

Abstract
Non-human primates provide genetic model systems biologically intermediate between humans and other mammalian model organisms. Populations of Caribbean vervet monkeys (Chlorocebus aethiops sabaeus) are genetically homogeneous and large enough to permit well-powered genetic mapping studies of quantitative traits relevant to human health, including expression quantitative trait loci (eQTL). Previous transcriptome-wide investigation in an extended vervet pedigree identified 29 heritable transcripts for which levels of expression in peripheral blood correlate strongly with expression levels in the brain. Quantitative trait linkage analysis using 261 microsatellite markers identified significant (n = 8) and suggestive (n = 4) linkages for 12 of these transcripts, including both cis- and trans-eQTL. Seven transcripts, located on different chromosomes, showed maximum linkage to markers in a single region of vervet chromosome 9; this observation suggests the possibility of a master trans-regulator locus in this region. For one cis-eQTL (at B3GALTL, beta-1,3-glucosyltransferase), we conducted follow-up single nucleotide polymorphism genotyping and fine-scale association analysis in a sample of unrelated Caribbean vervets, localizing this eQTL to a region of <200 kb. These results suggest the value of pedigree and population samples of the Caribbean vervet for linkage and association mapping studies of quantitative traits. The imminent whole genome sequencing of many of these vervet samples will enhance the power of such investigations by providing a comprehensive catalog of genetic variation.
INTRODUCTION
Non-human primates (NHPs) fill a critical need for biomedical models that are more directly relevant to human biology and disease than rodents or other commonly employed model systems, and they also permit longitudinal or invasive investigations that are infeasible in humans (1). The advent of next-generation sequencing has created the opportunity to assay cost-effectively genome-wide genetic variation in NHPs. This possibility has raised interest in using large-scale genetic and genomic investigations of NHPs to elucidate the biological basis of diverse human traits, including HIV/AIDS, cardiometabolic diseases and disorders of brain and behavior (2–4). However, the perceived lack of suitable and sizable NHP samples has thus far limited the implementation of such investigations. We propose here a strategy for mapping complex traits that leverages both the large pedigrees available in NHP research colonies and extensive samples available from wild NHP populations. As we suggest in this report, the Caribbean vervet (also termed the African green monkey, AGM, Chlorocebus aethiops sabaeus) offers particular advantages for the implementation of this approach.
The Caribbean vervet population descends from very small numbers of West African sabaeus monkeys transported to the islands of St Kitts, Nevis and Barbados during the early colonial era (5) (Fig. 1). These monkeys rapidly established feral populations that, in the absence of natural predators, expanded dramatically over a period of ∼80–100 generations. Current estimates place the total size of the feral populations on these three islands as high as 50 000–100 000 (Alexis Nisbett, personal communication). Although such island-wide population estimates are imprecise, detailed surveys of a single location in St Kitts has demonstrated the dramatic expansion in feral vervet population size that can occur there over short periods (nearly 4-fold growth over a single decade from 1971 to 1981) (6).
Figure 1.

Vervet population history from Africa to the VRC. The African vervet meta-population (Chlorocebus aethiops) consists of five major sub-species, with estimated ranges as shown: sabaeus (red), tantalus (purple), aethiops (yellow), cynosurus (green) and pygerythrus (blue). Facial images (35, adapted from 36) represent African subspecies. The current Caribbean vervet population [inhabiting the islands of Saint Kitts, Nevis, and Barbados (red frame)] has expanded more than 100-fold from a bottleneck that occurred about 80–100 generations ago when a small number of African vervets (believed to be West African sabaeus monkeys) were transported to these islands. The growth of the VRC pedigree (from Caribbean 57 founders to a current size of > 450 monkeys) represents a more recent expansion from a bottleneck.
In the 1970s and 1980s, 57 monkeys trapped on the neighboring islands of St Kitts and Nevis were transported to UCLA to found the Vervet Research Colony (VRC). The VRC, which was relocated to the Wake Forest Primate Center in 2008, has been continuously managed as a single extended pedigree (now eight generations deep) and has included more than 2000 monkeys maintained in species-appropriate social housing. The VRC pedigree has been evaluated for a wide range of heritable quantitative traits, including brain and behavior measures (4,7–11), metabolic phenotypes (12) and gene expression levels (13), which have been assayed in multiple tissues.
The size and structure of the VRC pedigree suggest that it should be a powerful resource for initial localization of quantitative trait loci (QTL), given the many opportunities that the extended pedigree provides for observing the transmission of both trait measures and marker alleles. Available evidence supports this expectation; we have reported previously the mapping in the VRC of a locus for central dopamine metabolism (4) and report here the mapping of several expression QTL (eQTL), despite the fact that current studies rely on a sparse genetic map comprised of human microsatellites (14). Genetic investigations of the VRC will soon become considerably more powerful, given whole genome sequencing (WGS) now underway on a pedigree-wide basis.
The 57 monkeys utilized to establishment the VRC were trapped at several locations spanning St Kitts and Nevis. Given that the St Kitts and Nevis vervet population expanded dramatically from a small number of founders, we therefore reasoned that the VRC pedigree must be genetically broadly representative of the island populations, and that most variants that contribute to quantitative traits in the VRC pedigree also likely contribute to such traits in the feral St Kitts and Nevis vervet populations. Exceptions to this expectation could include loci for which there has been extensive genetic drift since the separation of the VRC and island populations, and loci for which de novo mutations have occurred in either group since that time.
We further expected that VRC monkeys with similar values on quantitative traits would share, identical-by-descent (IBD), long chromosomal segments containing variants contributing to these traits, and that St Kitts monkeys would share shorter IBD segments containing such variants. This expectation reflects the fact that, in the VRC, as in pedigrees generally, individuals measured for any given trait are separated from their most recent common ancestors by at most a few meiotic steps, whereas in the population sample the individuals measured for traits have been sampled from several dozen distinct locations and therefore are likely separated from their most recent common ancestor by a much larger number of meiotic steps. The predictable relationship between the number of meiotic steps to a common ancestor and the length of a shared segment around a disease gene has been extensively utilized in the design of human genetic mapping studies since early in the positional cloning era, that is, the progression from coarse-scale linkage mapping in pedigrees to fine-scale association mapping in population samples (15). Human studies have also established the importance, to detection of IBD sharing, of demographic history; rapidly expanding founder populations, such as that of the St Kitts and Nevis vervets, typically display particularly extensive and predictable patterns of linkage disequilibrium (LD), compared with more heterogeneous populations (16).
Based on our expectations for allele and segment sharing between the VRC pedigree and island populations, we designed an initial test of a two-stage strategy for genetic mapping of complex traits in the Caribbean vervet. The first stage in this approach consists of genome-wide linkage mapping in the VRC (establishing the strongest coarse-scale regions for follow-up and providing a priori evidence in support of the second-stage association studies). The second stage consists of association analyses that target the candidate regions established in the linkage stage, utilizing large numbers of independent feral vervet monkeys sampled on St Kitts and Nevis to fine-map the linked loci.
In this study, we focus on mapping loci contributing to gene expression phenotypes assayed by genome-wide microarrays. While the long-term focus of genomic investigation of the vervet is primarily on phenotypes hypothesized to directly inform our understanding of human diseases, identification of eQTL has proven invaluable in the genetic dissection of a wide range of complex traits (17). For example, eQTL are significantly overrepresented among single nucleotide polymorphisms (SNPs) that that are associated with complex traits in genome wide association studies (18). In human studies, variation in gene expression is generally limited to readily accessible tissues, particularly blood, fibroblasts and lymphoblast cell lines (19). In the vervet, by contrast, it is feasible to obtain, in selected individuals, high-quality RNA from any tissue and then to identify transcripts whose expression patterns in that tissue have a strong correlation with their expression patterns in blood or fibroblasts (which are available in most members of the VRC and in large numbers of individuals from population samples). We previously used such an approach to identify 29 vervet transcripts that display expression patterns in blood that are stable, heritable and highly correlated with their expression patterns in the brain (9).
We report here the genetic mapping in the VRC of loci linked to the blood expression levels of these 29 transcripts (eQTL). Among these loci, several markers in a single genome region show linkage to transcripts located on several different vervet chromosomes, suggesting the presence of a master trans-regulator locus. We then describe an association analysis conducted in independently ascertained animals from St Kitts, genotyped with multiple SNPs from one of the cis-eQTL regions [for B3GALTL, a glycosyltransferase which, when recessively mutated results in Peters plus syndrome, MIM#261540, a developmental delay disorder (20)] that narrowed this region to a chromosome segment of ∼170 kb.
RESULTS
QTL linkage results in the VRC for 29 expression traits
We first conducted QTL linkage analysis of 29 gene expression traits (9) using genotypes from 261 genome-wide microsatellite markers in 347 monkeys from the VRC pedigree (see Materials and Methods, and Supplementary Material, Table S1). As shown in Table 1, eight loci (all apparently cis-eQTL) exceeded a threshold for genome-wide significance (LOD > 4.78) obtained by applying the method of Conneely and Boehnke (21) to correct for the analysis of 29 traits. An additional four loci (two cis- and two trans-) exceeded a threshold for suggestive linkage (LOD > 3.27) determined using the same method. These suggestive and significant loci are estimated to account for between ∼0.25 and 0.65 of the overall heritability for the specific transcripts to which they are linked (Table 1).
Table 1.
Results of linkage analysis of gene expression traits to microsatellite loci, in monkeys from the VRC pedigree
| Gene symbol | Transcript probe location (Chr)a | Transcript level trait heritabilityb | Marker with maximal LOD scorec | Locus-specific heritability | Max LOD | Max LOD location (Chr)a | Max LOD location (cM)a |
|---|---|---|---|---|---|---|---|
| TMEM57 | 20 | 0.57 | D10S1646 | 0.557 | 4.00 | 9 | 62.19 |
| TMEM111 | 22 | 0.76 | D3S3589 | 0.580 | 20.55 | 22 | 33.76 |
| RAB5A | 15 or 22 | 0.24 | D10S1646 | 0.324 | 2.01 | 9 | 62.19 |
| TMEM14C | 17 | 0.70 | D6S1653 | 0.427 | 6.18 | 17 | 8.08 |
| BAT1 | 17 | 0.60 | D6S273 | 0.611 | 21.82 | 17 | 30.07 |
| CDKN1A | 17 | 0.46 | D6S273 | 0.381 | 5.00 | 17 | 30.07 |
| GUSB | 21 | 0.64 | D19S591 | 0.294 | 1.60 | 6 | 0 |
| LOC389541 | 21 | 0.29 | D17S923 | 0.278 | 2.90 | 16 | 55.73 |
| STOM | 12 | 0.44 | D9S258 | 0.288 | 3.42 | 12 | 36.12 |
| PRTFDC1 | 9 | 0.43 | D10S1432 | 0.228 | 1.95 | 9 | 61.12 |
| TSPAN14 | 9 | 0.39 | D10S1677 | 0.380 | 9.05 | 9 | 58.90 |
| SLC3A2 | 1 | 0.46 | D16S402 | 0.323 | 2.83 | 5 | 100.26 |
| SUV420H1 | 1 | 0.43 | D6S1705 | 0.360 | 2.59 | 13 | 33.25 |
| HSPA8 | 1 | 0.13 | D7S1820 | 0.231 | 1.62 | 21 | 35.24 |
| TUBA1B | 11 | 0.62 | D12S1701 | 0.642 | 20.24 | 11 | 56.67 |
| B3GALTL | 3 | 0.73 | D13S1233 | 0.513 | 8.65 | 3 | 22.17 |
| ZFYVE1 | 24 | 0.41 | D3S3610 | 0.182 | 1.78 | 22 | 32.73 |
| RAB11A | 26 | 0.33 | D1S2884 | 0.361 | 2.92 | 20 | 88.87 |
| TMED3 | 26 | 0.64 | D15S206 | 0.428 | 7.62 | 26 | 0 |
| LOC440589 | 5 | 0.18 | D3S2398 | 0.210 | 1.44 | 15 | 91.34 |
| ERAL1 | 16 | 0.46 | D17S923 | 0.232 | 2.63 | 16 | 55.73 |
| NME1-NME2 | 16 | 0.20 | D21S2051 | 0.187 | 1.50 | 8 | 66.90 |
| SLC25A23 | 6 | 0.43 | D10S1432 | 0.460 | 4.17 | 9 | 61.12 |
| C19orf62 | 6 | 0.52 | D19S222 | 0.238 | 2.29 | 6 | 45.21 |
| DEDD2 | 6 | 0.55 | D10S564 | 0.225 | 2.43 | 9 | 81.09 |
| SMOX | 2 | 0.39 | D20S116 | 0.248 | 3.39 | 2 | 67.59 |
| MTMR3 | 19 | 0.26 | D10S1646 | 0.223 | 0.98 | 9 | 62.19 |
| GRIPAP1 | X | 0.33 | D4S1629 | 0.291 | 2.25 | 7 | 31.20 |
| TFE3 | X | 0.28 | D21S364 | 0.218 | 1.25 | 2 | 21.93 |
Using the method of Conneely et al. (21) to correct for analysis of multiple, correlated phenotypes, thresholds were determined for significant linkage (LOD > 4.78) and suggestive linkage (LOD > 3.27).
All physical locations are according to hg19.
Significant and suggestive LOD scores are shown in bold.
aVervet chromosome location.
bAll heritabilities are significant at P < 0.001, see (13).
cDue to pedigree complexity, we were unable to test for linkage to the X chromosome in SOLAR.
The still incomplete state of the vervet reference genome assembly complicates analysis of the eQTL linkage results, particularly in regions where synteny between the human and vervet genomes is uncertain. Specifically, while the sparse vervet genetic map reflects genotypes obtained from the VRC pedigree (14), the information on the physical position of the transcripts currently derives from the position in the human assembly of the microarray probes used to assay them. It is therefore not yet possible to directly compare the genetic and physical locations of eQTL in any of the several regions that display large-scale rearrangements between the vervet genome (which has 29 autosomes) and the human genome (14,22). An example of such a region is an ∼22 cM segment of vervet chromosome 9 that is syntenic with a segment of ∼25 Mb on human chromosome 10 (Fig. 2). Seven of the transcripts that we assessed display their peak genome-wide LOD score to markers that map to this region. The strongest linkage evidence (peak LOD scores ≥ 4 for TSPAN14, SLC25A23 and TMEM57) was observed at three microsatellite markers located within a genetic interval of ∼3 cM, suggesting that a single locus may be predominantly responsible for the eQTL signal in this region. The location of SLC25A23 and TMEM57 on vervet chromosomes 6 and 20, respectively, further suggests that this eQTL may be a master trans-regulator (23,24). Detailed analysis of this region, however, is currently precluded by the fact that the segment of maximal linkage, although internally syntenic with the corresponding human region, is inverted in the vervet genome compared with that of the human, and high-resolution analysis of the structure of this region in the vervet genome is not yet complete. Additionally, the sparseness of the current genetic marker map makes it difficult to define the inversion region genetically.
Figure 2.

A putative master trans-regulator locus on vervet chromosome 9. The peak LOD scores observed for transcripts sited on several different vervet chromosomes map to a single region on vervet chromosome 9 (syntenic to human chromosome 10), suggesting the location of a master trans-regulator locus. The linked microsatellites span a region of ∼22cM from D10S1677–D10S564. Two of the linked transcripts are located on chromosome 9 (PRFDC1 and TSPAN14) while five other linked transcripts (SLC25A23, TMEM57, RAB5A, MTMR3 and DEDD2) are located on other vervet chromosomes. Interpretation of the linkage results to this region is complicated by the sparseness of the microsatellite-based vervet genetic map and by an inversion in the human genome (relative to the vervet and macaque genomes) that includes the four markers demonstrating maximal linkage to the transcript measures. Marker order in human and vervet chromosomes is shown in units of genetic distance (Kosambi centiMorgans, cM). Approximate gene locations (blue stars) are shown based on their known physical location in the human genome relative to microsatellite markers.
Evaluation of genetic characteristics of the VRC and St Kitts population samples in relation to fine-mapping of a single eQTL (B3GALTL)
To assess the potential utility of independent vervets from the Caribbean population for follow-up of QTL mapping results obtained in the VRC, we conducted an initial genotyping study using wild-trapped monkeys from St Kitts. We considered eQTL for follow-up genotyping based on three criteria: genome-wide significant linkage findings in the VRC (providing strong a priori evidence for the existence of trait-associated variation in the follow-up region), the availability of vervet sequence in the linked genome regions at the time that we were designing follow-up studies and cross-platform validation [by reverse transcription–quantitative polymerase chain reaction (RT–qPCR)] of the expression results. A region on vervet chromosome 3 that included B3GALTL met these criteria. Significant linkage to B3GALTL expression level was observed for two microsatellite markers spanning a distance of >10 cM (D13S1493 and D13S1233, peak LOD scores of 5.76 and 8.65, respectively, Supplementary Material, Fig. S1) and nearly complete vervet sequence data were available for the B3GALTL gene region. RT–qPCR assays of this transcript provided clear experimental evidence indicating that the expression levels measured using the microarray accurately reflected inter-individual variation in expression of this gene among animals of the VRC (validation P-value = 1.08 × 10−5, Wilcoxon test). Based on these results, we chose to test a vervet sample from the St Kitts population for association with SNPs from the B3GALTL gene region.
Candidate SNPs for fine-mapping of B3GALTL were identified from preliminary data of the Vervet Genome Sequencing Project (VGSP, see Materials and Methods). We selected SNPs that included B3GALTL and flanking regions of ∼100 kb, a total of 15 SNPs within a gene region of ∼330 kb. We genotyped the 15 B3GALTL SNPs in 279 independent wild-born vervets from St Kitts, sampled at the St Kitts Biomedical Research Foundation, SKBRF (see Materials and Methods and Fig. 3). In order to determine the suitability of the St Kitts samples for following up the linkage result at this locus from the VRC, we compared LD patterns and allele frequencies between the VRC pedigree and St Kitts population samples; for this comparison, we evaluated monkeys from each sample chosen to be as independent from one another as possible. From St Kitts, we selected one monkey from each of the 45 different trapping sites from which they were acquired, and from the VRC we selected 45 monkeys, all of whom were separated from each other by at least four meiotic steps. Thirteen of the 15 SNPs typed in the St Kitts animals were successfully genotyped in the VRC pedigree monkeys.
Figure 3.

Genetic investigation of populations of feral Caribbean vervets. The wild-born animals (n = 279) used in the fine-mapping association study presented here were sampled from several sites on St Kitts. Pin size corresponds to the number of animals trapped from a given location.
The correlation in allele frequencies between the pedigree and the population samples is strong [0.804, 95% confidence intervals of 0.454–0.939, minor allele frequency range = 6.7–42.2% (St Kitts) and 7.8–41.1% (VRC)] and significant (P = 9.3 × 10−4), consistent with the assumption that the VRC broadly reflects the genetic diversity of the St Kitts population. The degree to which fine-mapping association studies can resolve the location of a trait-related variant depends upon the number of recombination events that have occurred around the locus of interest since the variant was introduced into the population. The statistic D′ is an indicator of historical recombination between two loci within a given study sample (25). Evaluation of D′ values for all of the pairwise comparisons between the 13 SNPs genotyped in both study samples (Supplementary Material, Fig. S2) revealed a significantly larger proportion of comparisons within the VRC for which D′ = 1 (Chi-square = 4.76, P = 0.03), indicating that less historical recombination has occurred at B3GALTL in the VRC than in the St Kitts population sample. This observation is consistent with the hypothesis that, among St Kitts monkeys, IBD sharing will extend, on average over shorter chromosomal distances than among VRC monkeys; accordingly, fine-mapping association studies in St Kitts samples should narrow the candidate region of loci mapped in the VRC.
Association analysis in St Kitts samples of the B3GALTL eQTL
Four of the 15 SNPs typed in the 279 St Kitts monkeys displayed significant association with B3GALTL expression levels after a Bonferroni correction (0.05/15 = 0.003) for multiple comparisons (Table 2). Three of these four SNPs (B3GALTL 2, 4 and 12) were in tight LD with one another (r2 > 0.8). The fourth SNP (B3GALTL 16) LD pattern displayed an r2 of ∼0.65 with the first three SNPs; however, modeling the expression versus a linear combination of the four SNPs suggests that all four are detecting a single association signal. The peak association (P = 7.28 × 10−6) observed at B3GALTL_SNP_2 corresponds to position 31 689 187 bp on human chromosome 13, located ∼85 kb upstream of B3GALTL [hg19]. Additionally, two significantly associated SNPs (P < 0.00005) lie in sequences syntenic to introns of this gene, and another associated SNP lies ∼15 kb downstream of the gene boundary as predicted from the human sequence. The SNPs genotyped at B3GALTL were insufficient (in terms both of density and of the extent of the genomic region that they cover) to define precisely the boundaries of the association signal at this locus.
Table 2.
Association fine mapping of the B3GALTL eQTL on vervet chromosome 3 (human chromosome 13) in a vervet Caribbean population sample
| Position in hg19 (bp) | SNP ID | BETA | SE | R2 | T | P-value* |
|---|---|---|---|---|---|---|
| 31 676 914 | B3GALTL_0 | −0.1438 | 0.0592 | 0.02086 | −2.429 | 0.01576 |
| 31 689 187 | B3GALTL_2 | 0.2865 | 0.06264 | 0.07092 | 4.573 | 7.28E − 06 |
| 31 758 316 | B3GALTL_3 | −0.1159 | 0.05773 | 0.01445 | −2.008 | 0.04565 |
| 31 758 727 | B3GALTL_4 | 0.2773 | 0.0629 | 0.06558 | 4.409 | 1.49E − 05 |
| 31 761 667 | B3GALTL_5 | −0.09483 | 0.1279 | 0.001988 | −0.7415 | 0.459 |
| 31 834 058 | B3GALTL_11 | 0.1201 | 0.07922 | 0.008235 | 1.517 | 0.1305 |
| 31 836 303 | B3GALTL_12 | 0.2623 | 0.0602 | 0.06416 | 4.358 | 1.85E − 05 |
| 31 837 552 | B3GALTL_13 | −0.03341 | 0.08569 | 0.000551 | −0.3899 | 0.6969 |
| 31 855 094 | B3GALTL_15 | 0.1059 | 0.06451 | 0.009813 | 1.642 | 0.1018 |
| 31 860 398 | B3GALTL_16 | 0.2342 | 0.05671 | 0.05918 | 4.129 | 4.86E − 05 |
| 31 906 160 | B3GALTL_19 | −0.0279 | 0.1278 | 0.000172 | −0.2184 | 0.8273 |
| 31 932 565 | B3GALTL_20.1 | −0.0279 | 0.1278 | 0.000172 | −0.2184 | 0.8273 |
| 31 948 748 | B3GALTL_21 | −0.02533 | 0.13 | 0.000138 | −0.1949 | 0.8456 |
| 31 953 645 | B3GALTL_22 | −0.04565 | 0.06007 | 0.00208 | −0.7599 | 0.448 |
| 32 005 195 | B3GALTL_24 | −0.1161 | 0.06103 | 0.01293 | −1.902 | 0.05827 |
*Association P-values shown here were calculated for regression of expression on SNP genotype, assuming an additive model. Beta is the regression coefficient, SE the standard error of beta, R2 the coefficient of determination from the regression model and T is the t-statistic testing the null hypothesis that Beta = 0. Significant association P-values based on Bonferroni correction (P < 0.003) are shown in boldface. Three of the four significantly associated SNPs are in very tight LD (r2> 0.80), and clearly represent the same signal. The fourth SNP (SNP _16) has r2 of ∼0.65 with the other three SNPs; however, modeling the expression phenotype versus a linear combination of the four SNPs suggests that the signals are not independent.
DISCUSSION
We conducted linkage and association analyses of multiple expression traits to assess the potential of the vervet system for large-scale genomic investigations of various quantitative traits. We focused on expression phenotypes based on the assumption that they could have a relatively simple genetic basis, and believed that we could gain substantial empirical information about this potential despite the sparseness of the genetic marker sets currently available in this system. In particular, we expected to observe significant eQTL for transcripts sited in close proximity to informative markers from the vervet microsatellite map. The very strong linkage evidence observed for several cis-eQTL (e.g. three transcripts displayed peak LOD scores > 20, all at microsatellites located at a distance of 2Mb or less) confirms this expectation.
The linkage results further indicate that despite the extensive bottlenecks in the population from which the VRC founders were drawn, the pedigree maintains sufficient genetic variation to provide excellent power for initial QTL mapping. Consideration of results for the entire set of transcripts and comparison with prior studies further suggest that the availability of polymorphisms that adequately cover the genome will enable trait mapping in the VRC to be routinely successful. We previously identified a significant linkage for brain dopamine metabolism (4) by using SNPs to saturate the most promising region (peak LOD = 2.3) in a microsatellite screening study of the VRC. In the current study, the microsatellite screen identified evidence of at least one locus of this magnitude or greater for 21 of the 29 transcripts evaluated. Given that the microsatellites in the vervet map are individually not nearly as informative as the markers used to construct human linkage maps, and considering the paucity of markers available in the regions containing several of the transcripts (five of the transcripts for which neither suggestive nor significant linkage was observed had no marker within 5 Mb of the probe), we anticipate identifying highly significant eQTL for virtually all of these transcripts, once a next-generation marker map is available, based on WGS data.
It has been proposed that trans-eQTL exert more important biological effects than cis-eQTL (23,26), but they are more challenging to detect. As such they may provide a clearer indication of the power of a genetic system for the elucidation of complex traits. The observation that almost 25% of the highly selected transcripts assessed here demonstrated their peak linkage signals in a single small chromosomal region on vervet chromosome 9 suggests that future investigations of the vervet system will provide an excellent opportunity to uncover additional loci with comparable trans effects. This expectation is strengthened by the fact that even the most significant cis-eQTL linkages reported here leave a substantial proportion of trait heritability unaccounted for. Extending the approach used to select these transcripts, establishing that their expression patterns in peripheral blood correlated strongly to their expression pattern in brain, to other typically inaccessible tissues may facilitate the discovery of regulatory loci that are particularly relevant to the function of specific tissue or organ systems.
The localization of the chromosome 9 trans-QTL to a region that includes a large inversion between the vervet and human genome highlights the potential importance of the many structural variations between the vervet genome and that of other primates, as a basis for identifying phenotypic differences between species. Most of the Catarrhini (Old World Monkeys and Hominidae) are characterized by exceptional genome structural stability. In contrast, the Cercopithecini tribe to which the vervet belongs, like the Hylobatidae (gibbons), displays a dramatically accelerated rate of chromosome evolution, with 2N karyotype varying between 54 and 72 (for vervet 2N = 60), reflecting multiple non-centromeric fissions from the ancestral karyotype (27). Analyses of breakpoint regions from numerous structural rearrangements between human and gibbon genomes have suggested that many of these rearrangements could have functional significance, for example by direct interruption of genes (28,29). Unlike gibbons, however, which as endangered species are not available for large-scale investigations of gene expression, epigenetics or higher order phenotypes, vervet pedigree and population samples are ideally suited for comparative phenotypic-structural genomic investigations with both humans and other NHP models such as baboons and macaques.
Although QTL analysis in pedigrees is a powerful means for initial localization of variants contributing to complex phenotypes, the resolution of such analyses is inherently low. Pinpointing the signal identified through QTL linkage studies has proven a major challenge in many species, including mice (30), reflecting the paucity of sufficient genetically independent phenotyped individuals for fine-scale association analyses. The vervet is unique among NHP models in that its most intensively investigated pedigree sample (the VRC) descends entirely from a large but closed ancestral population (St Kitts and Nevis). A common set of alleles is responsible for most genetic variation in both study samples, but the sizable sample of independent individuals available in the island populations offers the opportunity to observe haplotypes at a trait locus that are substantially diminished in length compared with the haplotypes linked to the trait in the pedigree (15).
The high correlation in allele frequencies observed, for B3GALTL region SNPs, between the VRC samples and St Kitts samples drawn from across the island, is consistent with our assumption that the VRC incorporates most of the genetic variation present in the St Kitts population. The observation of greater D′ between SNPs in the VRC compared with the St Kitts samples is also consistent with our expectation of a greater number of historical recombinations in the independently ascertained monkeys from the island population. This expectation is reflected in the trait mapping findings, where the B3GALTL significant linkage signal extends over more than 10 Mb in the VRC, while association evidence in the St Kitts sample is limited to a region of <200 kb. More conclusive confirmation of our hypotheses regarding both the allelic similarity and the extent of haplotype conservation between the VRC and St Kitts population will require genome-wide data, as will be provided by WGS efforts now underway in both study samples.
The WGS of pedigree and population samples, together with the ongoing collection of larger study samples, will provide the opportunity to extend the two-stage pedigree-population approach reported here for eQTL to well-powered investigations of a wide range of quantitative phenotypes. The WGS underway in the VRC will ameliorate the impediments to genetic mapping represented by the inadequacy of current polymorphisms. These studies will provide comprehensive genotype data for >700 vervets; more than 600 monkeys were selected based on their having been phenotyped for multiple measures (from a set of 19 heritable brain and behavior, metabolic and morphometric traits) and nearly 100 monkeys were chosen because they will provide a vervet transcriptome data set comprising RNA sequencing data from 85 brain and peripheral tissues.
For several of the phenotypes assessed in the VRC, it will be possible to conduct follow-up studies in an expanded population sample that now consists of ∼1000 monkeys assessed on St Kitts and Nevis. For QTL in which the St Kitts and Nevis samples yield unsatisfactory resolution, it will be feasible to conduct even finer scale association analyses using large samples available from the vervet population of Barbados; preliminary comparative sequence analyses suggest that this population is more distantly related to the VRC than that of St Kitts and Nevis, but is more closely related to the VRC than that of the African populations from which the Caribbean vervets are presumed to derive (Y. Huang, unpublished data).
Traditionally, genetic investigation of NHPs has been limited to the relatively small samples that could be maintained in primate colonies. The development of technologies for tracking and repeat identification of wild or feral NHP populations now enables a wide range of longitudinal investigations of various biomedically important traits that may be relatively inexpensive to conduct compared with human research. International collaborative efforts have already enabled the collection of biomaterials and phenotypic data from more than 1 500 independent vervet monkeys from the Caribbean and African populations (Fig. 4), all of whom have been tagged by microchip and released, and are therefore available for longitudinal studies.
Figure 4.
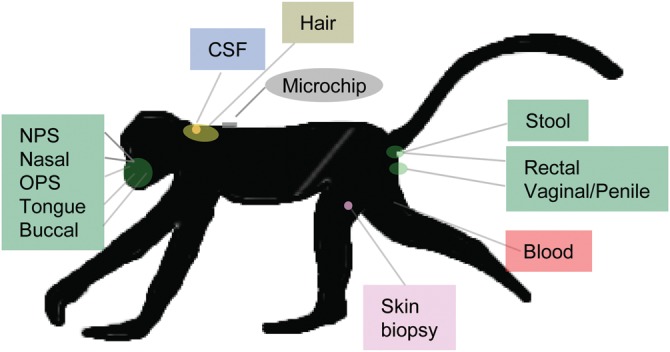
Biomaterial repository established from vervet population samples in the Caribbean and Africa. Animals have been sampled for skin biopsy specimens (to establish fibroblast cell lines), blood, cerebrospinal fluid, hair and specimens for assaying the natural microbiome populating the gastro-intestinal tract, reproductive system and oro-nasal cavity (including nasopharyngeal, NPS; oropharyngeal, OPS). Wild monkeys have been assigned unique identifiers using a microchip injected subcutaneously to enable long-term follow-up studies of each monkey. Global positioning coordinates for all monkeys will be available online at http://www.genomequebec.mcgill.ca/compgen/vervet_research/animal_resources/.
The data from numerous phenotyping assays and genome-wide sequencing, as well as a broad range of biological samples obtained from pedigreed and wild-caught Caribbean and African vervet monkeys, will be made widely available to the scientific community. The majority of the pedigreed animals from which these data were obtained are themselves available for further investigations by the scientific community, particularly for studies that do not interfere with the integrity of the pedigree or its social groups. Investigators can gain access to these materials by contacting the authors and, in the future, through a web-based database. The Integrated Vervet/AGM Research & Resources website provides access to a summary of international vervet phenotyping and genomics efforts and currently available results from VRC microsatellite genotyping, microarray-based gene expression studies and the vervet reference genome sequence, http://www.genomequebec.mcgill.ca/compgen/vervet_research/genomics_genetics/.
MATERIALS AND METHODS
Study samples
The 347 monkeys (all over 2 years of age) from the VRC have been utilized in previous genotyping and gene expression studies as described previously. From the SKBRF, we identified monkeys that had been trapped in independent social groups from sites dispersed throughout the island (Fig. 3). The sample of 279 monkeys included 273 females and 26 males (a preponderance of adult females reflects the natural social structure of vervet social groups) with mean (SD) ages of 11.95 (4.5) and 11.62 (4.8) years, respectively. For all of the investigated monkeys, genomic DNA and total RNA samples were obtained from peripheral blood using a PaxGene system (PreAnalyticX) as described previously (13).
Gene expression
Genome-wide gene expression in the VRC monkeys was measured, as described previously, using an Illumina HumanRef-8 v2 chip (GSE15301) (13). A detailed description of the microarray-based strategy used to identify the most promising candidate transcripts for eQTL mapping of transcripts relevant for brain function was presented in Jasinska et al. (13). Briefly, to identify eQTL for which expression patterns in blood are highly correlated with expression patterns in the brain, we created two data sets: (i) a set of matched tissues from eight brain regions and from peripheral blood of 12 VRC monkeys; (ii) a set of duplicate blood samples from 18 VRC monkeys, all collected at two time points. Criteria for selecting transcripts for eQTL analysis included correlated expression pattern between the brain and blood, a high degree of inter-individual variability in expression, longitudinal stability of expression pattern and heritability. Applying these criteria resulted in 29 transcripts for eQTL analyses (Supplementary Material, Table S2).
We validated array-based expression results by using preliminary sequence data from the VGSP to design RT–qPCR amplicons (Supplementary Material, Table S3). The whole-genome sequence data (∼4-fold coverage of Roche/454 data aligned to the human reference genome assembly) were derived from a VRC animal (1994–021) from which a vervet BAC library (CHORI-252) was previously created. We performed RT–qPCR using a validation sample set that included 20 monkeys with the most extreme (high and low) expression values from the microarray results. For this experiment, we followed guidelines described by Nolan et al. (31). Briefly, 500 ng of blood-isolated RNA was converted into cDNA using a High Capacity RNA-to-cDNA kit (Applied Biosystems, USA). RT–qPCR reactions were prepared using the SYBR® PCR Master Mix from Applied Biosystems and oligos from Invitrogen, and analyzed with the 7900HT Fast Real Time PCR System (Applied Biosystems). For cDNA amplification, 25 ng of cDNA and 300 nm primers were used per sample reaction and each sample was analyzed in triplicate. Expression changes for the transcript were quantified relative to glyceraldehyde 3-phosphate dehydrogenase (32), selected as an endogenous control, using the Pfaffl method (33). The medians of expression from the bottom tail and top tail as defined by microarray results were compared with RT–qPCR quantification results using a non-parametric Wilcoxon test. We assayed B3GALTL expression in the SKBRF monkeys using the RT–qPCR protocol described above (primers: F-5′-CTT TCA AGT GGG TGA TGA GC-3′, R-5′-AAT GCG TCT GGG AGT CAA TC-3′).
Genotyping
The VRC pedigree DNA samples were genotyped for genome-wide analyses in a previous study (14) using 261 microsatellite markers that constitute the first-generation vervet genetic map. These animals and the animals sampled from SKBRF were genotyped using SNPs from the B3GALTL region. SNPs were identified from the preliminary data generated by the VGSP, as described above, and were selected based on a minimum of two reads indicating each of two alleles in heterozygous loci, and based on the surrounding sequence being sufficiently complete for assay design.
Genotyping for the 15 B3GALTL SNPs was performed using Sequenom iPLEX technology (SEQUENOM). Multiplex PCR assays were designed using MassARRAY software (v3.1), with default settings of the software and no masking of repeats. Oligos for the PCR reactions and mass-extend reactions were ordered from Integrated DNA Technologies (USA). Reactions for the SNP genotyping were prepared according to the MassARRAY® iPLEX® Gold SNP Genotyping protocol from Sequenom. Multiple positive, negative and non-amplification controls were included.
All of the 279 SKBRF samples were genotyped for each of the 15 SNPs. Forty-five of the VRC monkeys were also genotyped for these SNPs for comparisons of LD and allele frequency with the SKBRF samples; for these monkeys, we obtained acceptable-quality genotypes for 13 of the 15 SNPs.
Statistical analyses
QTL linkage analysis was performed in the 347 VRC monkeys, to the probe intensities of the 29 transcripts; this analysis was conducted using SOLAR (34). As in Jasinska et al. (13), covariates of age, sex and batch (sets of samples run in the same microarray experiment) were considered in each analysis and those significant at P = 0.10 were retained. Correction for testing linkage for multiple expression traits was calculated using a modification of the method of Conneely and Boehnke (21), where the level of correlation among the traits was used to adjust the genome-wide significant LOD score threshold.
For the comparison of the VRC pedigree and population genotype data, we used the following methods: correlations in allele frequencies between the VRC pedigree and the population samples were assessed with a Pearson correlation. LD was measured as D′ between each pair of the SNPs.
We used regression analyses to test for association between SNP genotypes and overall gene expression (from both alleles) in the SKBRF population sample. Each genotype was coded as 0, 1 or 2 copies of the minor allele, and expression levels regressed on this variable, assuming an additive model of action of the minor allele.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
ACKNOWLEDGEMENTS
We gratefully acknowledge the expertise and technical assistance of Dr Gene Redmond and the entire staff of the St Kitts Biomedical Research Foundation, particularly Messrs. Alexis Nisbett, Ernell (Zyka) Nisbett and O'Neal Whattley. We also wish to acknowledge the contributions of Ms Jennifer Danzy, Dr Alison Grand, Dr Yu Huang, Mr Oliver (Pess) Morton and the Production Sequencing Group, The Genome Institute, Washington University School of Medicine.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by National Institutes of Health grants R01OD010980 and P40OD010965 from the Office of Research Infrastructure Programs/OD (formerly R01RR016300 and P40RR019963 from NCRR), PL1NS062410, UL1DEO19580, P30NS062691, RL1MH083270 and U54HG003079, Genome Quebec and Genome Canada.
REFERENCES
- 1.Bearden C.E., Jasinska A.J., Freimer N.B. Methodological issues in molecular genetic studies of mental disorders. Annu. Rev. Clin. Psychol. 2009;5:49–69. doi: 10.1146/annurev.clinpsy.032408.153545. [DOI] [PubMed] [Google Scholar]
- 2.Cox L.A., Glenn J., Ascher S., Birnbaum S., VandeBerg J.L. Integration of genetic and genomic methods for identification of genes and gene variants encoding QTLs in the nonhuman primate. Methods. 2009;49:63–69. doi: 10.1016/j.ymeth.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Degenhardt J.D., de Candia P., Chabot A., Schwartz S., Henderson L., Ling B., Hunter M., Jiang Z., Palermo R.E., Katze M., et al. Copy number variation of CCL3-like genes affects rate of progression to simian-AIDS in Rhesus Macaques (Macaca mulatta) PLoS Genet. 2009;5:e1000346. doi: 10.1371/journal.pgen.1000346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freimer N.B., Service S.K., Ophoff R.A., Jasinska A.J., McKee K., Villeneuve A., Belisle A., Bailey J.N., Breidenthal S.E., Jorgensen M.J., et al. A quantitative trait locus for variation in dopamine metabolism mapped in a primate model using reference sequences from related species. Proc. Natl Acad. Sci. USA. 2007;104:15811–15816. doi: 10.1073/pnas.0707640104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGuire M.T. Contributions to Primatology: The St.Kitts Vervet. 1974. Karger. [DOI] [PubMed] [Google Scholar]
- 6.Fedigan L.M., Fedigan L., Chapman C., McGuire M.T. A demographic model of colonization by a population of St. Kitts Vervets. Folia Primatol. 1984;42:194–202. [Google Scholar]
- 7.Fairbanks L.A., Bailey J.N., Breidenthal S.E., Laudenslager M.L., Kaplan J.R., Jorgensen M.J. Environmental stress alters genetic regulation of novelty seeking in vervet monkeys. Genes Brain Behav. 2011;10:683–688. doi: 10.1111/j.1601-183X.2011.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fairbanks L.A., Jorgensen M.J., Bailey J.N., Breidenthal S.E., Grzywa R., Laudenslager M.L. Heritability and genetic correlation of hair cortisol in vervet monkeys in low and higher stress environments. Psychoneuroendocrinology. 2011;36:1201–1208. doi: 10.1016/j.psyneuen.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fairbanks L.A., Newman T.K., Bailey J.N., Jorgensen M.J., Breidenthal S.E., Ophoff R.A., Comuzzie A.G., Martin L.J., Rogers J. Genetic contributions to social impulsivity and aggressiveness in vervet monkeys. Biol. Psychiatry. 2004;55:642–647. doi: 10.1016/j.biopsych.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 10.Fears S.C., Melega W.P., Service S.K., Lee C., Chen K., Tu Z., Jorgensen M.J., Fairbanks L.A., Cantor R.M., Freimer N.B., et al. Identifying heritable brain phenotypes in an extended pedigree of vervet monkeys. J. Neurosci. 2009;29:2867–2875. doi: 10.1523/JNEUROSCI.5153-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fears S.C., Scheibel K., Abaryan Z., Lee C., Service S.K., Jorgensen M.J., Fairbanks L.A., Cantor R.M., Freimer N.B., Woods R.P. Anatomic brain asymmetry in vervet monkeys. PLoS ONE. 2011;6:e28243. doi: 10.1371/journal.pone.0028243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kavanagh K., Fairbanks L.A., Bailey J.N., Jorgensen M.J., Wilson M., Zhang L., Rudel L.L., Wagner J.D. Characterization and heritability of obesity and associated risk factors in vervet monkeys. Obesity (Silver Spring) 2007;15:1666–1674. doi: 10.1038/oby.2007.199. [DOI] [PubMed] [Google Scholar]
- 13.Jasinska A.J., Service S., Choi O.W., DeYoung J., Grujic O., Kong S.Y., Jorgensen M.J., Bailey J., Breidenthal S., Fairbanks L.A., et al. Identification of brain transcriptional variation reproduced in peripheral blood: an approach for mapping brain expression traits. Hum. Mol. Genet. 2009;18:4415–4427. doi: 10.1093/hmg/ddp397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jasinska A.J., Service S., Levinson M., Slaten E., Lee O., Sobel E., Fairbanks L.A., Bailey J.N., Jorgensen M.J., Breidenthal S.E., et al. A genetic linkage map of the vervet monkey (Chlorocebus aethiops sabaeus) Mamm Genome. 2007;18:347–360. doi: 10.1007/s00335-007-9026-4. [DOI] [PubMed] [Google Scholar]
- 15.Houwen R.H., Baharloo S., Blankenship K., Raeymaekers P., Juyn J., Sandkuijl L.A., Freimer N.B. Genome screening by searching for shared segments: mapping a gene for benign recurrent intrahepatic cholestasis. Nat. Genet. 1994;8:380–386. doi: 10.1038/ng1294-380. [DOI] [PubMed] [Google Scholar]
- 16.Service S., DeYoung J., Karayiorgou M., Roos J.L., Pretorious H., Bedoya G., Ospina J., Ruiz-Linares A., Macedo A., Palha J.A., et al. Magnitude and distribution of linkage disequilibrium in population isolates and implications for genome-wide association studies. Nat. Genet. 2006;38:556–560. doi: 10.1038/ng1770. [DOI] [PubMed] [Google Scholar]
- 17.Goring H.H., Curran J.E., Johnson M.P., Dyer T.D., Charlesworth J., Cole S.A., Jowett J.B., Abraham L.J., Rainwater D.L., Comuzzie A.G., et al. Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nat. Genet. 2007;39:1208–1216. doi: 10.1038/ng2119. [DOI] [PubMed] [Google Scholar]
- 18.Nicolae D.L., Gamazon E., Zhang W., Duan S., Dolan M.E., Cox N.J. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dimas A.S., Deutsch S., Stranger B.E., Montgomery S.B., Borel C., Attar-Cohen H., Ingle C., Beazley C., Gutierrez Arcelus M., Sekowska M., et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lesnik Oberstein S.A., Kriek M., White S.J., Kalf M.E., Szuhai K., den Dunnen J.T., Breuning M.H., Hennekam R.C. Peters Plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am. J. Hum. Genet. 2006;79:562–566. doi: 10.1086/507567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conneely K.N., Boehnke M. So many correlated tests, so little time! Rapid adjustment of P values for multiple correlated tests. Am. J. Hum. Genet. 2007;81:1158–1168. doi: 10.1086/522036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finelli P., Stanyon R., Plesker R., Ferguson-Smith M.A., O'Brien P.C., Wienberg J. Reciprocal chromosome painting shows that the great difference in diploid number between human and African green monkey is mostly due to non-Robertsonian fissions. Mamm. Genome. 1999;10:713–718. doi: 10.1007/s003359901077. [DOI] [PubMed] [Google Scholar]
- 23.Rotival M., Zeller T., Wild P.S., Maouche S., Szymczak S., Schillert A., Castagne R., Deiseroth A., Proust C., Brocheton J., et al. Integrating genome-wide genetic variations and monocyte expression data reveals trans-regulated gene modules in humans. PLoS Genet. 2011;7:e1002367. doi: 10.1371/journal.pgen.1002367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Small K.S., Hedman A.K., Grundberg E., Nica A.C., Thorleifsson G., Kong A., Thorsteindottir U., Shin S.Y., Richards H.B., Soranzo N., et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat. Genet. 2011;43:561–564. doi: 10.1038/ng.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gabriel S.B., Schaffner S.F., Nguyen H., Moore J.M., Roy J., Blumenstiel B., Higgins J., DeFelice M., Lochner A., Faggart M., et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 26.Fehrmann R.S., Jansen R.C., Veldink J.H., Westra H.J., Arends D., Bonder M.J., Fu J., Deelen P., Groen H.J., Smolonska A., et al. Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLoS Genet. 2011;7:e1002197. doi: 10.1371/journal.pgen.1002197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moulin S., Gerbault-Seureau M., Dutrillaux B., Richard F.A. Phylogenomics of African guenons. Chromosome Res. 2008;16:783–799. doi: 10.1007/s10577-008-1226-6. [DOI] [PubMed] [Google Scholar]
- 28.Carbone L., Harris R.A., Vessere G.M., Mootnick A.R., Humphray S., Rogers J., Kim S.K., Wall J.D., Martin D., Jurka J., et al. Evolutionary breakpoints in the gibbon suggest association between cytosine methylation and karyotype evolution. PLoS Genet. 2009;5:e1000538. doi: 10.1371/journal.pgen.1000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Girirajan S., Chen L., Graves T., Marques-Bonet T., Ventura M., Fronick C., Fulton L., Rocchi M., Fulton R.S., Wilson R.K., et al. Sequencing human-gibbon breakpoints of synteny reveals mosaic new insertions at rearrangement sites. Genome Res. 2009;19:178–190. doi: 10.1101/gr.086041.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aylor D.L., Valdar W., Foulds-Mathes W., Buus R.J., Verdugo R.A., Baric R.S., Ferris M.T., Frelinger J.A., Heise M., Frieman M.B., et al. Genetic analysis of complex traits in the emerging Collaborative Cross. Genome Res. 2011;21:1213–1222. doi: 10.1101/gr.111310.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nolan T., Hands R.E., Bustin S.A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 2006;1:1559–1582. doi: 10.1038/nprot.2006.236. [DOI] [PubMed] [Google Scholar]
- 32.Lachance P.E., Chaudhuri A. Microarray analysis of developmental plasticity in monkey primary visual cortex. J. Neurochem. 2004;88:1455–1469. doi: 10.1046/j.1471-4159.2003.02274.x. [DOI] [PubMed] [Google Scholar]
- 33.Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Almasy L., Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am. J. Hum. Genet. 1998;62:1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elton S., Dunn J., Cardini A. Size variation facilitates population divergence but does not explain it all: an example study from a widespread African monkey. Biol. J. Linnean Soc. 2010;101:21. [Google Scholar]
- 36.Osman Hill W.C. Primates: Comparative Anatomy and Taxonomy Catarrhini, Cercopithecoidea, Cercopithecinae. Edinburgh: Edinburgh University Press; 1966. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.