached to the surface of chylomicrons and lipoproteins as well as in association with serum albumin.(40) Thus, we cannot rule out the thought that ASX would be in a favorable location to prevent protein oxidation rather than lipid oxidation.
Despite protection against protein oxidation, no ASX doses prevented the increase in serum SOD activity induced by hypercholesterolemia. Serum SOD is mainly synthesized and secreted from vascular smooth muscle cells and macrophages, playing a central role in cardiovascular antioxidant mechanisms.(41) SOD activity increased in HC rabbits most likely to remove superoxide anion generated during hypercholesterolemia,(42) however superoxide production seems to have exceeded the antioxidant capacity of SOD, since we observed LPO and protein oxidation even with increased SOD activity.
TrxR-1 is a redox-active selenoprotein that efficiently regenerates oxidized thioredoxin. Reduced thioredoxin maintains proteins, such as peroxiredoxins, in their reduced state in order to remove hydrogen peroxide (H2O2) and alkyl peroxides.(6) The increased TrxR-1 activity in HC rabbits may indicate that TrxR-1 plays a protective role against protein oxidation induced by hypercholesterolemia. In fact, overexpression and release of TrxR-1 occurs in oxidative events.(43) Also, it suggests a role of TrxR-1 in the decomposition of H2O2 generated by SOD.(44) Thus, ASX protection against protein oxidation could also explain its protective action on TrxR-1 changes, since ASX would remove ROS sparing this antioxidant enzyme. Although the protective effect of ASX against changes in TrxR-1 activity has previously been reported in the aortic tissue from atherosclerotic rabbits,(16) this is the first report on serum TrxR-1 activity. Thioredoxin levels are known to increase in plasma during oxidative stress,(45) but there are few data on the behavior of TrxR-1 in plasma and its relationship with cardiovascular diseases.(8,9) Since serum TrxR-1 seems to be secreted by circulating monocytes(43) and these cells are involved in the atherogenic process, it is possible that serum TrxR-1 can be modulated by cardiovascular events, such as hypercholesterolemia and atherosclerosis. A previous report(8) revealed that serum TrxR-1 increased in patients with coronary artery disease measured by coronary angiograms, when compared to healthy individuals. In line with this, atherosclerotic plaques had almost two-fold increase in the level of TrxR-1mRNA when compared to the surrounding healthy areas of the artery specimen taken from the same patients.(46) Thus, changes in TrxR-1 seem to be relevant to the pathology of cardiovascular diseases. Accordingly, dietary blueberries prevented changes in TrxR-1 and reduced the area of atherosclerotic lesion in the aorta of apolipoprotein E-deficient mice.(47)
PON1 is an enzyme almost exclusively located on serum HDL that is mainly responsible for the breakdown of lipid peroxides before they can accumulate in LDL.(10) Despite its importance, the enzymatic activity of PON1 is readily inactivated by oxidants.(10) Accordingly, PON1 activity was inhibited along with increased LDLox in the serum of HC rabbits in the present study. The decrease of serum PON1 activity seems to be important for the pathological changes in rabbits fed an atherogenic diet, because it was accompanied by the formation of atherosclerotic plaques.(48,49) All ASX doses prevented PON1 inhibition and this effect could be explained by the metabolism of xanthophylls, such as ASX, in the blood stream of Atlantic salmon since this process is similar in salmons and mammalians.(50) During its transportation in salmon, ASX is transferred to HDL from VLDL and LDL.(40) Thus, we suggest that ASX could be next to the attachment site of PON1 in HDL and exerting its antioxidant effects. Interestingly, although PON1 activity was restored by ASX, this carotenoid did not prevent LDL oxidation in HC rabbits. HC rabbits that received ASX had PON1 levels similar to those of control rabbits (no cholesterol diets), however, their serum levels of all lipoproteins were much higher than those of the control rabbits. Thus, it is possible that the inability of PON1 to prevent LDL oxidation in the HC rabbits that received ASX may be related to their lower PON1/HDL ratio (0.9–1.6) when compared to control groups (54.5–83.1). Alternatively, these findings suggest that factors other than PON1 may influence LDL oxidation and that these factors could not be completely controlled by ASX. In fact, HDL-associated enzymes other than PON1, such as lecithin/cholesterol acyltransferase (LCAT) and platelet-activating factor acyl hydrolase (PAF-AH) were also implicated in the antioxidative properties of HDL.(51) The protective effect of ASX on PON1 has been described here for the first time, but the effects of ASX on LCAT and PAF-AH still remain unknown. Although ASX did not prevent LDL oxidation in the present study, the prevention of changes in PON1 activity has been associated to the protective effect of antioxidants against atherosclerotic lesions in apolipoprotein E-deficient mice.(52,53) In addition, the hypocholesterolemic drug atorvastatin limited plaque formation and attenuated the inibition in serum PON1 activity caused by HC diet in New Zealand rabbits.(48)
Our cell-free in vitro assays revealed that ASX had no direct effect on PON1 activity, which contrasts with the in vivo effect of ASX in HC rabbits, i.e., increase of PON1 activity. Therefore, the protective effect of ASX against the PON1 decrease caused by hypercholesterolemia is not due to a direct stimulatory effect of this carotenoid on PON1. TrxR-1 data also indicate that the protective effect of ASX against the increase of TrxR-1 caused by hypercholesterolemia in vivo was not caused by a direct inhibitory effect of the carotenoid on enzyme activity, because ASX stimulated TrxR-1 activity in vitro. This stimulatory effect of ASX on TrxR-1 was not observed in vivo, possibly because it occurs at ASX concentrations (30 µg/ml onwards) higher than the peak concentration reached after dietary intake of the highest ASX dose (about 7 µg/ml serum). Although this serum ASX concentration was evaluated after the intake of a single meal, a previous study in other mammals (cats and dogs) showed that serum ASX concentration has a small increase after repeated daily oral doses when compared to a single oral dose (1.5-fold).(30) Thus, we can assume that the daily peak serum ASX concentrations during the treatment of HC rabbits with ASX were only slightly higher than those found in our single meal study.
All protective effects of ASX shown in the present study could be attributed to the high scavenger potential of ASX, which protects against free radicals generated by hypercholesterolemia, sparing antioxidant defenses. ASX stabilizes free radicals by adding them to its long double-bond chain rather than donating an atom or electron to the radical.(15) Consequently, it can resist chain reactions that occur when a FA is oxidized, thus allowing it to scavenge or quench longer than antioxidants that cannot stop this chain reaction.(13)
For these reasons, natural microalgae or shrimp shell extracts containing ASX have been approved by the Food and Drug Administration (FDA) as nutraceuticals, while derivatives of ASX are being developed for pharmaceutical applications in human disorders where oxidative stress-mediated damage may be important, including cardiovascular diseases.(14)
In conclusion, we demonstrated that ASX prevents changes in PON1 and TrxR-1 activities as well as protein oxidation in the serum from HC rabbits and this protection could be valuable for cardiovascular diseases therapy.
Acknowledgments
Authors thank to CNPq and FAPERGS for the financial support, to Fuji Chemical Industry Co. Ltd., Toyama for the kind donation of the astaxanthin and to Carlos Rubini Junior for valuable assistance in gas chromatography analysis. This study was also supported by Capes edital no. 11/2009 – Pró-equipamentos Institucional.
Abbreviations
- ASX
astaxanthin
- ANOVA
analysis of variance
- FA
fatty acids
- FDA
food and drug administration
- HC
hypercholesterolemic
- HDL
high-density lipoprotein
- HPLC
high performance liquid chromatography
- IDL
intermediate-density lipoprotein
- LCAT
lecithin/cholesterol acyltransferase
- LDL
low-density lipoprotein
- LDLox
oxidized low-density lipoprotein
- LDLoxAB
oxidized low-density lipoprotein antibodies
- LPO
lipid peroxidation
- MUFA
monounsaturated fatty acids
- PAF-AH
platelet-activating factor acyl hydrolase
- PON1
paraoxonase
- PUFA
polyunsaturated fatty acids
- ROS
reactive oxygen species
- SFA
saturated fatty acids
- SOD
superoxide dismutase
- TBARS
thiobarbituric acid reactive substances
- TC
total cholesterol
- TG
triglycerides
- TrxR-1
thioredoxin reductase
- UFA
unsaturated fatty acids
- VLDL
very low-density lipoprotein
Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Kruth HS. Lipoprotein cholesterol and atherosclerosis. Curr Mol Med. 2001;1:633–653. doi: 10.2174/1566524013363212. [DOI] [PubMed] [Google Scholar]
- 2.Prasad K, Kalra J. Oxygen free radicals and hypercholesterolemic atherosclerosis: effect of vitamin E. Am Heart J. 1993;125:958–973. doi: 10.1016/0002-8703(93)90102-f. [DOI] [PubMed] [Google Scholar]
- 3.Duarte MM, Rocha JB, Moresco RN, et al. Association between ischemia-modified albumin, lipids and inflammation biomarkers in patients with hypercholesterolemia. Clin Biochem. 2009;42:666–671. doi: 10.1016/j.clinbiochem.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 4.Berliner JA, Navab M, Fogelman AM, et al. Atherosclerosis: basic mechanisms. Oxidation, inflammation, and genetics. Circulation. 1995;91:2488–2496. doi: 10.1161/01.cir.91.9.2488. [DOI] [PubMed] [Google Scholar]
- 5.Kondo A, Murunaka Y, Ohta I, et al. Relationships between triglyceride concentrations and LDL size evaluated by malonadehyde-modified LDL. Clin Chem. 2001;47:893–900. [PubMed] [Google Scholar]
- 6.Nordberg J, Arnér ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med. 2001;31:1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 7.Furman C, Rundlöf AK, Larigauderie G, et al. Thioredoxin reductase 1 is upregulated in atherosclerotic plaques: specific induction of the promoter in human macrophages by oxidized low-density lipoproteins. Free Radic Biol Med. 2004;37:71–85. doi: 10.1016/j.freeradbiomed.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 8.Wu Y, Yang L, Zhong L. Decreased serum levels of thioredoxin in patients with coronary artery disease plus hyperhomocysteinemia is strongly associated with the disease severity. Atherosclerosis. 2010;212:351–355. doi: 10.1016/j.atherosclerosis.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 9.Jekell A, Hossain A, Alehagen U, Dahlström U, Rosé A. Elevated circulating levels of thioredoxin and stress in chronic heart failure. Eur J Heart Fail. 2004;6:883–890. doi: 10.1016/j.ejheart.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 10.Aviram M. Introduction to the serial review on paraoxonases, oxidative stress, and cardiovascular diseases. Free Radic Biol Med. 2004;37:1301–1303. doi: 10.1016/j.freeradbiomed.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 11.Mackness B, Durrington P, McElduff P, et al. Low paraoxonase activity predicts coronary events in the Caerphilly prospective study. Circulation. 2003;107:2775–2779. doi: 10.1161/01.CIR.0000070954.00271.13. [DOI] [PubMed] [Google Scholar]
- 12.Madrid AE, Vásques ZD, Levton AF, Mandiola C, Escobar FJA. Short-term Lycopersicum esculentum consumption may increase plasma high density lipoproteins and decrease oxidative stress. Rev Med Chil. 2006;134:855–862. doi: 10.4067/s0034-98872006000700008. [DOI] [PubMed] [Google Scholar]
- 13.Kurashige M, Okimasu E, Inoue M, Utsumi K. Inhibition of oxidative injury of biological membranes by astaxanthin. Physiol Chem Phys Med NMR. 1990;22:27–38. [PubMed] [Google Scholar]
- 14.Pashkow FJ, Watumull DG, Campbell CL. Astaxanthin: a novel potential treatment for oxidative stress and inflammation in cardiovascular disease. Am J Cardiol. 2008;22:580–680. doi: 10.1016/j.amjcard.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 15.Jacobsson LS, Yuan XM, Ziedén B, Olsson AG. Effects of α-tocopherol and astaxanthin on LDL oxidation and atherosclerosis in WHHL rabbits. Atherosclerosis. 2004;173:231–237. doi: 10.1016/j.atherosclerosis.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 16.Augusti PR, Conterato GM, Somacal S, et al. Astaxanthin reduces oxidative stress, but not aortic damage in atherosclerotic rabbits. J Cardiovasc Pharmacol Ther. 2009;14:314–322. doi: 10.1177/1074248409350136. [DOI] [PubMed] [Google Scholar]
- 17.Iwamoto T, Hosoda K, Hirano R, et al. Inhibition of low-density lipoprotein oxidation by astaxanthin. J Atheroscler Thromb. 2000;7:216–222. doi: 10.5551/jat1994.7.216. [DOI] [PubMed] [Google Scholar]
- 18.Li W, Hellsten A, Jacobsson LS, Blomqvist HM, Olsson AG, Yuan XM. Alpha-tocopherol and astaxanthin decrease macrophage infiltration, apoptosis and vulnerability in atheroma of hyperlipidaemic rabbits. J Mol Cell Cardiol. 2004;37:969–978. doi: 10.1016/j.yjmcc.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 19.Choi HD, Kang HE, Yang SH, Lee MG, Shin WG. Pharmacokinetics and first-pass metabolism of astaxanthin in rats. Br J Nutr. 2011;105:220–227. doi: 10.1017/S0007114510003454. [DOI] [PubMed] [Google Scholar]
- 20.Prasad K. Reduction of serum cholesterol and hypercholesterolemic atherosclerosis in rabbits by secoisolariciresinol diglucoside isolated from flaxseed. Circulation. 1999;99:1355–1362. doi: 10.1161/01.cir.99.10.1355. [DOI] [PubMed] [Google Scholar]
- 21.Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 22.Levine RL, Garland D, Oliver CN, et al. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990;186:464–478. doi: 10.1016/0076-6879(90)86141-h. [DOI] [PubMed] [Google Scholar]
- 23.Holvoet P, Stassen JM, Van Cleemput J, Collen D, Vanhaecke J. Oxidized low density lipoproteins in patients with transplant-associated coronary artery disease. Arterioscler Thromb Vasc Biol. 1998;18:100–107. doi: 10.1161/01.atv.18.1.100. [DOI] [PubMed] [Google Scholar]
- 24.Wu R, Lefvert AK. Autoantibodies against oxidized low density lipoprotein (OxLDL): characterization of antibody isotype, subclass, affinity and effect on the macrophage uptake of oxLDL. Clin Exp Immunol. 1995;102:174–180. doi: 10.1111/j.1365-2249.1995.tb06652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bolayirli IM, Aslan M, Balci H, Altug T, Hacibekiroglu M, Seven A. Effects of atorvastatin therapy on hypercholesterolemic rabbits with respect to oxidative stress, nitric oxide pathway and homocysteine. Life Sci. 2007;81:121–127. doi: 10.1016/j.lfs.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 26.Holmgren A, Björnstedt M. Thioredoxin and thioredoxin reductase. Methods Enzymol. 1995;252:199–208. doi: 10.1016/0076-6879(95)52023-6. [DOI] [PubMed] [Google Scholar]
- 27.Misra HP, Fridovich I. The role of superoxide anion in the auto-oxidation of epinephrine and a simple assay for superoxide-dismutase. J Biol Chem. 1972;247:3170–3175. [PubMed] [Google Scholar]
- 28.Lepage G, Roy CC. Direct transesterification of all classes of lipids in a one-step reaction. J Lipid Res. 1986;27:114–120. [PubMed] [Google Scholar]
- 29.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 30.Park JS, Kim HW, Mathison BD, et al. Astaxanthin uptake in domestic dogs and cats. Nutr Metab (Lond) 2010;7:52. doi: 10.1186/1743-7075-7-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coral-Hinostroza GN, Ytrestøyl T, Ruyter B, Bjerkeng B. Plasma appearance of unesterified astaxanthin geometrical E/Z and optical R/S isomers in men given single doses of a mixture of optical 3 and 3'R/S isomers of astaxanthin fatty acyl diesters. Comp Biochem Physiol C Toxicol Pharmacol. 2004;139:99–110. doi: 10.1016/j.cca.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 32.Ntambi JM. Regulation of stearoyl-CoA desaturase by polyunsaturated fatty acids and cholesterol. J Lipid Res. 1999;40:1549–1558. [PubMed] [Google Scholar]
- 33.Rose HG. Origin of cholesterol esters in the blood of cholesterol-fed rabbits: relative contributions of serum lecithin-cholesterol acyltransferase and hepatic ester synthesis. Biochim Biophys Acta. 1972;260:312–326. doi: 10.1016/0005-2760(72)90042-2. [DOI] [PubMed] [Google Scholar]
- 34.Farmer JA, Gotto AM., Jr Dyslipidemia and the vulnerable plaque. Prog Cardiovasc Dis. 2002;44:415–428. doi: 10.1053/pcad.2002.123474. [DOI] [PubMed] [Google Scholar]
- 35.Balkan J, Kanbağli O, Mehmetçik G, Mutlu-Türkoğlu U, Aykaç-Toker G, Uysal M. Increased lipid peroxidation in serum and low-density lipoproteins associated with aging in humans. Int J Vitam Nutr Res. 2002;72:315–320. doi: 10.1024/0300-9831.72.5.315. [DOI] [PubMed] [Google Scholar]
- 36.Bell JG, McEvoy J, Tocher DR, Sargent JR. Depletion of α-tocopherol and astaxanthin in Atlantic salmon (Salmo salar) affects autoxidative defense and fatty acid metabolism. J Nutr. 2000;130:1800–1808. doi: 10.1093/jn/130.7.1800. [DOI] [PubMed] [Google Scholar]
- 37.Wingerath T, Stahl W, Sies H. Beta-cryptoxanthin selectively increases in human chylomicrons upon ingestion of tangerine concentrate rich in beta-cryptoxanthin esters. Arch Biochem Biophys. 1995;324:385–390. doi: 10.1006/abbi.1995.0052. [DOI] [PubMed] [Google Scholar]
- 38.Shaw PX, Hörkkö S, Tsimikas S, et al. Human-derived anti-oxidized LDL autoantibody blocks uptake of oxidized LDL by macrophages and localizes to atherosclerotic lesions in vivo. Arterioscler Thromb Vasc Biol. 2001;21:1333–1339. doi: 10.1161/hq0801.093587. [DOI] [PubMed] [Google Scholar]
- 39.Augusti PR, Conterato GM, Somacal S, et al. Effect of astaxanthin on kidney function impairment and oxidative stress induced by mercuric chloride in rats. Food Chem Toxicol. 2008;46:212–219. doi: 10.1016/j.fct.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 40.Aas GH, Bjerkeng B, Storebakken T, Ruyter B. Blood appearance, metabolic transformation and plasma transport proteins of 14C-astaxanthin in Atlantic salmon (Salmo salar L.) Fish Physiol Biochem. 1999;21:325–334. [Google Scholar]
- 41.Strålin P, Karlsson K, Johansson BO, Marklund SL. The interstitium of the human arterial wall contains very large amounts of extracellular superoxide dismutase. Arterioscler Thromb Vasc Biol. 1995;15:2032–2036. doi: 10.1161/01.atv.15.11.2032. [DOI] [PubMed] [Google Scholar]
- 42.Ohara Y, Peterson TE, Harrison DG. Hypercholesterolemia increases endothelial superoxide anion production. J Clin Invest. 1993;91:2546–2551. doi: 10.1172/JCI116491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Söderberg A, Sahaf B, Rosén A. Thioredoxin reductase, a redoxactive selenoprotein, is secreted by normal and neoplastic cells: presence in human plasma. Cancer Res. 2000;60:2281–2289. [PubMed] [Google Scholar]
- 44.Björnstedt M, Hamberg M, Kumar S, Xue J, Holmgren A. Human thioredoxin reductase directly reduces lipid hydroperoxides by NADPH and selenocystine strongly stimulates the reaction via catalytically generated selenols. J Biol Chem. 1995;270:11761–11764. doi: 10.1074/jbc.270.20.11761. [DOI] [PubMed] [Google Scholar]
- 45.Hokamaki J, Kawano H, Soejima H, et al. Plasma thioredoxin levels in patients with unstable angina. Int J Cardiol. 2005;99:225–231. doi: 10.1016/j.ijcard.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 46.Furman C, Rundlöf AK, Larigauderie G, et al. Thioredoxin reductase 1 is upregulated in atherosclerotic plaques: specific induction of the promoter in human macrophages by oxidized low-density lipoproteins. Free Radic Biol Med. 2004;37:71–85. doi: 10.1016/j.freeradbiomed.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 47.Wu X, Kang J, Xie C, et al. Dietary blueberries attenuate atherosclerosis in apolipoprotein E-deficient mice by upregulating antioxidant enzyme expression. J Nutr. 2010;140:1628–1632. doi: 10.3945/jn.110.123927. [DOI] [PubMed] [Google Scholar]
- 48.Sezer EB, Sozmen EY, Nart D, Onat T. Effect of atorvastatin therapy on oxidant-antioxidant status and atherosclerotic plaque formation. Vasc Health Risk Manag. 2011;7:333–343. doi: 10.2147/VHRM.S17781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mackness M, Boullier A, Hennuyer N, et al. Paraoxonase activity is reduced by a pro-atherosclerotic diet in rabbits. Biochem Biophys Res Commun. 2000;269:232–236. doi: 10.1006/bbrc.2000.2265. [DOI] [PubMed] [Google Scholar]
- 50.Rajasingh H, Øyehaug L, Våge DI, Omholt SW. Carotenoid dynamics in Atlantic salmon. BMC Biol. 2006;4:10. doi: 10.1186/1741-7007-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mackness MI, Durrington PN. HDL, its enzymes and its potential to influence lipid peroxidation. Atherosclerosis. 1995;115:243–253. doi: 10.1016/0021-9150(94)05524-m. [DOI] [PubMed] [Google Scholar]
- 52.Kwon EY, Do GM, Cho YY, Park YB, Jeon SM, Choi MS. Anti-atherogenic property of ferulic acid in apolipoprotein E-deficient mice fed Western diet: comparison with clofibrate. Food Chem Toxicol. 2010;48:2298–2303. doi: 10.1016/j.fct.2010.05.063. [DOI] [PubMed] [Google Scholar]
- 53.Leckey LC, Garige M, Varatharajalu R, et al. Quercetin and ethanol attenuate the progression of atherosclerotic plaques with concomitant up regulation of paraoxonase1 (PON1) gene Eexpression and PON1 activity in LDLR−/− mice. Alcohol Clin Exp Res. 2010;34:1535–1542. doi: 10.1111/j.1530-0277.2010.01238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]