

Abstract
Herpesvirus nucleocapsids are translocated from their assembly site in the nucleus to the cytosol by acquisition of a primary envelope at the inner nuclear membrane which subsequently fuses with the outer nuclear membrane. This transport through the nuclear envelope requires homologs of the conserved herpesviral pUL31 and pUL34 proteins which form the nuclear egress complex (NEC). In its absence, 1,000-fold less virus progeny is produced. We isolated a UL34-negative mutant of the alphaherpesvirus pseudorabies virus (PrV), PrV-ΔUL34Pass, which regained replication competence after serial passages in cell culture by inducing nuclear envelope breakdown (NEBD) (B. G. Klupp, H. Granzow, and T. C. Mettenleiter, J. Virol. 85:8285–8292, 2011). To test whether this phenotype is unique, passaging experiments were repeated with a UL31 deletion mutant. After 60 passages, the resulting PrV-ΔUL31Pass replicated similarly to wild-type PrV. Ultrastructural analyses confirmed escape from the nucleus via NEBD, indicating an inherent genetic disposition in herpesviruses. To identify the mutated viral genes responsible for this phenotype, the genome of PrV-ΔUL34Pass was sequenced and compared to the genomes of parental PrV-Ka and PrV-ΔUL34. Targeted sequencing of PrV-ΔUL31Pass disclosed congruent mutations comprising genes encoding tegument proteins (pUL49, pUL46, pUL21, pUS2), envelope proteins (gI, pUS9), and protease pUL26. To investigate involvement of cellular pathways, different inhibitors of cellular kinases were tested. While induction of apoptosis or inhibition of caspases had no specific effect on the passaged mutants, roscovitine, a cyclin-dependent kinase inhibitor, and U0126, an inhibitor of MEK1/2, specifically impaired replication of the passaged mutants, indicating involvement of mitosis-related processes in herpesvirus-induced NEBD.
INTRODUCTION
Herpesviruses exhibit a complex replication cycle involving nuclear and cytoplasmic compartments of their target cells. Target cells vary, and in particular, members of the subfamily Alphaherpesvirinae are able to infect a wide range of different cells of various origins. Even highly differentiated, nondividing cells like neurons can be productively infected, indicating that the virus has adapted to replication in cells at different stages of differentiation. Although herpesviruses encode enzymes for genome replication and nucleotide metabolism, they rely on the host cell machinery in other aspects of virus production and release (53).
Intranuclear stages of herpesvirus replication include viral transcription, genome replication, capsid formation, and genome packaging. Newly formed nucleocapsids leave the nucleus by budding at the inner nuclear membrane, thereby acquiring a primary envelope which subsequently fuses with the outer nuclear membrane to release nucleocapsids for continuing maturation in the cytosol. This transport through the nuclear membranes mediated by a vesicular structure, the primary envelope, is unique in cell biology (reviewed in references 21, 47, 48, and 49).
Regulated, vesicle-mediated nuclear egress requires the presence of two viral proteins which are conserved in the three subfamilies of the Herpesviridae. They are homologs of the pUL34 and pUL31 proteins of herpes simplex virus 1 (HSV-1). pUL34 is a type II tail-anchored membrane protein which is targeted to the nuclear envelope and interacts at the inner nuclear membrane (INM) with pUL31, forming the nuclear egress complex (NEC) (15, 54). Whereas coexpression of the two NEC partners is sufficient for the formation of primary envelopes in the absence of nucleocapsids (32), interaction of NEC with nucleocapsids results in primary envelopment by the INM (35, 66). In the absence of the NEC, the formation of infectious progeny is ca. 1,000-fold reduced but not abolished, indicating that alternative mechanisms for nuclear egress exist. Suggested alternative pathways include exit of capsids from the nucleus via dilated nuclear pores (36, 65) or via remodelling of the nuclear envelope by a process designated pseudomitosis, which has been reported for human cytomegalovirus (HCMV) (18, 19). Cyclin-dependent kinase 1 (cdk1) plays a major role in pseudomitosis, but its inhibition had no influence on HCMV replication (18). Neither mechanism has been verified to play a role in nuclear egress.
Other viruses have also been shown to impair integrity of the nuclear envelope. Parvoviruses were reported to hijack cellular caspase-3 for local dissolution of the nuclear envelope and the underlying lamina, thus creating gaps through which capsids gain access to the nucleus during entry (10). Disruptions of the nuclear envelope are also induced by human immunodeficiency virus (HIV) vpr. Expression of vpr in HeLa cells induced transient herniations in the nuclear envelope which burst sporadically but reseal within minutes (12, 59).
The alphaherpesvirus pseudorabies virus (PrV) has a broad host range both in cell culture and in vivo. It is the causative agent of Aujeszky's disease, and the pig is the only susceptible species which can survive infection (reviewed in reference 46). The approximately 143-kb genome codes for 70 different proteins, of which ca. 40 constitute core proteins, i.e., gene products conserved within the Herpesviridae (29, 40). Approximately half of the genes encode structural components of the virion. Also, about half of the genes are dispensable for viral replication in cell culture (29) and, with only a few exceptions, also in model animal hosts (26), which greatly hampers their functional characterization. Assigning functions to specific proteins is further complicated by functional redundancy, implicating that similar functions can be accomplished by more than one viral protein.
For functional characterization, the gene of interest is mutated or deleted and the effect on virus replication is tested. In reversion assays, mutant viruses with impaired but not completely blocked replication competence can be serially passaged in cell culture, which repeatedly resulted in phenotypic rescue mutants exhibiting enhanced replication kinetics. Analysis of the revertants and identification of second-site mutations allowed conclusions on the function of the native proteins and putative interaction partners. Using this approach, we isolated a replication-competent PrV mutant lacking the receptor-binding envelope glycoprotein D (gD), PrV-gD−Pass, which is competent to infect host cells using non-gD-specific receptors (58). By the same approach, a replication-competent PrV mutant lacking envelope glycoprotein L (gL), which normally complexes with glycoprotein H (gH) and is involved in mediating penetration, could be obtained, regaining infectivity via generation of a gD-gH chimeric protein (27).
We recently applied reversion analysis to elucidate alternative pathways for primary envelope-mediated nuclear egress. To this end, we serially passaged a PrV mutant with a UL34 deletion (PrV-ΔUL34) in rabbit kidney cells (RK13), resulting in wild-type virus-like titers after 90 passages. The stepwise increase in infectivity pointed to acquisition of more than one compensatory mutation, ultimately leading to efficient replication (31). Ultrastructural analysis showed that in PrV-ΔUL34Pass-infected RK13 cells, the integrity of the nuclear envelope was impaired in approximately half of the cells, which was not observed in either PrV wild-type or PrV-ΔUL34 infection. Thus, capsids of PrV-ΔUL34Pass reached the cytosol for further maturation through the fragmented nuclear envelope (31). However, the viral genomic mutations necessary to induce this nuclear envelope breakdown (NEBD) and the cellular pathways involved remained unclear.
It also remained to be tested whether the observed phenotype could be reproduced in an independent assay starting with a different viral mutant, i.e., to analyze whether the observed NEBD is a bona fide alternative pathway whose induction is intrinsic in the herpesvirus replication repertoire. Deletion of either partner of the NEC resulted in a similar phenotype with a block in nuclear egress but residual replication capacity (15, 30) allowing serial passage of a PrV mutant lacking the other component of the NEC, pUL31. To unravel compensating mutations, the complete genomes of PrV-ΔUL34Pass and its parental strains, PrV-Ka and PrV-ΔUL34, were sequenced and compared. Moreover, by targeted sequencing of the PrV-ΔUL31Pass genome regions and comparison of the sequences with those identified to be altered in PrV-ΔUL34Pass, we observed congruent mutations in both passaged viruses. Lastly, using several inhibitors of cellular kinases involved in mitosis and/or signal transduction, we probed which cellular pathways are targeted in the passaged mutants inducing NEBD.
MATERIALS AND METHODS
Cells and viruses.
PrV strain Kaplan (PrV-Ka) (23) was used as the parental strain for the UL31- and UL34-deletion mutants (30, 31). PrV-ΔUL31G containing a green fluorescent protein (GFP) expression cassette at the deleted UL31 locus (31) was used for passaging, which was done as described for PrV-ΔUL34 (31). Plaques of the 60th passage were isolated, and one plaque isolate (PrV-ΔUL31Pass) was further characterized. RK13-DrdI cells, which contain a genomic fragment encompassing genes UL31 to UL35, have been described (32). RK13-UL46, RK13-UL21, and RK13-UL49 cells were established after transfection with pcDNA-UL46 (34), pcDNA-UL21 (33), or pcDNA-UL49 (16), followed by Geneticin (Life Technologies) selection. UL21-, UL26-, and UL46-deletion mutants of PrV-Ka or PrV-ΔUL34Pass were obtained after homologous recombination, followed by selection using autofluorescence. The deletion in UL21 was established as described previously (33), but the kanamycin resistance gene was replaced by GFP. To replace UL46 with GFP, pUC-UL27/47GFP (34) was used. For the isolation of PrV-ΔUL34Pass/ΔUL49, PrV-ΔUL34Pass was cloned as a bacterial artificial chromosome and UL49 was deleted using the plasmid pUC-ΔUL49T (16). Cloned BamHI fragment 9 of PrV-Ka encompassing the wild-type UL26 gene was cotransfected with pSV2neo to generate RK13-BamHI9 (Ka) cells. To delete UL26, the 1.4-kb XcmI/Tth111I subfragment of BamHI-9 was cleaved with NcoI and SalI, thereby deleting approximately 170 bp of the UL26 coding region immediately following the start codon and substituted by a GFP expression cassette.
Cloning and sequencing.
Genomic DNA isolated from PrV-Ka, PrV-ΔUL34, or PrV-ΔUL34Pass virions purified from infected cell supernatants by ultracentrifugation through a 30% sucrose cushion was used for sequencing. First, genome sequencing was done by a company (GATC, Konstanz, Germany) using GS FLX (454; Roche) titanium technology. Since the generated sequences did not cover the complete genomes, missing parts were sequenced in-house. To this end, PCR products were generated with sequence-specific primers flanking the sequence gaps. PCR products were sequenced either directly with the same primers or after cloning into pUC19 or pBluescript with vector-specific primers. Alternatively, suitable restriction fragments of viral DNA were cloned and sequenced by cycle sequencing (BigDye Terminator cycle sequencing kit; Applied Biosystems) and run on an ABI 3130 genetic analyzer (Applied Biosystems). Analysis was performed using the GCG software package (GCG, version 11.1; Accelrys Inc., San Diego, CA) programs pileup and pretty. Open reading frames (ORFs) found mutated in PrV-ΔUL34Pass (UL50, UL49, UL46, UL27, UL29 [partial], UL39, UL41, UL21, UL12, and US7; Table 1) were sequenced in the PrV-ΔUL31Pass genome with gene-specific primers.
Table 1.
Alterations in deduced proteins of PrV-ΔUL34Pass and PrV-ΔUL31Pass
| Gene | Protein | Mutation |
|
|---|---|---|---|
| PrV-ΔUL34Pass | PrV-ΔUL31Pass | ||
| UL50 | dUTPase (268)a | Thr 205 Ala | None |
| UL49 | Tegument protein (249) | Frameshift after codon 76 | Frameshift after codon 143 |
| UL46 | Tegument protein (693) | 87-bp/29-aa sequence duplication (aa 514–542) | 37-bp duplication; frameshift after codon 662 |
| UL27 | Glycoprotein gB (920) | Gln 148 Arg | None |
| UL29 | Single-strand DNA binding protein (1,177) | Val 778 Ala | None |
| UL39 | Ribonucleotide reductase (790) | Ile 547 Leu | None |
| UL41 | VHS (365) | Lys 8 Arg | None |
| UL26 | Protease (524) | Gly 82 Asp | Val 81 Ala |
| UL21 | Tegument protein (525) | Val 236 Ala | Arg 192 His |
| US1 | RSp40/ICP22 (364) | His 55 Tyr | None |
| US7 | Glycoprotein gI (365) | Frameshift after codon 221 | Frameshift after codon 221 |
| US8 | Glycoprotein gE (577) | Frameshift after codon 542 | Deleted |
| US9 | Tail-anchored membrane protein (98) | Ser 38 Leu | Deleted |
| US2 | Prenylated tegument protein (256) | Frameshift after codon 180 | Deleted |
Numbers in parentheses give the number of amino acids of wild-type PrV-Ka protein.
Western blotting.
For immunoblotting, RK13 cells were infected with PrV-Ka, PrV-ΔUL31, PrV-ΔUL31Pass, PrV-ΔUL34, or PrV-ΔUL34Pass at a multiplicity of infection (MOI) of 5 or remained noninfected. After 16 h, cells were scraped into the medium, pelleted by centrifugation, washed twice with phosphate-buffered saline (PBS), and lysed in sodium dodecyl sulfate (SDS) sample buffer. Cell lysates were separated on SDS–10 or 12% polyacrylamide gels and blotted onto nitrocellulose. Membranes were incubated with monospecific rabbit sera against pUL12 (unpublished), pUL21 (33), pUL26 (unpublished), pUL31 (15), pUL34 (30), pUL38 (unpublished), pUL39 (unpublished), pUL46 (34), pUL49 (3), pUS2 (unpublished), pUS9 (unpublished), or gI (3), and bound antibody was detected after incubation with peroxidase-conjugated goat anti-rabbit secondary antibodies (Dianova, Hamburg, Germany) or, after incubation with monoclonal antibodies against gB or gE (3), with peroxidase-conjugated goat anti-mouse secondary antibodies (Dianova, Hamburg, Germany). After incubation with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific), blots were analyzed with an imager (Intas ChemiCam).
One-step replication kinetics.
For one-step replication kinetics, RK13 or RK13-DrdI cells (32) were infected at an MOI of 5 and harvested at different times after low-pH treatment (45). Cells were scraped into the supernatant and frozen at −70°C. After thawing, titers were determined on RK13 and RK13-DrdI cells. Mean values for at least three independent assays were calculated and plotted. Corresponding standard deviations are given.
Inhibitor studies.
Inhibitors were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10 mM or 1 mM (staurosporine). To investigate the influence on virus replication, different dilutions were tested and effects on virus infection were calculated corresponding to a DMSO control. To this end, cells were infected at an MOI of 5 for 1 h on ice. After addition of prewarmed medium, they were further incubated for 1 h at 37°C to allow virus entry. Inhibitors were added at the indicated concentration 1 h later, and incubation was continued for 24 h. Viral titers in the supernatant were determined after freezing and thawing of the infected cell cultures. Mean values of three independent experiments and corresponding standard deviations are shown. Inhibitors tested included 3-amino-1H-pyrazolo-quinoxaline (Enzo), Akt inhibitor (Calbiochem), bisindolylmaleimide (Calbiochem), CDK2-inhibitor II (Merck Biosciences), FR180204 (Calbiochem), N9-isopropylolomoucine (Enzo), PD184352 (Santa Cruz), PNU112455A.HCl (Enzo), N-(2-quinolyl)valyl-aspartyl-(2,6-difluorophenoxy)methyl ketone (Q-VD-OPH; R&D Systems), RO-3306 (Calbiochem), roscovitine (Calbiochem), SB203580 (Calbiochem), staurosporine (Alexis), U0126 (Cell Signalling), and wortmannin (Calbiochem). For the specificities of the inhibitors, see Table 2.
Table 2.
Effect on viral replication of inhibitors of cellular signaling pathways
| Compound | Targeta | Fold reduction at 100 μM at 24 h p.i. compared to DMSO only |
||||
|---|---|---|---|---|---|---|
| PrV-Ka | PrV-ΔUL34Pass | PrV-ΔUL31Pass | PrV-ΔUL31 | PrV-ΔUL34 | ||
| 3-Amino-1H-pyrazolo-quinoxaline | cdk1, cdk5, GSK-3β | 3 | 2 | 2 | NDb | ND |
| Akt inhibitor | Akt, (PI3K) | 1 | 1 | 1 | ND | ND |
| Bisindolylmaleimide I (Gö6850) | PKC, (PKA) | 18 | 50 | 100 | ND | ND |
| CDK inhibitor II | cdk1, cdk2, (cdk4) | 1 | 1 | 1 | ND | ND |
| FR180204 | ERK1/2, (p38a) | 1 | 1 | 1 | ND | ND |
| N9-Isopropylolomoucine | cdk1, cdk5 | 8 | 800 | 100 | ND | ND |
| PD184352 | MEK1/2 | 1 | 1.5 | 2 | ND | ND |
| PNU112455A · HCl | cdk2, cdk5, (ERK2) | 1 | 1 | 3 | ND | ND |
| RO-3306 | cdk1, (cdk2, cdk4, PKCδ, SGK) | 2 | 10 | 3.5 | ND | ND |
| Roscovitine | cdk1, cdk2, cdk5, (ERK1/2) | 80 | 2 × 106 | 1 × 104 | >6 × 104 | >5 × 103 |
| SB203580 | p38 | 6 | 3 | 9 | ND | ND |
| U0126 | MEK1/2 | 200 | 1 × 105 | 7 × 105 | >6 × 104 | >5 × 103 |
| Wortmannin | PI3K, PLD, (MLCK, PI4K) | 5 | 10 | 30 | ND | ND |
Targets which are affected only at higher concentrations are given in parentheses. PKC and PKA, protein kinases C and A; SGK, serum- and glucocorticoid-induced protein kinase; MLCK, myosin light-chain kinase.
ND, not determined.
Electron microscopy.
RK13 cells were infected with PrV-Ka, PrV-ΔUL31, PrV-ΔUL31Pass, or PrV-ΔUL34Pass at an MOI of 1 in the absence or presence of 50 μM roscovitine. At 14 h postinfection (p.i.), cells were fixed and processed for electron microscopy as described previously (30).
Nucleotide sequence accession numbers.
Genome sequences have been deposited in GenBank under accession numbers JQ809328 for PrV-Ka, JQ809329 for PrV-ΔUL34, and JQ809330 for PrV-ΔUL34Pass.
RESULTS
Isolation of PrV-ΔUL31Pass.
After serial passaging of PrV-ΔUL34 in rabbit kidney (RK13) cells, we isolated PrV-ΔUL34Pass, which replicated with wild-type kinetics and was able to exit the nucleus through a fragmented nuclear envelope (31). To test whether this phenotype could be reproduced, passaging experiments were repeated using a PrV mutant lacking the complex partner of pUL34, pUL31 (31). First, RK13 cells were infected with approximately 0.001 PFU of PrV-ΔUL31G in a 6-well culture dish. After 4 to 6 days, infected cells were trypsinized and 1/3 of the cells were reseeded, further incubated, and again diluted 1:3 after trypsinization. Titers of the supernatants were determined on RK13 cells. After a few passages, increased infectivity in the supernatant could be observed, and the passage regime was changed to transfer of infectious supernatant to freshly seeded RK13 cells. After approximately 60 passages, infectivity in the supernatant reached titers of between 106 and 107 PFU/ml. Several single plaque isolates of this passage were tested, and one plaque isolate, designated PrV-ΔUL31Pass, was further characterized in detail.
One-step growth kinetics (Fig. 1) showed that PrV-ΔUL31Pass exhibited significantly increased replication on RK13 cells compared to the parental mutant and titers were similar to those exhibited by PrV-ΔUL34Pass. Infection of RK13-DrdI cells had no impact on the replication of either passaged mutant, while viral titers of the nonpassaged mutants increased to wild-type levels (Fig. 1B). These data indicated that both passaged mutants lacking either pUL34 or pUL31 replicated independently of the NEC.
Fig 1.

Replication kinetics of PrV-ΔUL31 and PrV-ΔUL34 mutants. RK13 (A) or RK13-DrdI (B) cells carrying a genomic fragment encompassing PrV genes UL31 to UL35 were infected with PrV-Ka, PrV-ΔUL31, PrV-ΔUL31Pass, PrV-ΔUL34, or PrV-ΔUL34Pass at an MOI of 5. At the indicated times postinfection, infected cells were scraped into the supernatant and virus progeny titers were determined. Mean values of three independent assays and corresponding standard deviations are given.
Electron microscopic analysis of RK13 cells infected with PrV-ΔUL31Pass (Fig. 2) revealed a fragmented nuclear envelope in approximately 50% of the infected cells observed at 14 h p.i. (Fig. 2B and C). This phenotype resembled the phenotype seen in PrV-ΔUL34Pass-infected cells (31) but had not been observed in any of the other numerous mutants tested in our laboratory, including PrV-ΔUL31 (Fig. 2A) and PrV-Ka. As in PrV-ΔUL34Pass-infected RK13 cells, different capsid forms, empty A-, scaffold-containing B-, and genome-containing C-type capsids, of PrV-ΔUL31Pass could be detected in the cytoplasm undergoing secondary envelopment (Fig. 2C), indicating that nuclear envelope fragmentation was not the result of mechanical or osmotic stress during the preparation for electron microscopy. Thus, in independent assays, mutants lacking either component of the NEC gained replication competence independently of the NEC via induction of NEBD.
Fig 2.
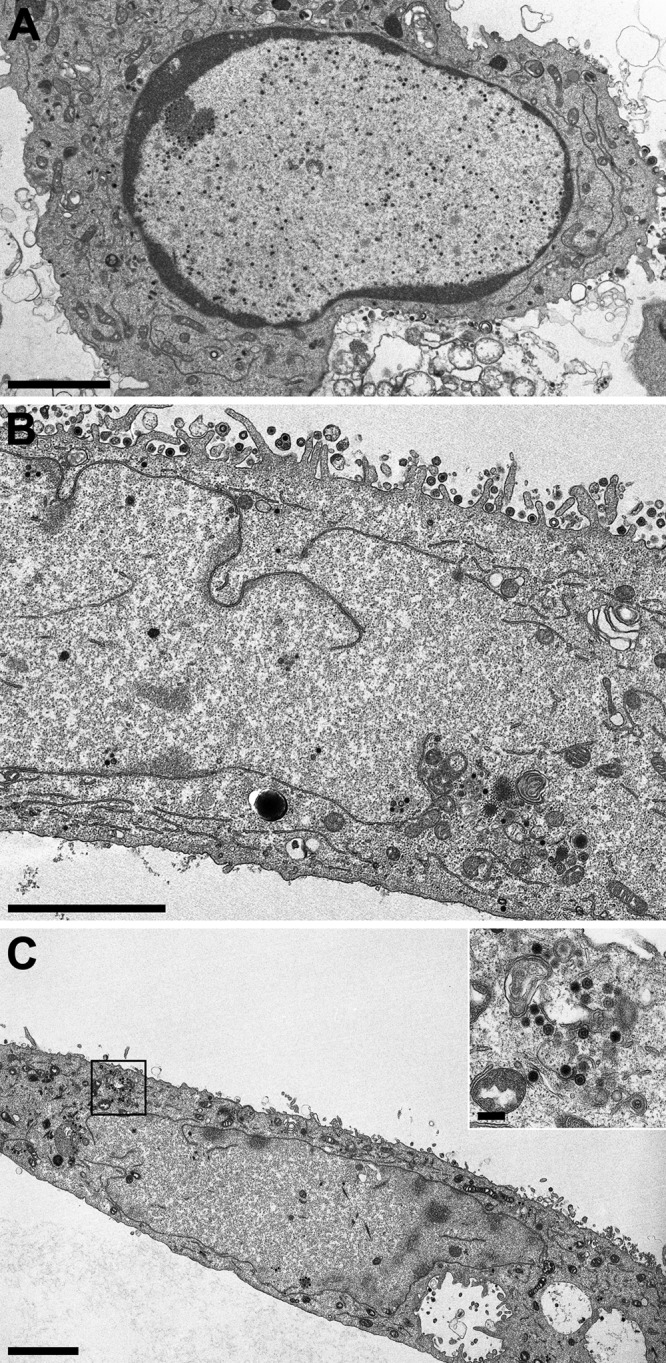
Ultrastructure of cells infected with PrV-ΔUL31 or PrV-ΔUL31Pass. RK13 cells were infected at an MOI of 1 with PrV-ΔUL31 (A) or PrV-ΔUL31Pass (B and C) and processed for electron microscopic analysis at 14 h p.i. The inset in panel C shows capsids undergoing secondary envelopment in the cytoplasm. Bars, 3 μm (A to C) and 250 nm (inset, panel C).
Sequence comparison of PrV-Ka, PrV-ΔUL34, and passaged mutants.
Since both independently passaged mutants, PrV-ΔUL34Pass and PrV-ΔUL31Pass, showed a similar phenotype, congruent compensatory mutations in their genomes might have occurred. To identify viral genes which are mutated in the passaged mutants, the complete genome sequences of the parental strains PrV-Ka and PrV-ΔUL34 as well as the genome sequence of PrV-ΔUL34Pass were determined. Sequencing was outsourced (GATC, Konstanz, Germany), and remaining gaps were filled by in-house sequencing either after PCR amplification or after cloning. PrV contains, scattered throughout the genome, a number of direct and indirect perfect and imperfect repeat sequences (29, 60) which are highly variable in number even within strains (64) and were therefore excluded from further analysis. Besides these variations and the intended deletion of UL34 coding sequences and insertion of the GFP expression cassette, differences between PrV-ΔUL34Pass and PrV-Ka were found at 36 loci. These included insertions, deletions, and nucleotide alterations in coding and noncoding regions. Five of them also appeared to be highly variable within the different PrV strains sequenced and were not further inspected. Three mutations in coding regions were already present in the parental deletion mutant PrV-ΔUL34, comprising a mutation in UL39 leading to an amino acid change from isoleucine to leucine at position 547, a silent mutation in UL12, and a frameshift affecting the cytoplasmic domain of gE after codon 542 shortening the protein product to 557 amino acids (aa) compared to the native 577 aa in PrV-Ka. Mutations specific for PrV-ΔUL34Pass affecting coding regions were found in UL50, UL49, UL46, UL27 (gB), UL29, UL41, UL26, UL21, US1, US7 (gI), US9, and US2 (summarized in Table 1).
Open reading frames found mutated in PrV-ΔUL34Pass were analyzed by targeted sequencing of corresponding regions in the PrV-ΔUL31Pass genome. Gene-specific primers were used to amplify ORFs UL50, UL49, UL46, UL27, UL29 (partial), UL39, UL41, UL21, UL12, and US7. In addition, restriction enzyme analysis of genomic DNA revealed that PrV-ΔUL31Pass contained a large deletion in the unique short (US) region comprising genes encoding gE (partial), US9, and US2 (deletion corresponding to nucleotides [nt] 123733 to 126667; GenBank accession number BK001744) (29). While wild-type sequences were found at the relevant positions in PrV-ΔUL31Pass for UL50, UL27, UL29, UL39, UL41, and UL12, genes encoding tegument proteins pUL49, pUL46, and pUL21, envelope protein gI, and the protease pUL26 showed mutations similar to those found in PrV-ΔUL34Pass (Table 1).
A single nucleotide deletion in UL49 led to frameshifts after codon 76 in PrV-ΔUL34Pass and after codon 143 in PrV-ΔUL31Pass, resulting in predicted 80-aa and 199-aa products, respectively, compared to the 249-aa wild-type protein. The UL46 open reading frame comprised sequence duplications in PrV-ΔUL34Pass and PrV-ΔUL31Pass. While the mutation in PrV-ΔUL34Pass was in frame, resulting in duplication of a highly charged amino acid sequence (DEEEEEDDRFPRRDGDAGASTSSSQESAA), in PrV-ΔUL31Pass the sequence duplication was out of frame after codon 662 and resulted in an extension of the protein by 32 aa. pUL26, the maturational protease, exhibited an amino acid exchange from glycine to aspartic acid at position 82 in PrV-ΔUL34Pass, while in PrV-ΔUL31Pass position 81 was mutated from valine to alanine. A point mutation at codon 236 of UL21 resulted in a valine-to-alanine exchange in pUL21 of PrV-ΔUL34Pass, whereas in PrV-ΔUL31Pass arginine 192 was altered to histidine. Gene US7, encoding gI, carried a frameshift in both mutants at exactly the same position (codon 221), although in PrV-ΔUL34Pass a cytidine had been added, while a cytidine had been deleted in PrV-ΔUL31Pass. However, in both mutants the frameshift resulted in a loss of the hydrophobic membrane anchor sequence of gI. The US9 ORF carried an amino acid exchange from serine to leucine at position 38 in PrV-ΔUL34Pass and was missing completely in PrV-ΔUL31Pass due to the large deletion, as was pUS2. In PrV-ΔUL34Pass pUS2 contained a frameshift after codon 180, resulting in an enlarged predicted protein product with 304 aa compared to the predicted 256-aa protein in PrV-Ka. Remarkably, the frameshift led to loss of the prenylation motif CAAX, which is present in the C-terminal part of wild-type pUS2 (9).
Protein expression of mutant viruses.
To analyze the effects of the mutations detected by genome sequencing at the protein level, cells were infected with the different deletion mutants and compared to cells infected with PrV-Ka. One day after infection, cell lysates were separated on SDS–10 or 12% polyacrylamide gels and blotted onto nitrocellulose. As shown in Fig. 3, pUL34 is missing in cells infected by PrV-ΔUL34 and PrV-ΔUL34Pass, while pUL31 was not detectable in PrV-ΔUL31- and PrV-ΔUL31Pass-infected cells. Slightly reduced amounts of pUL31 were present in the UL34-deletion mutants. Sequence analysis predicted an approximately 9-kDa pUL49 in PrV-ΔUL34Pass-infected cells, which, however, was not detected even on higher-percentage polyacrylamide gels (data not shown). In contrast, a truncated pUL49 was found in PrV-ΔUL31Pass-infected cells and corresponds to the predicted 199-aa pUL49 compared to the 249-aa product in wild-type PrV. As expected, the mutated pUL46 of PrV-ΔUL34Pass and PrV-ΔUL31Pass showed slightly decreased electrophoretic mobility, but smaller protein species were also detectable in all samples, indicating instability of the protein. In both passaged virus mutants, the gI-encoding US7 carried a frameshift eliminating the hydrophobic transmembrane region. However, in PrV-ΔUL31Pass-infected cells, no gI-specific signal was observed, which might be due to the concomitant loss of gE, while in PrV-ΔUL34Pass-infected cells, a smaller gI which still might be able to interact with its complex partner, gE, was expressed. pUS2 was not detectable in cells infected with either passaged mutant, which was not surprising for PrV-ΔUL31Pass, where the US8, US9, and US2 gene region was deleted. However, in PrV-ΔUL34Pass-infected cell lysates, a larger product was expected. Possibly, the frameshift mutation after position 180, resulting in loss of the prenylation motif which is thought to tether wild-type pUS2 to membranes (39), might lead to efficient secretion or rapid degradation of PrV-ΔUL34Pass pUS2. Despite the identified point mutations, no differences in electrophoretic mobility were found for pUL21, pUL26, pUL39, or pUL12. Capsid protein pUL38 and glycoprotein B were used as loading controls (Fig. 3).
Fig 3.

Western blot analysis. RK13 cells were infected at an MOI of 5 with PrV-Ka (lanes 2), PrV-ΔUL31 (lanes 3), PrV-ΔUL31Pass (lanes 4), PrV-ΔUL34 (lanes 5), and PrV-ΔUL34Pass (lanes 6) and harvested at 24 h p.i. In lanes 1, noninfected cells were used as a negative control. Cell lysates were separated in SDS–10% or 12% polyacrylamide gels and incubated with the antisera indicated on the right. Locations and molecular masses of marker proteins are given on the left.
Whereas deletions in the US region encompassing the nonessential US7, US8, US9, and US2 genes are known to occur regularly during passaging of PrV (1, 50, 64), UL21, UL26, UL46, and UL49 are not normally mutated as a result of serial passage. To analyze whether any of these genes is involved in the observed phenotype, mutants of PrV-Ka and PrV-ΔUL34Pass with deletion of either of these genes were isolated and analyzed comparatively for replication on cell lines expressing the wild-type versions of the proteins. Unfortunately, none of the tested combinations yielded any significantly different phenotypes concerning viral titers or plaque sizes (data not shown).
Pathways involved in apoptosis are not affected in the passaged PrV mutants.
Since NEBD is known to occur during apoptosis, we first tested whether caspases might be involved in the observed phenotype. Therefore, infected cells were treated with the pan-caspase inhibitor N-(2-quinolyl)valyl-aspartyl-(2,6-difluorophenoxy)methyl ketone (Q-VD-OPH), which is reported to decrease caspase-dependent cell death more efficiently and to be less cytotoxic than other broad-spectrum caspase inhibitors like Z-Val-Ala-Asp-fluoromethylketone (Z-VAD-fmk) (8). However, addition of 100 μM Q-VD-OPH to either wild-type PrV- or PrV-ΔUL34Pass-infected cells decreased viral titers to the same extent (data not shown). Similar results were observed when apoptosis was induced by staurosporine (Fig. 4A). Parallel curve progressions were found for PrV-Ka and the passaged mutants, indicating that apoptosis-associated pathways are not involved in NEBD.
Fig 4.

Effect of inhibitors of cellular signaling pathways on viral replication. Cells were infected at an MOI of 5 with PrV-Ka, PrV-ΔUL34Pass, or PrV-ΔUL31Pass and subsequently incubated in medium containing either DMSO or inhibitors dissolved in DMSO at the indicated concentrations. Samples were harvested after 24 h and titrated on RK13 cells. Given are mean values of three independent experiments and the corresponding standard deviations. Diamonds, PrV-Ka; quadrangles, PrV-ΔUL34Pass; triangles, PrV-ΔUL31Pass.
Cell cycle and cell signaling pathways are affected by the passaged mutants.
Since neither induction of apoptosis by staurosporine nor inhibition of caspases showed a specific effect on replication of the passaged mutants, we analyzed whether pathways involved in mitosis might be targeted. Roscovitine is a potent and selective inhibitor affecting cdk1/cyclin B (inhibitory concentration [IC] = 650 nM), cdk2/cyclin A (IC = 700 nM), cdk2/cyclin E (IC = 700 nM), and cdk5/p35 (IC = 200 nM) complexes by competing for the ATP binding domains of the kinases, but it also inhibits the related extracellular signal-regulated kinase 1 (ERK1) and ERK2, albeit at higher concentrations (43). With its selective binding, roscovitine blocks mitosis at the G1/S and G2/M phases of the cell cycle. In addition, roscovitine has been shown to inhibit herpesvirus replication, although the mechanism is not completely understood (56, 57). As reported for HSV-1 and varicella-zoster virus (VZV) (13, 61), addition of roscovitine to infected cells reduced infectious titers of PrV. While PrV-Ka showed approximately 80-fold reduced titers at 100 μM concentrations, titers of the passaged mutants were more drastically reduced and dropped to nearly 0 at the same concentration (Fig. 4B; Table 2). In ultrastructural analyses (Fig. 5), NEBD induced by PrV-ΔUL34Pass (Fig. 5A) was completely blocked by addition of 50 μM roscovitine (Fig. 5B), resulting in the phenotype of the nonpassaged parent PrV-ΔUL34 (30). In contrast, replication of wild-type PrV-Ka was unaffected ultrastructurally at this concentration (Fig. 5C and D).
Fig 5.

Roscovitine blocks herpesvirus-induced NEBD. RK13 cells were infected at an MOI of 1 with PrV-ΔUL34Pass (A and B) or PrV-Ka (C and D) in either the absence (A and C) or presence (B and D) of 50 μM roscovitine. Cells were fixed and processed for electron microscopy at 14 h p.i. Bars, 5 μm (A, C, and D), 3 μm (B), 500 nm (insets, panels A and D), and 200 nm (insets, panels B and C).
To further pinpoint which cyclin-dependent kinase might be targeted, more specific inhibitors were used. RO-3306 acts as a potent inhibitor of cdk1 (IC50, 35 nM and 110 nM for cdk1/B1 and cdk1/A, respectively) and results in G2/M cell cycle arrest (62). However, addition of different concentrations of RO-3306 to infected cells had only a marginal effect on replication of either PrV-Ka or the passaged mutants (Fig. 4C). The same was found for a cdk2-specific inhibitor (Fig. 4D). Involvement of cdk5 was tested using different compounds, including 3-amino-1H-pyrazolo-quinoxaline (cdk1, cdk5, glycogen synthase kinase 3β [GSK-3β]), N9-isopropylolomoucine (cdk1, cdk5), and PNU112455A · HCl (cdk2, cdk5, ERK2). Of these, only N9-isopropylolomoucine had an effect on the titers of PrV-ΔUL34Pass (800-fold reduction) and PrV-ΔUL31Pass (100-fold reduction) greater than the 8-fold reduction for PrV-Ka (Table 2).
To test for an indirect effect on mitosis independent of direct cdk targeting, we assayed for inhibition of ERK1/2 and the upstream kinases MEK1/MEK2, which are involved in the mitogen-activated protein kinase (MAPK) cascade controlling cell growth and differentiation. U0126 is a highly active inhibitor of MEK1 and MEK2 (14). Like roscovitine, U0126 drastically reduced the titers of the passaged mutants while exhibiting only a moderate effect on PrV-Ka (Fig. 4E), indicating an involvement of the RAS/RAF/MEK/ERK signaling pathway in virus-induced NEBD. However, specific blocking of ERK1/2 with FR180204, which is an ATP-competitive inhibitor of ERK1/2, had no effect on infection by either PrV-Ka or the passaged viruses (Table 2).
Interestingly, the residual infectivity of both PrV-ΔUL34 and PrV-ΔUL31 was completely blocked by addition of either roscovitine or U0126 (Table 2).
DISCUSSION
Herpesvirus nucleo capsids leave the nucleus by a mechanism involving vesicle formation, fission, and fusion mediated by the NEC containing pUL31 and pUL34 (reviewed in references 21, 47, 48, and 49). This vesicle-mediated transport through the nuclear envelope is unique in cell biology. However, successful isolation of PrV mutants lacking either pUL31 (this paper) or pUL34 (31), which regained wild-type-like replication competence in the absence of the NEC after extensive cell culture passage, indicated other possibilities for capsids to leave the nucleus. This raises the question on the biological basis of the complex, vesicle-mediated, and highly regulated nuclear egress. In both passaged mutants, approximately half of the infected cells showed a fragmented nuclear envelope, thereby allowing direct access of capsids to the cytosol for further maturation. NEBD had not been observed before in wild-type virus-infected cells or in cells infected with numerous PrV deletion mutants investigated over the years, including PrV-ΔUL31 and PrV-ΔUL34 (15, 30). The integrity of the nuclear envelope usually remains intact even at late times after herpesvirus infection (20). However, it is unclear how nuclear integrity is maintained despite high-level viral replication. Possibly, herpesviruses encode proteins which are actively involved in maintaining an intact nucleus, and these proteins may be altered or deleted in the passaged PrV mutants exhibiting NEBD.
To assay whether the same phenotype observed in PrV-ΔUL34Pass (31) could be reproduced in an independent assay, we serially passaged a mutant virus lacking the other partner of the NEC, pUL31. PrV with a UL31 deletion behaves like PrV-ΔUL34 in vitro and in vivo (15, 26, 30), demonstrating that the functional unit for nuclear egress is the NEC. A cell-type dependence of the impact of pUL31 deletion has also been noted (37). As observed for PrV-ΔUL34, infectious titers of PrV-ΔUL31 gradually rose until reaching wild-type-like levels after 60 passages. Ultrastructurally, NEBD was observed in ca. 50% of infected cells, as was observed after infection with PrV-ΔUL34Pass (31), indicating that both are mechanistically similar if not identical. Thus, elimination of either pUL34 or pUL31 can be compensated for by NEBD after serial passaging in RK13 cells. These data show that NEBD is a bona fide compensatory mechanism to allow nucleocapsid egress independent of the NEC.
To identify candidate proteins which may be involved in induction of NEBD or, in their wild-type forms, in maintaining nuclear integrity, PrV-ΔUL34Pass as well as parental PrV-Ka and PrV-ΔUL34 genomes were completely sequenced. Sequence comparison yielded a limited number of differences between the unpassaged and the passaged deletion mutant. Most of them were located either in regions of reiterated sequences, which are known to be highly variable (1, 64), or in intergenic regions. Since changes in coding regions were considered most likely to be involved in the observed pUL34-independent nuclear egress, we also sequenced the corresponding regions in PrV-ΔUL31Pass, which has been passaged and isolated independently of PrV-ΔUL34Pass. Surprisingly congruent mutations were found in genes encoding the tegument proteins pUL49, pUL46, and pUL21, in glycoprotein gI, as well as in the protease-encoding part of UL26. While only single amino acid changes were found for pUL21 and pUL26, frameshifts resulted in more dramatic changes in protein products of UL49, UL46, and US7 (gI). US8 (gE), US9, and US2 were altered in PrV-ΔUL34Pass by either frameshifts (US8, US2) or single amino acid substitutions (US9), while the complete region was deleted in PrV-ΔUL31Pass. This large deletion in the unique short region of PrV-ΔUL31Pass comprises genes encoding gE, US9, and US2 (corresponding to nt 123733 to 126667; GenBank accession number BK001744) (29). Similar deletions in the US region have been observed before after extensive passaging of PrV mutants in avian cells or embryonated eggs (38, 50). UL21, UL26, UL46, and UL49 are not normally mutated as a result of serial passage. Therefore, we analyzed whether any of these genes is involved in the observed phenotype by assessing replication of mutants of PrV-Ka and PrV-ΔUL34Pass with a deletion in either of these genes on cell lines expressing the wild-type versions of the proteins. However, none of the tested combinations yielded any phenotypes significantly different in viral titers or plaque sizes (data not shown). Thus, as already indicated by the reversion analyses, a combination of these mutations appears to be necessary to allow NEC-independent nuclear escape.
All proteins which were found to be mutated or deleted in the passaged mutants, except the protease pUL26, are nonessential and can be deleted from the PrV genome without seriously impairing virus replication (4, 9, 11, 26, 28, 33, 34). Except for pUL26 and pUL21, the mutated genes are conserved only within the alphaherpesviruses, indicating virus subfamily-specific functions. Most of the gene products mutated in the passaged mutants seem to be membrane associated. While gE/gI and pUS9 are integral membrane proteins, pUS2, pUL46, and pUL49 have also been reported to bind to membranes either directly or indirectly (5, 9, 51). However, none of these proteins was demonstrated in the nuclear envelope of PrV-infected cells.
Two of the mutated proteins especially warrant further investigation, since they are involved in cell signaling cascades related to nuclear envelope remodelling. pUL46 is one of the most abundant tegument proteins and, in HSV-1, is required for virus-induced activation of the phosphoinositide-3-kinase (PI3K)-Akt signaling cascade. pUL46 is a substrate of Lck or other Src family kinases (SFKs), and activated pUL46 might modulate Akt targets. Akt controls many biological functions, including cell survival, cell motility, and translation (63). In addition, pUL46 enhances the transactivating function of pUL48 (24, 41, 67). However, a PrV deletion mutant lacking genes UL49 to UL46 proved to be replication competent in cultured cells (17). Interestingly, the varicella-zoster virus ORF12 protein, which is homologous to pUL46, triggers phosphorylation of ERK1/2 and inhibits apoptosis (37a).
PrV pUS2 is prenylated in infected cells and localizes to membranes (9). It has been reported to act as a spatiotemporal regulator of cellular ERK1/2 and tethers ERK1/2 at the plasma membrane, thereby preventing translocation of activated ERK1/2 to the nucleus (39). Prenylation is not required to anchor pUS2 at the plasma membrane since it was shown that nonprenylated mutant forms of PrV pUS2 (22) as well as the pUS2 homolog of equine herpesvirus 1 (EHV-1) (44), which lacks the prenylation motif, are membrane associated, probably by interaction with other viral membrane proteins. The PrV, EHV-1, and Marek's disease virus (MDV) US2-deletion mutants replicate with wild-type kinetics in cell culture, although entry kinetics appear to be delayed (6, 9, 44, 52). PrV, HSV, and MDV US2-deletion mutants retain their pathogenicity in animals (6, 25, 42), indicating that sequestration of ERK1/2 at the plasma membrane is not required for efficient virus replication in vitro or in vivo. No ultrastructural analyses have been reported for any of these mutants.
In noninfected cells, NEBD is known to occur during either apoptosis or mitosis. Neither induction of apoptosis by staurosporine nor its prevention by addition of a pan-caspase inhibitor had any specific effects on the titers of the passaged PrV mutants, which suggests that apoptosis-associated pathways do not play a role in herpesvirus-induced NEBD. In contrast, NEBD was efficiently inhibited by roscovitine, with titers of the passaged mutants dropping to nearly 0, while the effect on wild-type PrV was significantly less and in the range reported for HSV-1 and VZV (13, 61). Roscovitine is a selective inhibitor of cdk's (especially cdk1, cdk2, and cdk5), indicating that cdk's are involved in herpesvirus-induced NEBD. However, more specific inhibitors of cdk1, cdk2, and cdk5 or different combinations thereof had no effect on viral replication, which might be due to the functional redundancy and promiscuity of cdk's (L. Schang, personal communication). We are currently performing studies using dominant-negative versions of cdk's which might provide further insight into interaction of any of the viral proteins with cdk's.
In addition to its effect on cdk's, roscovitine also acts on ERK1/2. ERK1/2 is part of the extracellular signal-regulated kinase cascade which, within other MAPK cascades, signals from the outside of cells to the nucleus, thereby inducing processes such as cell growth and differentiation. To test whether the RAS/RAF/MEK/ERK pathway is involved in herpesvirus-induced NEBD, we used the MEK-specific inhibitor U0126. Addition of U0126 drastically reduced titers of PrV-ΔUL31Pass and PrV-ΔUL34Pass, while the effect on wild-type PrV was much less pronounced. The known substrate for MEK1/2 is ERK1/2 (7). The role of ERK activity during G2/M transition is not well understood, but ERK seems to function through a complex and indirect role on a small subset of G2/M phosphoproteins (55), and the downstream effectors of ERK, which are required for mitosis progression, remain to be determined (2). However, a specific inhibitor of ERK1/2 (FR180204) had no influence on the titers of either the passaged mutants or wild-type PrV. Interestingly, the residual infectivity of nonpassaged PrV-ΔUL31 and PrV-ΔUL34 is also abolished by roscovitine and U0126, indicating a similar mechanism for NEC-independent nuclear escape.
In summary, our results show that herpesvirus-induced NEBD is an alternative pathway that can be used in the absence of regulated nuclear egress due to defects in either of the two conserved components of the NEC. We identified congruent alterations in the genomes of PrV-ΔUL34Pass and PrV-ΔUL31Pass in a limited set of proteins which might be relevant for herpesvirus-induced NEBD. Our studies also indicate that mitosis-related pathways are involved in herpesvirus-induced NEBD. Whether any putative function of pUL34 beyond nuclear egress, e.g., in cell-cell spread, has also been targeted by the compensatory mutations that we observed is currently under study.
ACKNOWLEDGMENTS
We thank C. Meinke, D. Werner, A. Carnitz, and P. Meyer for expert technical assistance and M. Jörn for photographic help.
This study was supported by the Deutsche Forschungsgemeinschaft Priority Program SPP1175, grant Me 854/8-2.
Footnotes
Published ahead of print 4 April 2012
REFERENCES
- 1. Ben-Porat T, Kaplan AS. 1985. Molecular biology of pseudorabies virus, p 105–173 In Roizman B. (ed), The herpesviruses. Plenum Press, New York, NY [Google Scholar]
- 2. Bodart JFL. 2010. Extracellular-regulated kinase-mitogen-activated protein kinase cascade: unsolved issues. J. Cell. Biochem. 109:850–857 [DOI] [PubMed] [Google Scholar]
- 3. Brack AR, et al. 2000. Role of the cytoplasmic tail of pseudorabies virus glycoprotein E in virion formation. J. Virol. 74:4004–4016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brideau AD, Card JP, Enquist LW. 2000. Role of pseudorabies virus Us9, a type II membrane protein, in infection of tissue culture cells and the rat nervous system. J. Virol. 74:834–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brignati MJ, Loomis JS, Wills JW, Courtney RJ. 2003. Membrane association of VP22, a herpes simplex virus type 1 tegument protein. J. Virol. 77:4888–4898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cantello JL, Anderson AS, Francesconi A, Morgan RW. 1991. Isolation of a Marek's disease virus (MDV) recombinant containing the lacZ gene of Escherichia coli stably inserted within the MDV US2 gene. J. Virol. 65:1584–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chambard J, Lefloch R, Pouysségur J, Lenormand P. 2007. ERK implication in cell cycle regulation. Biochim. Biophys. Acta 1773:1299–1310 [DOI] [PubMed] [Google Scholar]
- 8. Chauvier D, Ankri S, Charriaut-Marlangue C, Casimir R, Jacotot E. 2007. Broad-spectrum caspase inhibitors: from myth to reality? Cell Death Differ. 14:387–391 [DOI] [PubMed] [Google Scholar]
- 9. Clase AC, et al. 2003. The pseudorabies virus Us2 protein, a virion tegument component, is prenylated in infected cells. J. Virol. 77:12285–12298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cohen S, Marr AK, Garcin P, Panté N. 2011. Nuclear envelope disruption involving host caspases plays a role in the parvovirus replication cycle. J. Virol. 85:4863–4874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. del Rio T, Werner HC, Enquist LW. 2002. The pseudorabies virus VP22 homologue (UL49) is dispensable for virus growth in vitro and has no effect on virulence and neuronal spread in rodents. J. Virol. 76:774–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Noronha CM, et al. 2001. Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr. Science 294:1105–1108 [DOI] [PubMed] [Google Scholar]
- 13. Diwan P, Lacasse JJ, Schang LM. 2004. Roscovitine inhibits activation of promoters in herpes simplex virus type 1 genomes independently of promoter-specific factors. J. Virol. 78:9352–9365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Favata MF, et al. 1998. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 273:18623–18632 [DOI] [PubMed] [Google Scholar]
- 15. Fuchs W, Klupp BG, Granzow H, Osterrieder N, Mettenleiter TC. 2002. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 76:364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fuchs W, et al. 2002. Physical interaction between envelope glycoproteins E and M of pseudorabies virus and the major tegument protein UL49. J. Virol. 76:8208–8217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fuchs W, Granzow H, Mettenleiter TC. 2003. A pseudorabies virus recombinant simultaneously lacking the major tegument proteins encoded by the UL46, UL47, UL48, and UL49 genes is viable in cultured cells. J. Virol. 77:12891–12900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hertel L, Mocarski ES. 2004. Global analysis of host cell gene expression late during cytomegalovirus infection reveals extensive dysregulation of cell cycle gene expression and induction of pseudomitosis independent of US28 function. J. Virol. 78:11988–12011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hertel L, Chou S, Mocarski ES. 2007. Viral and cell cycle-regulated kinases in cytomegalovirus-induced pseudomitosis and replication. PLoS Pathog. 3:e6 doi:10.1371/journal.ppat.0030006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hofemeister H, O'Hare P. 2008. Nuclear pore composition and gating in herpes simplex virus-infected cells. J. Virol. 82:8392–8399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394 [DOI] [PubMed] [Google Scholar]
- 22. Kang MH, Banfield BW. 2010. Pseudorabies virus tegument protein Us2 recruits the mitogen-activated protein kinase extracellular-regulated kinase (ERK) to membranes through interaction with the ERK common docking domain. J. Virol. 84:8398–8408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaplan AS, Vatter AE. 1959. A comparison of herpes simplex and pseudorabies viruses. Virology 7:394–407 [DOI] [PubMed] [Google Scholar]
- 24. Kato K, et al. 2000. Synthesis, subcellular localization and VP16 interaction of the herpes simplex virus type 2 UL46 gene product. Arch. Virol. 145:2149–2162 [DOI] [PubMed] [Google Scholar]
- 25. Kimman TG, et al. 1992. Contribution of single genes within the unique short of Aujeszky's disease virus (suid herpesvirus type 1) to virulence, pathogenesis and immunogenicity. J. Gen. Virol. 73:243–251 [DOI] [PubMed] [Google Scholar]
- 26. Klopfleisch R, et al. 2006. Influence of pseudorabies virus proteins on neuroinvasion and neurovirulence in mice. J. Virol. 80:5571–5576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klupp BG, Mettenleiter TC. 1999. Glycoprotein gL-independent infectivity of pseudorabies virus is mediated by a gD-gH fusion protein. J. Virol. 73:3014–3022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Klupp BG, Lomniczi B, Visser N, Fuchs W, Mettenleiter TC. 1995. Mutations affecting the UL21 gene contribute to avirulence of pseudorabies virus vaccine strain Bartha. Virology 212:466–473 [DOI] [PubMed] [Google Scholar]
- 29. Klupp BG, Hengartner CJ, Mettenleiter TC, Enquist LW. 2004. Complete, annotated sequence of the pseudorabies virus genome. J. Virol. 78:424–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klupp BG, Granzow H, Mettenleiter TC. 2000. Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J. Virol. 74:10063–10073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Klupp BG, Granzow H, Mettenleiter TC. 2011. Nuclear envelope breakdown can substitute for primary envelopment-mediated nuclear egress of herpesviruses. J. Virol. 85:8285–8292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klupp BG, et al. 2007. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc. Natl. Acad. Sci. U. S. A. 104:7241–7246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Klupp BG, Böttcher S, Granzow H, Kopp M, Mettenleiter TC. 2005. Complex formation between the UL16 and UL21 tegument proteins of pseudorabies virus. J. Virol. 79:1510–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kopp M, Klupp BG, Granzow H, Fuchs W, Mettenleiter TC. 2002. Identification and characterization of the pseudorabies virus tegument proteins UL46 and UL47: role for UL47 in virion morphogenesis in the cytoplasm. J. Virol. 76:8820–8833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leelawong M, Guo D, Smith GA. 2011. A physical link between the pseudorabies virus capsid and the nuclear egress complex. J. Virol. 85:11675–11684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leuzinger H, et al. 2005. Herpes simplex virus 1 envelopment follows two diverse pathways. J. Virol. 79:13047–13059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liang L, Tanaka M, Kawaguchi Y, Baines JD. 2004. Cell lines that support replication of a novel herpes simplex virus 1 UL31 deletion mutant can properly target UL34 protein to the nuclear rim in the absence of UL31. Virology 329:68–76 [DOI] [PubMed] [Google Scholar]
- 37a. Liu X, Li Q, Dowdell K, Fischer ER, Cohen J. 2012. Varicella-zoster virus ORF12 protein triggers phosphorylation of ERK1/2 and inhibits apoptosis. J. Virol. 86:3143–3151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lomniczi B, Blankenship ML, Ben-Porat T. 1984. Deletions in the genomes of pseudorabies virus vaccine strains and existence of four isomers of the genomes. J. Virol. 49:970–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lyman MG, Randall JA, Calton CM, Banfield BW. 2006. Localization of ERK/MAP kinase is regulated by the alphaherpesvirus tegument protein Us2. J. Virol. 80:7159–7168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McGeoch DJ, Rixon FJ, Davison AJ. 2006. Topics in herpesvirus genomics and evolution. Virus Res. 117:90–104 [DOI] [PubMed] [Google Scholar]
- 41. McKnight JL, Pellett PE, Jenkins FJ, Roizman B. 1987. Characterization and nucleotide sequence of two herpes simplex virus 1 genes whose products modulate alpha-trans-inducing factor-dependent activation of alpha genes. J. Virol. 61:992–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Meignier B, Longnecker R, Mavromara Nazos P, Sears AE, Roizman B. 1988. Virulence of and establishment of latency by genetically engineered deletion mutants of herpes simplex virus 1. Virology 162:251–254 [DOI] [PubMed] [Google Scholar]
- 43. Meijer L, et al. 1997. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 243:527–536 [DOI] [PubMed] [Google Scholar]
- 44. Meindl A, Osterrieder N. 1999. The equine herpesvirus 1 Us2 homolog encodes a nonessential membrane-associated virion component. J. Virol. 73:3430–3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mettenleiter TC. 1989. Glycoprotein gIII deletion mutants of pseudorabies virus are impaired in virus entry. Virology 171:623–625 [DOI] [PubMed] [Google Scholar]
- 46. Mettenleiter TC. 2000. Aujeszky's disease (pseudorabies) virus: the virus and molecular pathogenesis—state of the art, June 1999. Vet. Res. 31:99–115 [DOI] [PubMed] [Google Scholar]
- 47. Mettenleiter TC. 2002. Herpesvirus assembly and egress. J. Virol. 76:1537–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mettenleiter TC, Klupp BG, Granzow H. 2006. Herpesvirus assembly: a tale of two membranes. Curr. Opin. Microbiol. 9:423–429 [DOI] [PubMed] [Google Scholar]
- 49. Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res. 143:222–234 [DOI] [PubMed] [Google Scholar]
- 50. Mettenleiter TC, Zsak L, Kaplan AS, Ben-Porat T, Lomniczi B. 1988. Host cell-specific growth advantage of pseudorabies virus with a deletion in the genome sequences encoding a structural glycoprotein. J. Virol. 62:12–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Murphy MA, Bucks MA, O'Regan KJ, Courtney RJ. 2008. The HSV-1 tegument protein pUL46 associates with cellular membranes and viral capsids. Virology 376:279–289 [DOI] [PubMed] [Google Scholar]
- 52. Parcells MS, Anderson AS, Cantello JL, Morgan RW. 1994. Characterization of Marek's disease virus insertion and deletion mutants that lack US1 (ICP22 homolog), US10, and/or US2 and neighboring short-component open reading frames. J. Virol. 68:8239–8253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pellett P, Roizman B. 2007. The family Herpesviridae: a brief introduction, p 2479–2499 In Knipe DM, et al. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 54. Reynolds AE, et al. 2001. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 75:8803–8817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Roberts EC, Hammond K, Traish AM, Resing KA, Ahn NG. 2006. Identification of G2/M targets for the MAP kinase pathway by functional proteomics. Proteomics 6:4541–4553 [DOI] [PubMed] [Google Scholar]
- 56. Schang LM, Rosenberg A, Schaffer PA. 2000. Roscovitine, a specific inhibitor of cellular cyclin-dependent kinases, inhibits herpes simplex virus DNA synthesis in the presence of viral early proteins. J. Virol. 74:2107–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schang LM, Coccaro E, Lacasse JJ. 2005. Cdk inhibitory nucleoside analogs prevent transcription from viral genomes. Nucleosides Nucleotides Nucleic Acids 24:829–837 [DOI] [PubMed] [Google Scholar]
- 58. Schmidt J, Klupp BG, Karger A, Mettenleiter TC. 1997. Adaptability in herpesviruses: glycoprotein D-independent infectivity of pseudorabies virus. J. Virol. 71:17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Segura-Totten M, Wilson KL. 2001. Virology. HIV—breaking the rules for nuclear entry. Science 294:1016–1017 [DOI] [PubMed] [Google Scholar]
- 60. Szpara ML, et al. 2011. A wide extent of inter-strain diversity in virulent and vaccine strains of alphaherpesviruses. PLoS Pathog. 7:e1002282 doi:10.1371/journal.ppat.1002282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Taylor SL, Kinchington PR, Brooks A, Moffat JF. 2004. Roscovitine, a cyclin-dependent kinase inhibitor, prevents replication of varicella-zoster virus. J. Virol. 78:2853–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vassilev LT, et al. 2006. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. U. S. A. 103:10660–10665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wagner MJ, Smiley JR. 2009. Herpes simplex virus requires VP11/12 to induce phosphorylation of the activation loop tyrosine (Y394) of the Src family kinase Lck in T lymphocytes. J. Virol. 83:12452–12461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wathen MW, Pirtle EC. 1984. Stability of the pseudorabies virus genome after in vivo serial passage. J. Gen. Virol. 65:1401–1404 [DOI] [PubMed] [Google Scholar]
- 65. Wild P, et al. 2005. Impairment of nuclear pores in bovine herpesvirus 1-infected MDBK cells. J. Virol. 79:1071–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yang K, Baines JD. 2011. Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc. Natl. Acad. Sci. U. S. A. 108:14276–14281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang Y, Sirko DA, McKnight JL. 1991. Role of herpes simplex virus type 1 UL46 and UL47 in alpha TIF-mediated transcriptional induction: characterization of three viral deletion mutants. J. Virol. 65:829–841 [DOI] [PMC free article] [PubMed] [Google Scholar]