

Abstract
Microbial communities exhibit exquisitely complex structure. Many aspects of this complexity, from the number of species to the total number of interactions, are currently very difficult to examine directly. However, extraordinary efforts are being made to make these systems accessible to scientific investigation. While recent advances in high-throughput sequencing technologies have improved accessibility to the taxonomic and functional diversity of complex communities, monitoring the dynamics of these systems over time and space - using appropriate experimental design - is still expensive. Fortunately, modeling can be used as a lens to focus low-resolution observations of community dynamics to enable mathematical abstractions of functional and taxonomic dynamics across space and time. Here we review the approaches for modeling bacterial diversity at both the very large and the very small scales at which microbial systems interact with their environments. We show that modeling can help to connect biogeochemical processes to specific microbial metabolic pathways.
INTRODUCTION
To understand microbial systems, it is necessary to consider the scales at which they interact with their environment. These scales range spatially from microns to kilometers and temporally from eons to hours. Accounting for 350-550 billion tons of extant biomass (Whitman, et al., 1998), microbes are the principal form of life on Earth, and they have dominated Earth’s evolutionary history. Prokaryotes, the oldest lineage on the tree of life, first appeared about 3.8 billion years ago (Mojzsis, et al., 1996) and have been detected in virtually every environment that has been investigated, from boiling lakes (Barns, et al., 1994, Hugenholtz, et al., 1998), to the atmosphere (Fierer, et al., 2008, Bowers, et al., 2009), to deep in the planet’s crust (Takai, et al., 2001, Fisk, et al., 2003, Edwards, et al., 2006, Teske & Sorensen, 2008). Microbial metabolism contributes to biogeochemical cycles (O’dor, et al., 2009, Hoegh-Guldberg, 2010), and has both direct and indirect impacts on Earth’s climate (Bardgett, et al., 2008, Graham, et al., 2012). Indeed, marine microbial activity has even been implicated as a correlate in earlier mass species extinction events (Baune & Bottcher, 2010). The concept that living processes drive changes the physical environment at the global scale is not new. The “Gaia Hypothesis”, which postulates that living processes help maintain atmospheric homeostasis, was published nearly forty years ago (Lovelock, et al., 1974) and there is mounting evidence that this is indeed the case (Charlson, et al., 1987, Cicerone & Oremland, 1988, Gorham, 1991). Use of next generation high-throughput data, however, has only recently made possible direct investigations of the specific molecular mechanisms and microbial consortia responsible for the planet’s dynamic equilibrium.
While their effects may be global, microbial systems interact with their environments at microscopic scales. A single gram of soil might contain around 109 microbial units (Torsvik & Ovreas, 2002) and an average milliliter of seawater will contain approximately a million bacterial cells. The wide taxonomic diversity of these populations (Pedros-Alio, 2006) is fostered, at least in part, by myriad micro-environments accessible to the bacteria. In soil and marine systems, the majority of microbial diversity is represented in the minority of biomass (Pedros-Alio, 2006, Sogin, et al., 2006, Ashby, et al., 2007, Elshahed, et al., 2008). Generally, in highly diverse microbial communities, a few abundant taxa predominate, with a long tail of low abundance taxa (Sogin, et al., 2006). These low abundance taxa in particular are crucial to our understanding of microbial ecosystems, as they represent the vast functional diversity that can rapidly blossom to high abundance under the appropriate environmental conditions (e.g. (Caporaso, et al., 2011, Gilbert, et al., 2011)).
Microbial systems can be described using environmental DNA sequence information and contextual metadata, which reveal dynamic taxonomic and functional diversity across gradients of natural or experimental variation (Tyson, et al., 2004, Venter, et al., 2004, DeLong, et al., 2006, Gilbert, et al., 2010, Delmont, et al., 2011). Taxonomic diversity is a measure of the community species composition, which is maintained or altered via interactions and adaptations between each species and its environment. Functional diversity is a measure of the frequency and the type of predicted enzyme functions encoded in a community’s metagenome, and represents the potential to express a phenotype that interacts with a particular environmental state. Increasing depth from continuing advances in sequencing technologies has enabled whole genomes to be re-assembled from metagenomic data, which permits appropriate descriptions of the taxonomic and functional potential of individual species imbedded within each community (Woyke, et al., 2010, Hess, et al., 2011, Iverson, et al., 2012). While the goal of this mini review is not to highlight the impact of these studies on defining the relationships between microbial communities and their environments (which is covered in other reviews, e.g. (Torsvik & Ovreas, 2002, Fierer & Jackson, 2006, Falkowski, et al., 2008, Wooley, et al., 2010, Gilbert & Dupont, 2011)), it is important to state that each community, whether embedded in a desiccated soil particle, or in a biofilm attached to a hermit crab in a coral sea, presents a potentially unique set of interactions with the ecosystem. Here, we summarize current approaches used to generate predictive models that incorporate taxonomic and functional diversity at the metabolic, microbial interaction, community composition, and ecosystem scales of microbial ecology.
MICROBIAL COMMUNITY SAMPLING EFFORTS
Metagenomics is the capture and analysis of genomic information from a volume of environmental sample (Figure 1) (Handelsman, et al., 1998, Gilbert & Dupont, 2011). Recent advances in direct sequencing of DNA from an environmental sample have generated prodigious amounts of sequence information, resulting in a data bonanza (Field, et al., 2011). Equally important as the collection of metagenomic data, however, is the concurrent collection of associated metadata (i.e. the chemical and physical characteristics of the environment undergoing metagenomic analysis). To generate hypotheses regarding the interactions within a community that result in observed patterns in diversity and richness, the relevant physical, chemical and biological factors must be measured. Probes can quantify various parameters, such as temperature, pH, ammonia, silicate, and oxygen concentration at approximately the scale experienced by individual microbes (Debeer, et al., 1992, Zhang, et al., 1995, Rani, et al., 2007, Stewart & Franklin, 2008). Metabolomic techniques such as near- and mid-infrared diffuse reflectance spectroscopy (Forouzangohar, et al., 2009), nuclear magnetic resonance or gas chromatography-mass spectrometry (Viant, et al., 2003, Viant, 2008, Wooley, et al., 2010) can provide measurements for very small volumes of environmental samples, but they only provide for a fraction of the thousands of metabolites potentially present (Viant, 2008). At the opposite end of the physical scale, remote sensing, recognized as the only tool for gathering data over extensive spatial and temporal scales (Graetz, 1990), collects data measuring electromagnetic radiation reflected or emitted from earth’s surface, without direct physical contact with objects or phenomena under investigation. Remotely sensed imagery can provide a synoptic view of landscapes, enabling data acquisition over large expanses and/or physically inaccessible areas. Recent technological advances permit acquisition of imagery with spatial resolution as fine as 60 cm2 and temporal resolution as high as once a day when using a satellite platform.
Figure 1. Metagenomic analysis.
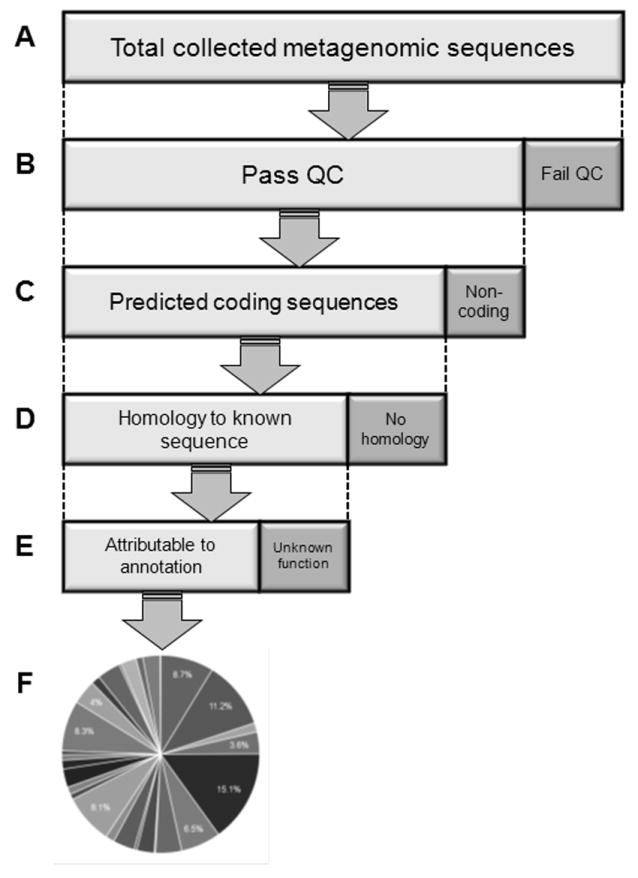
DNA is extracted directly from a volume of environmental sample. The specific outline featured here is specific to the frequently used MG-RAST metagenomic analysis pipeline (Meyer, et al., 2008), but can be generalized, with variations, to a broad range of metagenomic analysis approaches. (A) The width of this bar represents 100% of the entire DNA sequence data collected from an environmental sample. (B) DNA sequences are subjected to quality control, such as removing sequences that contain ambiguous base calls or are technical duplicates. (C) For sequences that pass quality control, the most likely protein-coding frame is identified. (D) For the predicted protein sequences from sequences that have a likely coding frame, the best homology to proteins in a large database of protein sequences is identified. Given the potentially large number of predicted protein sequences from the metagenomic dataset and the size of the database of known proteins, this step can require considerable computational time. (E) Not every predicted protein that has homology to a known protein will be to a protein of known or predicted function. At the end of metagenomic analysis, only a fraction of initial sequence reads may have generated hits to proteins of known functions or taxonomic identity. (F) Collected annotations, and their relative distributions across metagenomic datasets, are the principle input data for downstream modeling of microbial community structure and function.
Ongoing environmental monitoring projects that focus on using high-throughput sequencing techniques and continuous collection of contextual metadata to explore microbial life (e.g. The Global Ocean Survey (http://www.jcvi.org/cms/research/projects/gos), Tara Oceans (http://oceans.taraexpeditions.org), the Hawaiian Ocean Time Series (http://hahana.soest.hawaii.edu/hot), the Bermudan Ocean Time Series (http://bats.bios.edu), Western Channel Observatory (http://www.westernchannelobservatory.org) and The National Ecological Observatory Network (NEON; http://www.neoninc.org)) are generating huge quantities of data on the dynamics of microbial communities in ecosystems across local, continental and global scales. Recently, studies of coastal marine systems (Gilbert, et al., 2010, Caporaso, et al., 2011, Gilbert, et al., 2011), the human microbiome (Caporaso, et al., 2011), animal rumen (Hess, et al., 2011) and Arctic tundra (Graham, et al., 2011, Mackelprang, et al., 2011) provide examples of the data density (both sequencing-based and contextual metadata) required to characterize microbial community structure in complex ecosystems.
SCALES OF MICROBIAL COMMUNITY MODELS
Modeling approaches to microbial ecosystems can be grouped into four broad categories (Figure 2). While the specific boundaries in time or space that separate one scale of microbial modeling from another are somewhat arbitrary, modeling approaches can be grouped by their distinct approaches to representing microbial processes and their relationships with their environments.
Figure 2. Microbial systems at log scale.
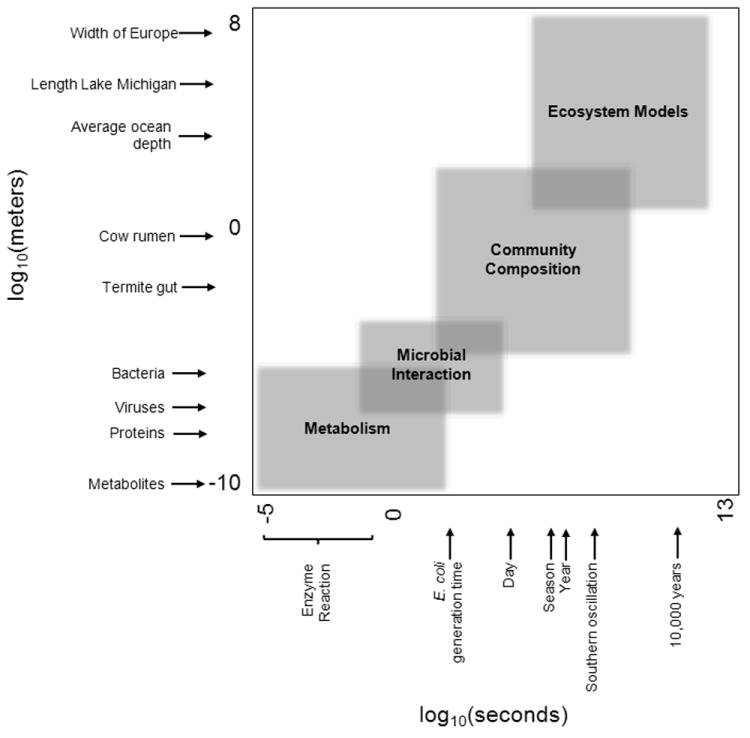
In this figure, time and physical scales of different categories of microbial interactions are arranged on log 10 scales. Placements of reference points of interest on figure are approximate. Not featured on this figure, time since the origin of microbial life on earth at ~17.1 log10(seconds).
Metabolic
Metabolic models investigate how a single microbial cell interacts with its environment. The ultimate single cell model is one that encapsulates the full potential biochemical reactions within the cell that result in its phenotype and interactions with environmental factors and available nutrients. Recently, developments in the prediction of flux-balance models for individual genomes (Henry, et al., 2010, Henry, et al., 2011), have enabled these models to be generated in a high throughput manner for tens of thousands of microbial genomes. This approach is becoming increasingly relevant as draft quality genomes of the most abundant organisms in a microbial community can be assembled from metagenomic data (Mackelprang, et al., 2011, Luo, et al., 2012) (Woyke, et al., 2010, Hess, et al., 2011, Iverson, et al., 2012). In particular, Mackelprang et al. (2011) found that the most abundant organism present in Alaskan permafrost soil was a novel methanogen and that modeling its metabolism from the assembled draft genome provided direct insight into how the thawing permafrost will contribute methane, a powerful greenhouse gas, to the atmosphere.
Microbial Interaction
Microbial interaction models predict how the metabolisms of two or more microbial taxa interact with one another and their environment. Flux balance models, which have been proven to be successful, are now being taken a step further to enable the development of simple interaction models between multiple individual flux-balance models for different genomes (Freilich, et al., 2011). Individual-based models represent space as a discrete lattice and each lattice element can contain microbial cells and measures of environmental parameter levels. Each microbial cell in the model is an individual, and can have various capacities to interact with environmental parameters (O’Donnell, et al., 2007). Applying individual based methods to entire microbial communities requires highly detailed, very accurate information about microbial metabolism and the nature of the microenvironment (Ferrer, et al., 2008, Freilich, et al., 2011). Fortunately, there are computational techniques for describing multiphase transport in complex, porous media like soil, such as the Lattice-Boltzmann method (i.e. (Zhang, et al., 2005)), which is a class of computational fluid dynamics techniques. Using these methods it may be possible to model the dynamic movement of soil, and then overlay this with biological information regarding the dynamics of the microbiome in that system; however, this has not yet been validated. Because this form of modeling can be computationally intensive, some methodological innovations, such as the use of super-individuals, have been advocated (Scheffer, et al., 1995). The first study using individual-based modeling to predict the behavior of a microbial community simulated the accumulation of nitrate by nitrifying bacteria in different soil types (Ginovart, et al., 2005). Recently, Gras and colleagues (2010) modeled the metabolism and dynamics of organic carbon and nitrogen in three different types of Mediterranean soil. The model incorporated specific parameters for growth and decay of microbial biomass, temporal evolution of mineralized intermediate carbon and nitrogen, mineral nitrogen in ammonium and nitrate, carbon dioxide, and O2. A good empirical fit of the model was observed using data from laboratory incubation experiments.
An alternate approach to modeling micro-scale dynamics over relatively short-time scales rather than across very small physical spaces is the Lotka-Volterra type predator-prey models, or so called “kill-the-winner” models (Rodriguez-Brito, et al., 2010). In the case of microbial life, the predators are viruses. In “kill-the-winner”, as abundances of particular taxa increase, so does their vulnerability to predation by viruses, leading to populations that are structurally stable over coarse-grained intervals but marked by rapid fluctuations in structure at the fine-grained level.
Two examples of ecologically relevant microbial interactions for modeling are complex microbial structures like biofilms (Chen, et al., 2004, Diaz, 2012) or microbial mats (Heidelberg, et al., 2009, Liu, et al., 2011). In both these types of microbial communities, certain properties of microbial interaction would not be predictable from the metabolic capacity of any of its constituent members.
Community Composition
Community Models are concerned with how local environmental conditions shape the compositions of microbial populations. There are currently a number of niche-based techniques that link environmental parameters with microbial community structure (Bowers, et al., 2011, Fierer, et al., 2011, Fierer & Lennon, 2011, Jutla, et al., 2011, Steele, et al., 2011, Barberan, et al., 2012). An extension of this idea is the development of predictive bioclimatic models (i.e. envelope models, ecological niche models or species distribution models) that enable the estimation of the geographic and temporal ranges of organisms as a function of environment (Risto K. Heikkinen 2006; Jeschke and Strayer 2008). Logistic regression uses generalized linear models (Bolker, et al., 2009) to fit the presence or absence of a species against climatic variables as a linear function. Generalized additive models (GAM) model species as an additive combination of functions of independent variables (Hastie & Tibshirani, 1990). Climate envelope models like BIOCLIM (Busby, 1991), DOMAIN (Carpenter, et al., 1993), and HABITAT (Walker & Cocks, 1991)) fit the minimal envelope that defines an organism’s possible habitat in multidimensional space, but use presence-only data rather than presence/absence. Maximum entropy models (MaxEnt (Phillips et al., 2006)) minimize the relative information entropy (dispersion) between two probability densities defined in covariate space (Elith et al., 2011). The classification and regression tree technique models communities as a binary decision tree in which the decision rules at each node use one or more independent environmental parameter variables (Che, et al., 2011). Neural network approaches, such as the genetic algorithm for rule-set prediction (Stockwell & Noble, 1992, Stockwell & Peters, 1999), have powerful predictive capabilities, but only model organism distributions as present or absent as a function of environmental parameters. These niche-based bioclimatic models interpolate species distributions without mechanistic information, based on observed species occurrences in the environment. This indirectly takes into account competition between species, barriers to distribution, and other historical factors a postori, which cannot be physiologically predicted. Niche models yield the realized (actual) niche, rather than the fundamental (theoretical) niche predicted by process-based models (Guisan and Zimmermann, 2000; Morin and Thuiller, 2009). These models can underestimate complex biotic interactions and do not necessarily allow for varying distributions of the same organisms in different environmental conditions. Therefore, a myriad of tools exist to model the dynamics of microbial community structure. However, few if any have attempted to predict the relative abundance of the many thousands of potential species observed in complex systems (Caporaso, et al., 2011, Caporaso, et al., 2011).
One particular example of relevant modeling at this scale is for animal associated microbial communities. Variation in the human gut microbiome has been linked to human health (Burcelin, et al., 2011, Marchesi, 2011, Wu, et al., 2011). In addition, microbial communities that live within other organisms, such the termite gut or the cow rumen, have potential applications in deriving biofuels from lignocellulosic plant materials (Hongoh, 2010, Hess, et al., 2011).
Ecosystem
Ecosystem models of microbial communities span large environments, up to the entire biosphere. The one ocean model (O’dor, et al., 2009) represents the global marine ecosystem at the largest possible scale: as a single circular ocean with a 10,000 km radius and a uniform 4 km depth. This model system is used to explore the potential for biodiversity dispersal. In the case of bacteria, a single “species” could transverse the whole ocean in only 10,000 years. However, there are complications to such a simple theoretical model, such as barriers to dispersal. While continents may be the most obvious, currents are just as potent. The MIT General Circulation Model (Marshall, et al., 1997) is a mathematical description of the motions that control oceanic and atmospheric currents. Combining these physical models with microbial diversity models, in which a number of microbial phenotypes are initialized and their interaction with the modeled environment determines their relative fitness, should enable accurate prediction of both dispersal, limits of dispersal and species fitness (Bruggeman & Kooijman, 2007, Follows, et al., 2007, Merico, et al., 2009). For example, using diversity based models with the high-resolution general circulation model (Marshall, et al., 1997) enables the generation of several dozen parameterized phytoplankton models (Follows, et al., 2007, Dutkiewicz, et al., 2009). In these ocean-wide model systems, the fitness of modeled organisms responding through a combination of light, nutrient, and temperature adaptations corresponds well to the fitness of laboratory cultures under similar conditions.
PREDICTING TRENDS IN MICROBIAL COMMUNITY MODELING
This is a golden age for microbial ecology. We are generating datasets that could lay the foundation of the next phase in microbial ecosystem modeling. As greater spatial and temporal resolution is achieved, the finer details of community structure will be elucidated, enabling biological, chemical and physical relationships to be described with mathematical formalisms. The next generation of micro-scale, bottom-up models will focus on imposing more accurate metabolic models to define flux rates of enzymatic reactions for biological units that interact in massively parallel computational arrays (e.g. http://systems.cs.uchicago.edu/projects/bhive.html). These systems, built of cellular and biochemical components, rely on a mechanistic understanding, which must be a focus for future microbial research. Without an improved knowledge of the biochemical nature of metabolism, metabolic interactions cannot be accurately described. A challenge for such systems will be to integrate physical and chemical disturbance into the model environment. As has been shown with macro-scale models of the global ocean, the physical currents, once modeled, enable significantly improved accuracy of prediction for community structure and biomass of individual taxonomic units.
It may be that microbial ecosystems, similar to life at macro-scales, are fundamentally fractal in nature (Gisiger, 2001, Brown, et al., 2002), displaying statistical self-similarity across multiple scales. If everything were in fact everywhere, then every sampled microbial population would contain a representation of the whole. Patterns of changing abundance in a milliliter of seawater might then mimic the patters observed in entire oceans. Fractal and multifractal systems have been applied to ecological patters in the past (Borda-de-Agua, et al., 2002, Brown, et al., 2002) and these tools may be valuable in modeling microbial systems as well. As understanding of microbial ecosystems continues to grow, the connections between the micro and the macro scales will become more apparent.
The ability to observe the taxonomic and functional diversity of microbial systems is still a very new technology and microbial ecosystems are ancient. For a largely immortal organism that takes only 10,000 years to move across the globe and can be safely embedded in solid rock to await the geochemical conditions suitable to resume growth, a few years of observations might be insufficient to grasp the true dynamics of these ecosystems. Perhaps for some microbial taxa, the passing of the seasons are less important than the cycles of El Niño/La Niña, or even the coming and going of ice ages. Microbial ecosystem models are the only lens through which the full scope of microbial ecology can be observed, and provide opportunities for researchers to make predictions of microbial taxonomic and functional structure that extend far beyond the current range of possible observations.
Acknowledgments
Funding for SMG was provided by NIH Training Grant 5T32EB009412.
References
- Ashby MN, Rine J, Mongodin EF, Nelson KE, Dimster-Denk D. Serial analysis of rRNA genes and the unexpected dominance of rare members of microbial communities. Appl Envir Microbiol. 2007;73:4532–4542. doi: 10.1128/AEM.02956-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberan A, Bates ST, Casamayor EO, Fierer N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012;6:343–351. doi: 10.1038/ismej.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardgett RD, Freeman C, Ostle NJ. Microbial contributions to climate change through carbon cycle feedbacks. ISME J. 2008;2:805–814. doi: 10.1038/ismej.2008.58. [DOI] [PubMed] [Google Scholar]
- Barns SM, Fundyga RE, Jeffries MW, Pace NR. Remarkable archaeal diversity detected in a Yellowstone National Park hot spring environment. Proc Natl Acad Sci USA. 1994;91:1609–1613. doi: 10.1073/pnas.91.5.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baune C, Bottcher ME. Experimental investigation of sulphur isotope partitioning during outgassing of hydrogen sulphide from diluted aqueous solutions and seawater. Isotopes in Env and Health Stud. 2010;46:444–453. doi: 10.1080/10256016.2010.536230. [DOI] [PubMed] [Google Scholar]
- Bolker B, Brooks M, Clark C, Geange S, Poulsen J, Stevens M, White J. Generalized linear mixed models: a practical guide for ecology and evolution. Trends Ecol Evol. 2009;3:171–193. doi: 10.1016/j.tree.2008.10.008. [DOI] [PubMed] [Google Scholar]
- Borda-de-Agua L, Hubbell SP, McAllister M. Species-area curves, diversity indices, and species abundance distributions: A multifractal analysis. Amer Nat. 2002;159:138–155. doi: 10.1086/324787. [DOI] [PubMed] [Google Scholar]
- Bowers RM, Sullivan AP, Costello EK, Collett JL, Jr, Knight R, Fierer N. Sources of bacteria in outdoor air across cities in the midwestern United States. Appl Environ Microbiol. 2011;77:6350–6356. doi: 10.1128/AEM.05498-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers RM, Lauber CL, Wiedinmyer C, et al. Characterization of airborne microbial communities at a high-elevation site and their potential to act as atmospheric ice nuclei. Appl Environ Microbiol. 2009;75:5121–5130. doi: 10.1128/AEM.00447-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JH, Gupta VK, Li BL, Milne BT, Restrepo C, West GB. The fractal nature of nature: power laws, ecological complexity and biodiversity. Philos Trans of the Royal Soc of London Series B, Biol Sci. 2002;357:619–626. doi: 10.1098/rstb.2001.0993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggeman J, Kooijman SALM. A biodiversity-inspired approach to aquatic ecosystem modeling. Limn and Ocean. 2007;52:1533–1544. [Google Scholar]
- Burcelin R, Serino M, Chabo C, Blasco-Baque V, Amar J. Gut microbiota and diabetes: from pathogenesis to therapeutic perspective. ACTA Diabetol. 2011;48:257–273. doi: 10.1007/s00592-011-0333-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busby JR. In: BIOCLIM - A Bioclimatic Analysis and Prediction System. Margules CR, A MP, editors. 1991. pp. 64–68. [Google Scholar]
- Caporaso JG, Paszkiewicz K, Field D, Knight R, Gilbert JA. The Western English Channel contains a persistent microbial seed bank. ISME J. 2011 doi: 10.1038/ismej.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Costello EK, et al. Moving pictures of the human microbiome. Genome Biol. 2011;12:R50. doi: 10.1186/gb-2011-12-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter G, Gillison AN, Winter J. DOMAIN: a flexible modelling procedure for mapping potential distributions of plants and animals. Biodiver and Cons. 1993:667–680. [Google Scholar]
- Charlson RJ, Lovelock JE, Andreae MO, Warren SG. Oceanic Phytoplankton, Atmospheric Sulfur, Cloud Albedo and Climate. Nature. 1987;326:655–661. [Google Scholar]
- Che DS, Liu Q, Rasheed K, Tao XP. Decision Tree and Ensemble Learning Algorithms with Their Applications in Bioinformatics. Soft Tools and Algo for Biol Syst. 2011;696:191–199. doi: 10.1007/978-1-4419-7046-6_19. [DOI] [PubMed] [Google Scholar]
- Chen CL, Liu WT, Chong ML, Wong MT, Ong SL, Seah H, Ng WJ. Community structure of microbial biofilms associated with membrane-based water purification processes as revealed using a polyphasic approach. Appl Microbiol and Biotech. 2004;63:466–473. doi: 10.1007/s00253-003-1286-7. [DOI] [PubMed] [Google Scholar]
- Cicerone RJ, Oremland RS. Biogeochemical aspects of atmospheric methane. Global Biogeochem Cycles. 1988;2:299–327. [Google Scholar]
- Debeer D, Huisman JW, Vandenheuvel JC, Ottengraf SPP. The Effect of Ph Profiles in Methanogenic Aggregates on the Kinetics of Acetate Conversion. Water Research. 1992;26:1329–1336. [Google Scholar]
- Delmont TO, Malandain C, Prestat E, Larose C, Monier JM, Simonet P, Vogel TM. Metagenomic mining for microbiologists. ISME J. 2011;5:1837–1843. doi: 10.1038/ismej.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong EF, Preston CM, Mincer T, et al. Community genomics among stratified microbial assemblages in the ocean’s interior. Science. 2006;311:496–503. doi: 10.1126/science.1120250. [DOI] [PubMed] [Google Scholar]
- Diaz PI. Microbial diversity and interactions in subgingival biofilm communities. Frontiers of oral biology. 2012;15:17–40. doi: 10.1159/000329669. [DOI] [PubMed] [Google Scholar]
- Dutkiewicz S, Follows MJ, Bragg JG. Modeling the coupling of ocean ecology and biogeochemistry. Global Biogeoche Cycles. 2009;23 [Google Scholar]
- Edwards RA, Rodriguez-Brito B, Wegley L, et al. Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics. 2006;7 doi: 10.1186/1471-2164-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshahed MS, Youssef NH, Spain AM, et al. Novelty and uniqueness patterns of rare members of the soil biosphere. Applied and Env Microbiol. 2008;74:5422–5428. doi: 10.1128/AEM.00410-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkowski PG, Fenchel T, Delong EF. The microbial engines that drive Earth’s biogeochemical cycles. Science. 2008;320:1034–1039. doi: 10.1126/science.1153213. [DOI] [PubMed] [Google Scholar]
- Ferrer J, Prats C, Lopez D. Individual-based modelling: an essential tool for microbiology. J of Biological Physics. 2008;34:19–37. doi: 10.1007/s10867-008-9082-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field D, Amaral-Zettler L, Cochrane G, et al. The Genomic Standards Consortium. Plos Biology. 2011;9 doi: 10.1371/journal.pbio.1001088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer N, Jackson RB. The diversity and biogeography of soil bacterial communities. Proc of the Nat Acad of Sci of the USA. 2006;103:626–631. doi: 10.1073/pnas.0507535103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer N, Lennon JT. The generation and maintenance of diversity in microbial communities. Amer J of Botany. 2011;98:439–448. doi: 10.3732/ajb.1000498. [DOI] [PubMed] [Google Scholar]
- Fierer N, Liu Z, Rodriguez-Hernandez M, Knight R, Henn M, Hernandez MT. Short-term temporal variability in airborne bacterial and fungal populations. Appl and Env Microbiol. 2008;74:200–207. doi: 10.1128/AEM.01467-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer N, Lauber CL, Ramirez KS, Zaneveld J, Bradford MA, Knight R. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 2011 doi: 10.1038/ismej.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk MR, Storrie-Lombardi MC, Douglas S, Popa R, McDonald G, Di Meo-Savoie C. Evidence of biological activity in Hawaiian subsurface basalts. Geochemistry Geophysics Geosystems. 2003;4 [Google Scholar]
- Follows MJ, Dutkiewicz S, Grant S, Chisholm SW. Emergent biogeography of microbial communities in a model ocean. Science. 2007;315:1843–1846. doi: 10.1126/science.1138544. [DOI] [PubMed] [Google Scholar]
- Forouzangohar M, Cozzolino D, Kookana RS, Smernik RJ, Forrester ST, Chittleborough DJ. Direct comparison between visible near- and mid-infrared spectroscopy for describing diuron sorption in soils. Env Sci & Tech. 2009;43:4049–4055. doi: 10.1021/es8029945. [DOI] [PubMed] [Google Scholar]
- Freilich S, Zarecki R, Eilam O, et al. Competitive and cooperative metabolic interactions in bacterial communities. Nat Commun. 2011;2:589. doi: 10.1038/ncomms1597. [DOI] [PubMed] [Google Scholar]
- Gilbert JA, Dupont CL. Microbial metagenomics: beyond the genome. Ann Rev Mar Sci. 2011;3:347–371. doi: 10.1146/annurev-marine-120709-142811. [DOI] [PubMed] [Google Scholar]
- Gilbert JA, Field D, Swift P, et al. The taxonomic and functional diversity of microbes at a temperate coastal site: a ‘multi-omic’ study of seasonal and diel temporal variation. PLoS One. 2010;5:e15545. doi: 10.1371/journal.pone.0015545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert JA, Steele JA, Caporaso JG, et al. Defining seasonal marine microbial community dynamics. ISME J. 2011 doi: 10.1038/ismej.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginovart M, Lopez D, Gras A. Individual-based modelling of microbial activity to study mineralization of C and N and nitrification process in soil. Nonlinear Analysis-Real World App. 2005;6:773–795. [Google Scholar]
- Gisiger T. Scale invariance in biology: coincidence or footprint of a universal mechanism? Biological Rev of the Camb Phil Soc. 2001;76:161–209. doi: 10.1017/s1464793101005607. [DOI] [PubMed] [Google Scholar]
- Gorham E. Biogeochemistry - Its Origins and Development. Biogeochem. 1991;13:199–239. [Google Scholar]
- Graetz RD. Remote sensing of terrestrial ecosystem structure: An ecologist’s pragmatic view. In: Mooney RJH, H A, editors. Remote Sensing of Biosphere Functioning. Springer-Verlag; New York: 1990. pp. 5–30. [Google Scholar]
- Graham DE, Wallenstein MD, Vishnivetskaya TA, et al. Microbes in thawing permafrost: the unknown variable in the climate change equation. ISME J. 2011;4:709–12. doi: 10.1038/ismej.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DE, Wallenstein MD, Vishnivetskaya TA, et al. Microbes in thawing permafrost: the unknown variable in the climate change equation. ISME J. 2012;6:709–712. doi: 10.1038/ismej.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gras A, Ginovart M, Portell X, Baveye PC. Individual-Based Modeling of Carbon and Nitrogen Dynamics in Soils: Parameterization and Sensitivity Analysis of Abiotic Components. Soil Sci. 2010;175:363–374. [Google Scholar]
- Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol. 1998;5:R245–249. doi: 10.1016/s1074-5521(98)90108-9. [DOI] [PubMed] [Google Scholar]
- Hastie T, Tibshirani R. Exploring the nature of covariate effects in the proportional hazards model. Biometrics. 1990;46:1005–1016. [PubMed] [Google Scholar]
- Heidelberg JF, Nelson WC, Schoenfeld T, Bhaya D. Germ warfare in a microbial mat community: CRISPRs provide insights into the co-evolution of host and viral genomes. PLoS One. 2009;4:e4169. doi: 10.1371/journal.pone.0004169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol. 2010;28:977–982. doi: 10.1038/nbt.1672. [DOI] [PubMed] [Google Scholar]
- Henry CS, Overbeek R, Xia F, et al. Connecting genotype to phenotype in the era of high-throughput sequencing. Biochim Biophys Acta. 2011;1810:967–77. doi: 10.1016/j.bbagen.2011.03.010. [DOI] [PubMed] [Google Scholar]
- Hess M, Sczyrba A, Egan R, et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331:463–467. doi: 10.1126/science.1200387. [DOI] [PubMed] [Google Scholar]
- Hoegh-Guldberg O. Dangerous shifts in ocean ecosystem function? ISME J. 2010;4:1090–1092. doi: 10.1038/ismej.2010.107. [DOI] [PubMed] [Google Scholar]
- Hongoh Y. Diversity and genomes of uncultured microbial symbionts in the termite gut. Biosci, Biotech, & Biochem. 2010;74:1145–1151. doi: 10.1271/bbb.100094. [DOI] [PubMed] [Google Scholar]
- Hugenholtz P, Pitulle C, Hershberger KL, Pace NR. Novel division level bacterial diversity in a Yellowstone hot spring. J Bacteriol. 1998;180:366–376. doi: 10.1128/jb.180.2.366-376.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson V, Morris RM, Frazar CD, Berthiaume CT, Morales RL, Armbrust EV. Untangling Genomes from Metagenomes: Revealing an Uncultured Class of Marine Euryarchaeota. Science. 2012;335:587–590. doi: 10.1126/science.1212665. [DOI] [PubMed] [Google Scholar]
- Jutla AS, Akanda AS, Griffiths JK, Colwell R, Islam S. Warming Oceans, Phytoplankton, and River Discharge: Implications for Cholera Outbreaks. The Amer J of Tropical Med and Hygine. 2011;85:303–308. doi: 10.4269/ajtmh.2011.11-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Klatt CG, Wood JM, et al. Metatranscriptomic analyses of chlorophototrophs of a hot-spring microbial mat. ISME J. 2011;5:1279–1290. doi: 10.1038/ismej.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovelock JE, Bowerchalke, Salisbury N, England W, Margulis L. Atmospheric homeostasis by and for the biosphere: the Gaia hypothesis. Tellus. 1974;26:1–2. [Google Scholar]
- Luo C, Tsementzi D, Kyrpides NC, Konstantinidis KT. Individual genome assembly from complex community short-read metagenomic datasets. ISME J. 2012;6:898–901. doi: 10.1038/ismej.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackelprang R, Waldrop MP, DeAngelis KM, et al. Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature. 2011;480:368–371. doi: 10.1038/nature10576. [DOI] [PubMed] [Google Scholar]
- Marchesi JR. Human distal gut microbiome. Env Microbiol. 2011;13:3088–3102. doi: 10.1111/j.1462-2920.2011.02574.x. [DOI] [PubMed] [Google Scholar]
- Marshall J, Adcroft A, Hill C, Perelman L, Heisey C. A finite-volume, incompressible Navier Stokes model for studies of the ocean on parallel computers. J of Geophysical Research-Oceans. 1997;102:5753–5766. [Google Scholar]
- Merico A, Bruggeman J, Wirtz K. A trait-based approach for downscaling complexity in plankton ecosystem models. Ecological Modelling. 2009;220:3001–3010. [Google Scholar]
- Meyer F, Paarmann D, D’Souza M, et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojzsis SJ, Arrhenius G, McKeegan KD, Harrison TM, Nutman AP, Friend CR. Evidence for life on Earth before 3,800 million years ago. Nature. 1996;384:55–59. doi: 10.1038/384055a0. [DOI] [PubMed] [Google Scholar]
- O’Donnell AG, Young IM, Rushton SP, Shirley MD, Crawford JW. Visualization, modelling and prediction in soil microbiology. Nature reviews Microbiology. 2007;5:689–699. doi: 10.1038/nrmicro1714. [DOI] [PubMed] [Google Scholar]
- O’dor RK, Fennel K, Vanden Berghe E. A one ocean model of biodiversity. Deep-Sea Research Part Ii-Topical Studies in Oceanography. 2009;56:1816–1823. [Google Scholar]
- Pedros-Alio C. Marine microbial diversity: can it be determined? Trends in Microbiol. 2006;14:257–263. doi: 10.1016/j.tim.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Rani SA, Pitts B, Beyenal H, et al. Spatial patterns of DNA replication, protein synthesis, and oxygen concentration within bacterial biofilms reveal diverse physiological states. J of Bacteriology. 2007;189:4223–4233. doi: 10.1128/JB.00107-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Brito B, Li L, Wegley L, et al. Viral and microbial community dynamics in four aquatic environments. ISME J. 2010;4:739–751. doi: 10.1038/ismej.2010.1. [DOI] [PubMed] [Google Scholar]
- Scheffer M, Baveco JM, Deangelis DL, Rose KA, Vannes EH. Super-Individuals a Simple Solution for Modeling Large Populations on an Individual Basis. Ecol Modelling. 1995;80:161–170. [Google Scholar]
- Sogin ML, Morrison HG, Huber JA, et al. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc of the Nat Acad of Sci of the USA. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele JA, Countway PD, Xia L, et al. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J. 2011;5:1414–25. doi: 10.1038/ismej.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart PS, Franklin MJ. Physiological heterogeneity in biofilms. Nature Rev Microbiol. 2008;6:199–210. doi: 10.1038/nrmicro1838. [DOI] [PubMed] [Google Scholar]
- Stockwell DRB, Noble IR. Induction of sets of rules from animal distribution data: A robust and informative method of analysis. Math and Comp in Sim. 1992:385–390. [Google Scholar]
- Stockwell DRB, Peters DP. The GARP modelling system: Problems and solutions to automated spatial prediction. International J of Geographic Infor Systems. 1999:143–158. [Google Scholar]
- Takai K, Moser DP, DeFlaun M, Onstott TC, Fredrickson JK. Archaeal diversity in waters from deep South African gold mines. Appl and Env Microbiol. 2001;67:5750–5760. doi: 10.1128/AEM.67.21.5750-5760.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teske A, Sorensen KB. Uncultured archaea in deep marine subsurface sediments: have we caught them all? ISME J. 2008;2:3–18. doi: 10.1038/ismej.2007.90. [DOI] [PubMed] [Google Scholar]
- Torsvik V, Ovreas L. Microbial diversity and function in soil: from genes to ecosystems. Cur Op in Microbiol. 2002;5:240–245. doi: 10.1016/s1369-5274(02)00324-7. [DOI] [PubMed] [Google Scholar]
- Tyson GW, Chapman J, Hugenholtz P, et al. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- Venter JC, Remington K, Heidelberg JF, et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- Viant MR. Recent developments in environmental metabolomics. Molecular Biosyst. 2008;4:980–986. doi: 10.1039/b805354e. [DOI] [PubMed] [Google Scholar]
- Viant MR, Rosenblum ES, Tieerdema RS. NMR-based metabolomics: a powerful approach for characterizing the effects of environmental stressors on organism health. Env Sci & Tech. 2003;37:4982–4989. doi: 10.1021/es034281x. [DOI] [PubMed] [Google Scholar]
- Walker PA, Cocks KD. HABITAT: a procedure for modelling a disjoint environmental envelope for a plant or animal species. Global Ecol and Biogeog Let. 1991;1:108–118. [Google Scholar]
- Whitman WB, Coleman DC, Wiebe WJ. Prokaryotes: the unseen majority. Proceedings of the Nat Acad of Sci of the USA. 1998;95:6578–6583. doi: 10.1073/pnas.95.12.6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooley JC, Godzik A, Friedberg I. A Primer on Metagenomics. Plos Computational Biology. 2010;6 doi: 10.1371/journal.pcbi.1000667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woyke T, Tighe D, Mavromatis K, et al. One bacterial cell, one complete genome. PLoS One. 2010;5:e10314. doi: 10.1371/journal.pone.0010314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GD, Chen J, Hoffmann C, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang TC, Fu YC, Bishop PL. Competition for substrate and space in biofilms. Water Env Res. 1995;67:992–1003. [Google Scholar]
- Zhang XX, Deeks LK, Bengough AG, Crawford JW, Young LM. Determination of soil hydraulic conductivity with the lattice Boltzmann method and soil thin-section technique. J of Hydrology. 2005;306:59–70. [Google Scholar]