

Abstract
In the title compound, C11H6N2O, the complete molecule is generated by the application of crystallographic twofold symmetry (the molecule is disordered about this axis). The prop-2-yn-1-yl residue is slightly twisted out of the plane of the benzene ring [C—O—C—C torsion angle = 173.1 (3)°] and is orientated away from the nitrile substituents. In the crystal, supramolecular chains along the a axis, arising from C—H⋯N interactions, are connected into stacks along the c axis by π–π interactions between the benzene rings [centroid–centroid distance = 3.6978 (6) Å = length of the c axis].
Related literature
For the solubilization and some applications of phthanocyanine dyes, see: Jiang et al. (2011 ▶); Sleven et al. (2001 ▶). For the synthesis of substituted phthalonitriles, see: Wöhrle et al. (1993 ▶); Wu et al. (1998 ▶); Li & Lieberman (2001 ▶); Sleven et al. (2001 ▶); Li et al. (2008 ▶); Seven et al. (2009 ▶); Foo et al. (2012 ▶).
Experimental
Crystal data
C11H6N2O
M r = 182.18
Monoclinic,
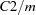
a = 11.4809 (9) Å
b = 22.2091 (16) Å
c = 3.6978 (6) Å
β = 91.304 (10)°
V = 942.62 (18) Å3
Z = 4
Mo Kα radiation
μ = 0.09 mm−1
T = 100 K
0.15 × 0.05 × 0.05 mm
Data collection
Agilent SuperNova Dual diffractometer with an Atlas detector
Absorption correction: multi-scan (CrysAlis PRO; Agilent, 2012 ▶) T min = 0.792, T max = 1.000
3258 measured reflections
1114 independent reflections
840 reflections with I > 2σ(I)
R int = 0.038
Refinement
R[F 2 > 2σ(F 2)] = 0.059
wR(F 2) = 0.163
S = 1.08
1114 reflections
86 parameters
12 restraints
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.41 e Å−3
Δρmin = −0.28 e Å−3
Data collection: CrysAlis PRO (Agilent, 2012 ▶); cell refinement: CrysAlis PRO; data reduction: CrysAlis PRO; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 (Farrugia, 1997 ▶) and DIAMOND (Brandenburg, 2006 ▶); software used to prepare material for publication: publCIF (Westrip, 2010 ▶).
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536812028309/hb6862sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812028309/hb6862Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812028309/hb6862Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C3—H3⋯N1i | 0.95 | 2.62 | 3.509 (3) | 155 |
Symmetry code: (i) 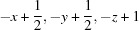 .
.
Acknowledgments
The authors gratefully acknowledge funding from the Brunei Research Council, and thank the Ministry of Higher Education (Malaysia) for funding structural studies through the High-Impact Research scheme (UM.C/HIR/MOHE/SC/12).
supplementary crystallographic information
Comment
Phthalocyanine dyes can be made soluble in water and organic solvents by addition of suitable alkoxy or aryloxy groups (Jiang et al., 2011; Sleven et al., 2001). This is most easily achieved from the correspondingly substituted phthalonitriles which, in turn, are either prepared by Sandmeyer reaction of alkyl or alkoxy functionalized dihalobenzenes (Li & Lieberman, 2001; Sleven et al., 2001) or aryloxy / alkoxy displacement of the corresponding halophthalonitrile (Wöhrle et al., 1993; Li et al., 2008; Foo et al., 2012) or 4-nitrophthalonitrile (Wu et al., 1998; Seven et al., 2009). The latter method is most suitable for preparing 4-alkoxyphthalonitriles and was used for preparing the title compound, 4-(prop-2-ylnyloxy)phthalonitrile (I).
In (I), Fig. 1, the complete molecule is generated by the application of 2-fold symmetry; the molecule is disordered about this axis. The O1 and C1 atoms lie -0.067 (3) and 0.059 (2) Å out of the plane through the benzene ring, respectively. The prop-2-yn-1-yl is twisted out of the plane of the benzene ring as seen in the value of the C4—O1—C5—C6 torsion angle of 173.1 (3)° and is orientated in the opposite direction to the nitrile substituents.
In the crystal packing, supramolecular chains along the a axis feature owing to C—H···N interactions, Table 1, and 10-membered {···HC3N}2 synthons, Fig. 2. Chains are connected into stacks along the c axis by π—π interactions between the benzene rings [inter-centroid distance = 3.6978 (6) Å = length of the c axis]. The layers inter-digitate along the b axis with no specific intermolecular interactions between them.
Experimental
The title compound was prepared by modification of literature procedures (Wu et al., 1998; Seven et al., 2009). Under a nitrogen atmosphere, anhydrous potassium carbonate (1.12 g, 8.1 mmol) was added in two portions at 1 h intervals to a solution of propargyl alcohol (1.5 ml, 26.0 mmol) and 4-nitrophthalonitrile (0.70 g, 4.04 mmol) in dry N,N-dimethylformamide (7 ml). After 96 h, the crude reaction mixture was poured into water (140 ml). The green precipitate was collected by vacuum filtration, washed with water and dried. The crude product was purified by silica gel column chromatography using dichloromethane as eluent to provide 0.4 g (63.9%) of a faintly coloured solid that was recrystallized from CH2Cl2 / hexane as colourless prisms. Melting point = 383 K. IR ν/cm-1: 3287, 3119, 3077, 2231, 2135, 1596, 1494, 1321, 1260. 1H NMR 400 MHz (CDCl3) δ: 7.75 (1H, d), 7.35 (1H, s), 7.28 (1H, d), 4.80 (2H, s), 2.62 (1H, s).
Refinement
With the exception of the acetylenic H-atom which was refined freely, carbon-bound H-atoms were placed in calculated positions [C—H = 0.95–0.99 Å, Uiso(H) = 1.2Ueq(C)] and were included in the refinement in the riding model approximation. The molecule is disordered over a 2-fold rotation axis in an exact 1:1 ratio. The anisotropic displacement parameters of the O1 and C4 atoms were tightly restrained to be nearly isotropic.
Figures
Fig. 1.

The molecular structure of (I) showing displacement ellipsoids at the 50% probability level. The molecule is disordered about the 2-fold axis - only one orientation is shown.
Fig. 2.

A view of the supramolecular chain along the a axis in (I). The C—H···N and interactions are shown as blue dashed lines.
Fig. 3.

A view of the supramolecular layer in the ac plane in (I). The C—H···O and π—π interactions are shown as blue and purple dashed lines, respectively.
Crystal data
| C11H6N2O | F(000) = 376 |
| Mr = 182.18 | Dx = 1.284 Mg m−3 |
| Monoclinic, C2/m | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -C 2y | Cell parameters from 938 reflections |
| a = 11.4809 (9) Å | θ = 3.3–27.5° |
| b = 22.2091 (16) Å | µ = 0.09 mm−1 |
| c = 3.6978 (6) Å | T = 100 K |
| β = 91.304 (10)° | Prism, colourless |
| V = 942.62 (18) Å3 | 0.15 × 0.05 × 0.05 mm |
| Z = 4 |
Data collection
| Agilent SuperNova Dual diffractometer with an Atlas detector | 1114 independent reflections |
| Radiation source: fine-focus sealed tube | 840 reflections with I > 2σ(I) |
| Mirror monochromator | Rint = 0.038 |
| Detector resolution: 10.4041 pixels mm-1 | θmax = 27.6°, θmin = 3.3° |
| ω scan | h = −14→14 |
| Absorption correction: multi-scan (CrysAlis PRO; Agilent, 2012) | k = −26→28 |
| Tmin = 0.792, Tmax = 1.000 | l = −4→4 |
| 3258 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.059 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.163 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.08 | w = 1/[σ2(Fo2) + (0.0718P)2 + 0.8054P] where P = (Fo2 + 2Fc2)/3 |
| 1114 reflections | (Δ/σ)max < 0.001 |
| 86 parameters | Δρmax = 0.41 e Å−3 |
| 12 restraints | Δρmin = −0.28 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | Occ. (<1) | |
| O1 | 0.1308 (2) | 0.45076 (11) | 0.1768 (8) | 0.0273 (7) | 0.50 |
| N1 | 0.15188 (16) | 0.19174 (9) | 0.2675 (6) | 0.0376 (6) | |
| C1 | 0.10982 (16) | 0.23658 (9) | 0.1829 (6) | 0.0260 (5) | |
| C2 | 0.05529 (16) | 0.29272 (8) | 0.0857 (5) | 0.0219 (5) | |
| C3 | 0.1103 (2) | 0.34676 (10) | 0.1663 (6) | 0.0349 (6) | |
| H3 | 0.1853 | 0.3469 | 0.2801 | 0.042* | |
| C4 | 0.0555 (3) | 0.40028 (10) | 0.0803 (7) | 0.0446 (7) | |
| H4 | 0.0936 | 0.4374 | 0.1309 | 0.054* | 0.50 |
| C5 | 0.2499 (3) | 0.44609 (17) | 0.3108 (13) | 0.0274 (10) | 0.50 |
| H5A | 0.2962 | 0.4211 | 0.1458 | 0.033* | 0.50 |
| H5B | 0.2522 | 0.4272 | 0.5535 | 0.033* | 0.50 |
| C6 | 0.2980 (3) | 0.5082 (7) | 0.3303 (12) | 0.030 (3) | 0.50 |
| C7 | 0.3440 (4) | 0.5544 (2) | 0.3459 (17) | 0.0463 (14) | 0.50 |
| H7 | 0.386 (5) | 0.595 (3) | 0.363 (16) | 0.054 (16)* | 0.50 |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0258 (13) | 0.0187 (12) | 0.0370 (15) | 0.0001 (10) | −0.0069 (11) | −0.0008 (11) |
| N1 | 0.0332 (10) | 0.0410 (11) | 0.0383 (12) | 0.0126 (9) | −0.0029 (8) | 0.0074 (9) |
| C1 | 0.0191 (9) | 0.0345 (12) | 0.0242 (11) | −0.0003 (8) | −0.0019 (8) | 0.0004 (8) |
| C2 | 0.0206 (10) | 0.0234 (10) | 0.0217 (10) | −0.0009 (7) | −0.0001 (8) | −0.0007 (7) |
| C3 | 0.0401 (12) | 0.0399 (13) | 0.0251 (12) | −0.0182 (10) | 0.0071 (9) | −0.0077 (9) |
| C4 | 0.0728 (15) | 0.0262 (10) | 0.0355 (13) | −0.0161 (10) | 0.0172 (11) | −0.0063 (9) |
| C5 | 0.0220 (19) | 0.0167 (19) | 0.043 (3) | 0.0005 (17) | −0.0104 (17) | −0.0001 (16) |
| C6 | 0.0262 (16) | 0.013 (9) | 0.051 (2) | −0.004 (2) | −0.0130 (15) | −0.001 (2) |
| C7 | 0.033 (3) | 0.027 (2) | 0.078 (4) | 0.001 (2) | −0.019 (2) | −0.001 (2) |
Geometric parameters (Å, º)
| O1—C5 | 1.447 (5) | C4—C4i | 1.393 (6) |
| O1—C4 | 1.455 (3) | C4—H4 | 0.9500 |
| N1—C1 | 1.147 (3) | C5—C6 | 1.487 (14) |
| C1—C2 | 1.437 (3) | C5—H5A | 0.9900 |
| C2—C3 | 1.385 (3) | C5—H5B | 0.9900 |
| C2—C2i | 1.406 (4) | C6—C7 | 1.155 (16) |
| C3—C4 | 1.379 (3) | C7—H7 | 1.02 (6) |
| C3—H3 | 0.9500 | ||
| C5—O1—C4 | 125.5 (3) | C3—C4—H4 | 119.8 |
| N1—C1—C2 | 178.4 (2) | C4i—C4—H4 | 119.8 |
| C3—C2—C2i | 119.96 (13) | O1—C5—C6 | 107.3 (3) |
| C3—C2—C1 | 120.26 (18) | O1—C5—H5A | 110.3 |
| C2i—C2—C1 | 119.77 (10) | C6—C5—H5A | 110.3 |
| C4—C3—C2 | 119.6 (2) | O1—C5—H5B | 110.3 |
| C4—C3—H3 | 120.2 | C6—C5—H5B | 110.3 |
| C2—C3—H3 | 120.2 | H5A—C5—H5B | 108.5 |
| C3—C4—C4i | 120.43 (14) | C7—C6—C5 | 174.6 (7) |
| C3—C4—O1 | 110.0 (2) | C6—C7—H7 | 178 (3) |
| C4i—C4—O1 | 129.56 (15) | ||
| C2i—C2—C3—C4 | 0.6 (4) | C5—O1—C4—C3 | 5.5 (5) |
| C1—C2—C3—C4 | −178.2 (2) | C5—O1—C4—C4i | −173.5 (4) |
| C2—C3—C4—C4i | 1.2 (4) | C4—O1—C5—C6 | 173.1 (3) |
| C2—C3—C4—O1 | −177.9 (2) |
Symmetry code: (i) −x, y, −z.
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C3—H3···N1ii | 0.95 | 2.62 | 3.509 (3) | 155 |
Symmetry code: (ii) −x+1/2, −y+1/2, −z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HB6862).
References
- Agilent (2012). CrysAlis PRO Agilent Technologies, Yarnton, Oxfordshire, England.
- Brandenburg, K. (2006). DIAMOND Crystal Impact GbR, Bonn, Germany.
- Farrugia, L. J. (1997). J. Appl. Cryst. 30, 565.
- Foo, C. C., Tan, A. L., Wimmer, F. L., Mirza, A. H., Young, D. J., Ng, S. W. & Tiekink, E. R. T. (2012). Acta Cryst. E68, o601. [DOI] [PMC free article] [PubMed]
- Jiang, X.-J., Yeung, S.-L., Lo, P.-C., Fong, W. P. & Ng, D. K. P. (2011). J. Med. Chem. 54, 320–330. [DOI] [PubMed]
- Li, H., Jensen, T. J., Fronczek, F. R. & Vicente, G. H. (2008). J. Med. Chem. 51, 502–511. [DOI] [PubMed]
- Li, Z. & Lieberman, M. (2001). Inorg. Chem. 40, 932–939.
- Seven, O., Dindar, B. & Gultekin, B. (2009). Turk. J. Chem. 33, 123–134.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sleven, J., Görller-Walrand, C. & Binnemans, K. (2001). Mater. Sci. Eng. C, 18, 229–238.
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
- Wöhrle, D., Eskes, M., Shigehara, K. & Yamada, A. (1993). Synthesis, pp. 194–196.
- Wu, Y., Tian, H., Chen, K., Liu, Y. & Zhu, D. (1998). Dyes Pigm. 37, 317–325.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536812028309/hb6862sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812028309/hb6862Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812028309/hb6862Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report