

Abstract
Background
During late differentiation, erythroid cells undergo profound changes involving actin filament remodeling. One of the proteins controlling actin dynamics is gelsolin, a calcium-activated actin filament severing and capping protein. Gelsolin-null (Gsn−/−) mice generated in a C57BL/6 background are viable and fertile.1
Design and Methods
We analyzed the functional roles of gelsolin in erythropoiesis by: (i) evaluating gelsolin expression in murine fetal liver cells at different stages of erythroid differentiation (using reverse transcription polymerase chain reaction analysis and immunohistochemistry), and (ii) characterizing embryonic and adult erythropoiesis in Gsn−/− BALB/c mice (morphology and erythroid cultures).
Results
In the context of a BALB/c background, the Gsn−/− mutation causes embryonic death. Gsn−/− embryos show defective erythroid maturation with persistence of circulating nucleated cells. The few Gsn−/− mice reaching adulthood fail to recover from phenylhydrazine-induced acute anemia, revealing an impaired response to stress erythropoiesis. In in vitro differentiation assays, E13.5 fetal liver Gsn−/− cells failed to undergo terminal maturation, a defect partially rescued by Cytochalasin D, and mimicked by administration of Jasplakinolide to the wild-type control samples.
Conclusions
In BALB/c mice, gelsolin deficiency alters the equilibrium between erythrocyte actin polymerization and depolymerization, causing impaired terminal maturation. We suggest a non-redundant role for gelsolin in terminal erythroid differentiation, possibly contributing to the Gsn−/− mice lethality observed in mid-gestation.
Keywords: erythropoiesis, erythroid maturation, actin dynamics, gelsolin
Introduction
During late maturation, erythroblasts undergo a complex sequence of differentiation events: in parallel with the accumulation of the specific gene products required for red blood cell (RBC) function, such as globin chains, these cells exit the cell cycle, their nucleus condenses and finally the cell is enucleated to give rise to a reticulocyte and, after the elimination of the residual organelles, to a fully mature RBC.2–4
Actin filament remodeling is critical for at least two crucial steps of erythrocyte maturation: enucleation and the proper assembly and maintenance of the red cell membrane. A so-called contractile actin ring is assembled immediately prior to enucleation, marking the boundary between the extruding nucleus and the incipient reticulocyte.2,5 The formation of this cytoskeletal structure is mediated by mDIA2, a formin required for nucleation of unbranched actin filaments.6
Moreover, actin filaments, together with several actin binding proteins (4.1, 4.9, adducins, tropomyosin, tropomodulin) constitute the core of the membrane skeletal junctions, to which the polygonal network of spectrin tetramers is anchored.7–9 Defects of components of the junctional complexes lead to cell fragility and are implicated in several forms of hemolytic anemia.10–12
Many of the actin-binding proteins participating in the junctional meshwork play a role in controlling the length of the polarized actin filaments, acting on polymerization, depolymerization and capping at the fast growing (barbed) and slow growing (pointed) ends. Their null mutations are associated with red cells that are mechanically weakened due to impaired cytoskeletal structure (for example, adducin13–15 and E-Tmod).16
Among various actin-remodeling proteins, gelsolin is a calcium-activated actin filament severing and capping protein found in many cell types, broadly expressed by cells of mesenchymal origin both in a cytoplasmic and in a plasma secreted form.1,17–21
Although gelsolin was suggested to be involved in the establishment of the actin-spectrin based membrane network during erythroid maturation,22 no evidence of its in vivo role in erythropoiesis has been provided so far. Gsn−/− mice generated in the C57BL/6 outbred genetic background were found to have impairments of specific aspects of cell motility, such as inflammation, although they are viable, fertile and with apparently normal hematopoiesis.1
Here we show that transferring the null Gelsolin allele into the BALB/c inbred genetic background results in defective erythroid maturation. These data suggest a non-redundant role for gelsolin in terminal erythroid differentiation, possibly contributing to the Gsn−/− mice lethality observed in mid-gestation.
Design and Methods
Generation of gelsolin null mice on a BALB/c congenic strain
Mice with a C57BL/6 outbred background1 homozygous for the mutation were crossed with mice of BALB/c inbred background. F1 heterozygous animals were crossed with mice of BALB/c inbred background to produce F2 progeny, among which only mice heterozygous for the mutation were used for the next generation. The same cycle was repeated until F10 mice were obtained. Heterozygous F10 mice were crossed to produce mice homozygous for the mutation, with a genetic background very close to the BALB/c inbred background. For timed pregnancies, BALB/c gelsolin heterozygous mice were mated overnight and noon of the day of vaginal plug appearance was considered day 0.5 post-coitum (E 0.5). Embryo dissections and genotyping were performed as previously described.1
All experiments and treatments in mice were approved by the Italian Ministry of Health and conducted using procedures designed to minimize animal stress and pain, in accordance with European Union guidelines.
Histology, antibodies and dyes
Embryos collected from timed pregnancies were analyzed. Details on histological staining, antibodies and dyes are provided in the Online Supplementary Design and Methods.
Immunocytochemistry and immunofluorescence analysis
Details on immunocytochemistry and immunofluorescence analysis are provided in the Online Supplementary Design and Methods.
Fetal liver cell purification and sorting
Freshly extracted mouse E13.5 fetal livers cells were disaggregated to single cells by gentle pipetting in phosphate-buffered saline containing 2mM EDTA and 0.5% bovine serum albumin. Cells were washed and incubated with the following labeled antibodies: allophycocyanin anti-mouse CD117 (c-Kit) and fluorescein isothiocyanate anti-mouse Ter119. Cells were sorted using a MoFlo (DAKO-Cytomation, Carpinteria, CA, USA) cell sorter. The purity of the cell populations obtained was greater than 95%.
Total embryonic blood cells were collected and counted as described by Kingsley et al.23 Each slide was prepared from 105 cells by cytospin centrifugation (400 rpm, 3 min) and slides were either air-dried or fixed for 5 min in ice-cold methanol.
Quantitative reverse transcriptase polymerase chain reaction analysis
Total RNA was purified from each cell population (105 cells) with TRI Reagent (Applied Biosystem, Carlsbad, CA, USA; AM9738) and retrotranscribed according to the manufacturer’s protocol. The primers used are listed in the Online Supplementary Design and Methods.
Hanging drop culture
E13.5 fetal livers were collected, disaggregated and resuspended in hanging drop medium as described by Gutiérrez et al.24 and detailed in the Online Supplementary Design and Methods. When indicated, Cytochalasin D (Sigma-Aldrich, C-8273) and Jasplakinolide (Sigma-Aldrich, C-5231) were dissolved in dimethyl sulfoxide (DMSO) (Sigma-Aldrich, D-2650) and added to the hanging drop medium at the final concentration of 50 nM. Quadruplicates - four drops - were analyzed for each condition.
Phenylhydrazine treatment
Age-matched mice were weighed and injected intraperitoneally for two consecutive days with 15 mg/Kg body weight of phenylhydrazine (PHZ, Sigma-Aldrich, P-6926). Two days after the second PHZ administration, animals were sacrificed and hematologic parameters and spleen morphology were analyzed.
Flow cytometry
Cells from blood or fetal liver or spleen were collected in Dulbecco’s modified Eagle’s medium, centrifuged and suspended in phosphate-buffered saline with 0.5% bovine serum albumin and stained with fluorescein isothiocyanate-conjugated anti-CD71 and with phycoerythrin-conjugated anti-Ter119. Samples were acquired using a FACS-Calibur (BD Bioscience) flow cytometer. Data were analyzed with Flow Jo software (Tree Star, Ashland, OR, USA).
Hematologic and biochemical analyses
Mice were bled into EDTA-containing tubes and the hematologic analysis was performed using a hemocytometer (Sysmex KX-21N Hematology Analyzer). Reticulocytes were scored by counting them on blood smears stained with new methylene blue (Sigma-Aldrich, R-4132), according to manufacturer’s protocol.
Osmotic fragility test
The osmotic fragility test was performed on fresh blood drawn from the tail veins of age-matched mice (6 months old), Gsn−/− mice (n=3) and wt mice (n=3). Blood was diluted in a series of hypotonic solutions with NaCl content starting from 160 mM and incubated for 30 min at 37°C. The concentration of proteins released into the supernatant was measured using a Bradford protein assay (BioRad, Hercules, CA, USA; cat. 500-0006). The osmotic fragility curve is obtained by plotting the measured absorbance at 595 nm for each solution against NaCl concentrations. Duplicates for each NaCl concentration point were read.
Results
Gelsolin expression increases during erythroid differentiation
To study the possible role of gelsolin in fetal erythropoiesis, we first analyzed its expression at different stages of erythroid differentiation. To this end, we FACS sorted three cell populations from E13.5 mouse fetal liver on the basis of their expression of c-Kit and Ter119 cell antigens: (i) c-Kit+Ter119− cells, which represent hematopoietic progenitors (including erythroid precursors); (ii) c-Kit+Ter119+ cells, which are erythroid committed early progenitors; and (iii) c-Kit−Ter119+ cells, which are more differentiated erythroblasts and mature erythrocytes (Figure 1A and25,26). Gelsolin expression was subsequently measured in these three sorted cell populations by quantitative reverse transcription polymerase chain reaction (RT-PCR) analysis, in parallel with that of glycophorin A (GpA) and CD44. While GpA increases during erythroid differentiation, the expression of the cell adhesion glycoprotein CD44 progressively decreases along with erythropoiesis,27 thus allowing a further staging of the degree of cellular differentiation. As shown in Figure 1B, gelsolin expression is slightly decreased in the erythroid committed progenitors (c-Kit+Ter119+) when compared to c-Kit+Ter119−cells and it is expressed at high levels in the more mature erythroid cells (c-Kit−Ter119+) in parallel with the accumulation of GpA and the simultaneous decrease in CD44 expression.
Figure 1.

Characterization of gelsolin expression during erythroid maturation. (A) FACS analysis of E13.5 mouse fetal liver cells for expression of c-Kit and Ter119 cell antigens. The three major cell populations are indicated. (B) Real-time PCR on Gsn, GpA and CD44 expression on cDNA from the sorted fetal liver cell populations indicated in (A) FL: cDNA from total fetal liver cells. Histograms show the levels of expression (mean SEM of at least 3 independent experiments) relative to hypoxanthine-phospho-ribosyltransferase (HPRT) considered as 1. Statistical significance is indicated above the chart. (C) Fetal liver cells cells were stained with O-dianosidine (bright orange) and counterstained with hematoxylin/eosin. Immunofluorescence analysis using an anti-Gsn antibody (green), an anti Ter119 antibody (red) and DAPI (blue). Bar: 25 μm. (D) The same analysis was carried out on FACS sorted cell populations. Single cells are shown as an example. Bars: 4 μm.
To visualize the protein distribution of cytoplasmic gelsolin within the same cell populations, we performed double immunocytochemical staining using an anti-gelsolin antibody (green) and anti-Ter119 antibody (red) (Figure 1C, D). Virtually all the most differentiated cells (recognizable by their small size and condensed nucleus, Figure 1C) were strongly positive for gelsolin staining, whereas more immature cells expressed lower gelsolin levels.
The differential expression of gelsolin during erythroid differentiation was confirmed by performing the same analysis on the three FACS-sorted populations described above (Figure 1D): the majority of c-Kit+Ter119−cells were negative or weakly positive (only few of them showed clear cytoplasmic positivity), whereas almost all cells expressing Ter119 (early erythroid c-Kit+Ter119+ and late erythroid c-Kit−Ter119+ cells) stained positively with gelsolin (Figure 1D, right panels). Co-expression of gelsolin and Ter119 was further confirmed by FACS analysis on permeabilized E13.5 fetal liver cells (data not shown).
Embryonic loss of gelsolin null mutant mice in a BALB/c background
The expression profile of gelsolin at the different stages of erythroid differentiation described above suggested a possible specific functional role of gelsolin in erythropoiesis.
Although Gsn knock-out mice generated on a C57BL/6 background are viable and fertile,1 when the Gsn−/− mutation was transferred into a BALB/c background almost no viable Gsn−/− offspring were born from heterozygous intercrosses. Timed matings revealed a normal proportion of homozygous mutant embryos until E12.5 (about 25%). Thereafter the recovery of viable null mutant embryos steadily decreased below the expected ratio with a significant reduction of null mutant embryos observed on day 13.5 (when Gsn−/− mice were about 8.8%, Online Supplementary Table S1). Finally, only 4% of homozygous mutant mice were found post-partum and reached adulthood.
Gelsolin null BALB/c mice show defects in embryonic red blood cells
The expression pattern of gelsolin during erythropoiesis, together with the timing of the first loss of mutant embryos during gestation (E12.5-E13.5 marks the transition between primitive and definitive erythropoiesis in liver), prompted us to analyze erythropoiesis in Gsn−/− mice in more detail. We first carried out hematoxylin/eosin staining of blood from wt and Gsn−/− embryos. At E13.5 no gross differences were visible (compare Figure 2A with 2B); however, at E.14.5 (Figure 2C-D) nucleated primitive erythroid cells (identified on the basis of their morphology, black arrows) persisted in mutant embryos, but not in normal ones. At the same developmental stage, in blood from Gsn−/− embryos we found small nucleated circulating cells, which we interpreted as immature definitive erythrocytes (blue arrow, Figure 2D). At E17.5 (Figure 2E-F) mutant (but not wild-type) embryos showed a consistent number (about 2.5%) of these cells in circulation, together with some primitive nucleated erythroid cells still present in both blood and fetal liver (Figure 2G-H, black arrows).
Figure 2.
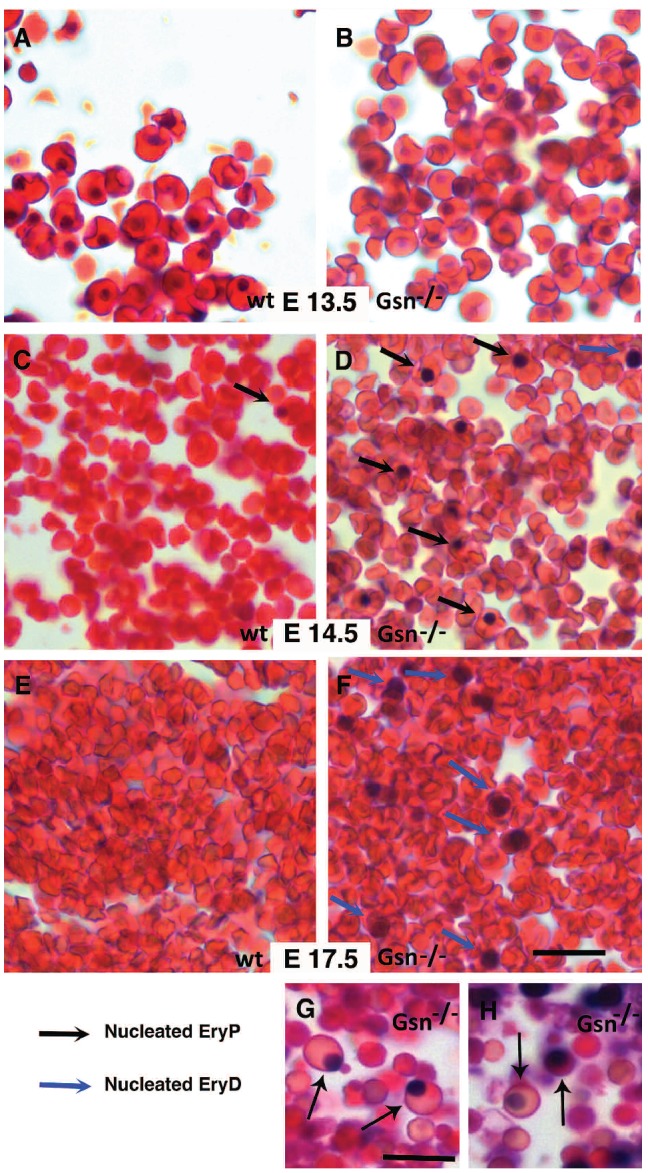
Gelsolin null BALB/c mice show defects in embryonic red blood cells. (A-H) Hematoxylin/eosin staining of blood from wt (A, C, E) and Gsn−/− (B, D, F, G, H) mice embryos showed no gross differences at E13.5 of gestation (panel A-B) and at E.14.5 of gestation (C-D). Some smaller nucleated cells, resembling immature definitive erythroid cells are present (blue arrows, D). At E17.5 of gestation (E-F) mutant (but not wild type) embryos showed a consistent number (about 2.5%) of nucleated definitive erythroid cells in the circulation, together with some primitive nucleated erythroid cells still present in both blood and fetal liver sections (black arrows, G-H). Bars: 24 μm (A-F); 12 μm (G, H).
Cytospins prepared from E17.5 wt (Figure 3, left column) and Gsn−/− (Figure 3, right column) blood samples were stained with Giemsa (Figure 3A-D) or DAPI (Figure 3E-F). Giemsa staining confirmed the presence in the circulation of nucleated cells (blue arrows), cells undergoing enucleation and binucleated cells (green arrows), together with many hypochromic cells (Figure 3A-D). DAPI staining (Figure 3E-F) confirmed the presence of a high number of nucleated cells in the circulation of Gsn−/− mice.
Figure 3.
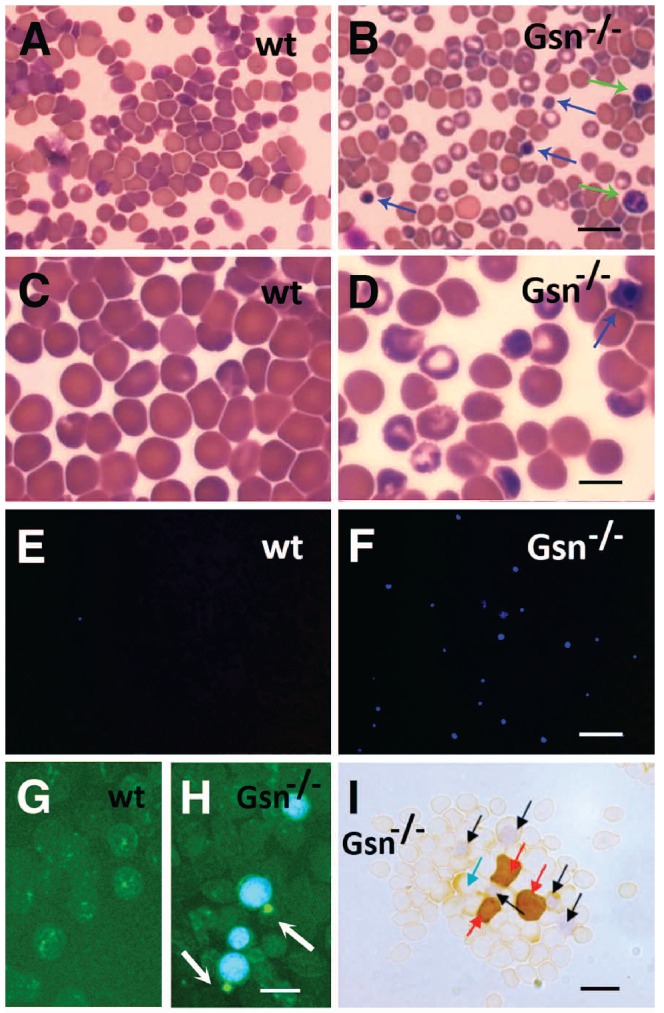
Increased number of circulating nucleated cells in Gsn−/− mice at E17.5. Cytospins from E17.5 wt (A, C, E, G) and Gsn−/− (B, D, F, H, I) blood samples were stained with Giemsa (A-D) or DAPI (E-F), Alexa Fluor 488–phalloidin (G, H) and βH1 (I). Bars: 20 μm (A, B, E, F); 10 μm (C, D, G, H);(I) 15 μm. (A-D) Blue arrows indicate nucleated cells while green arrows point to binucleated cells; (E, F) DAPI staining emphasizes the increased proportion of nucleated cells in blood samples from E17.5 Gsn−/− embryos; (G, H) white arrows evidence the contractile actin ring typical of cells undergoing enucleation; (I) red arrows indicate βH1+ cells, black arrows indicate βH1− nucleated cells and the blue arrow indicates a nucleated cell weakly positive for βH1 staining.
To visualize actin filaments, we treated fixed cells with Alexa Fluor 488–phalloidin (Figure 3G, H), which binds to F-actin. As shown in Figure 3H (white arrows), Gsn−/− blood contains circulating cells still undergoing enucleation, as indicated by the presence of the contractile actin ring, which forms immediately prior to expulsion of the nucleus, further supporting the observation of delayed erythroid maturation in Gsn−/− blood samples (Figure 3H) as compared to that in wt blood samples (Figure 3G).
At E17.5 primitive cells normally account for about 1% of circulating erythrocytes and their progressive transition to enucleated cells should be essentially completed.23 To evaluate the number of primitive cells in Gsn−/− mice blood, cytospins were also stained with an anti βH1-globin antibody (Figure 3I). We scored about 4% (against less than 1% in wt control samples) of large βH1-globin+ cells (Figure 3I, red arrows), and some weakly βH1-globin+ nucleated cells (Figure 3I, blue arrow), whereas many nucleated cells were not apparently expressing βH1-globin (Figure 3I, black arrows) and likely represented definitive erythroblasts.
Erythroid differentiation is impaired in gelsolin null embryos
On the basis of the above results, we analyzed the defective erythroid maturation observed in Gsn−/− embryos, focusing on E13.5, in more detail (Figure 4). At this time point Gsn−/− mice were moderately anemic, the total RBC count per embryo being reduced to approximately 75% of the normal value (Online Supplementary Figure S1). To monitor erythroid maturation, we stained blood cells from wt and mutant mice with anti-CD71 and anti-Ter119 antibodies (Figure 4A). The anti-CD71 antibody recognizes the transferrin receptor, which is highly expressed on proery-throblasts and early erythroblasts and less present in late erythroblasts and reticulocytes. The anti-Ter119 antibody binds a molecule associated with glycophorin A expressed from the erythroblast to mature erythrocyte stages. FACS analysis with the above antibodies identified three major distinct populations of progressively differentiating cells in the circulation: R1, CD71+Ter119+; R2, CD71+Ter119++; R3, CD71− Ter119++ (Online Supplementary Figure S1). Gsn−/− embryos fall in two broad categories, exemplified in Figure 3B by mut1 and mut2, which shows two representative embryos from the same litter: (i) in some embryos (as in mut1), the FACS profile is grossly similar to that of the wt littermates but with an increase in CD71− Ter119++ cells (R3, from about 11% in the wt animals to about 21% in mut1 mice), at the expense of the R1+R2 populations. A blood smear from a mut1 animal shows the accumulation of small, fragmented and misshaped cells (Figure 4B); (ii) in other embryos (as in mut2) a sharp loss in the R2 cell population (CD71+Ter119++ from about 27% in the wt mice to about 16% in the mut2 animals) is observed, suggesting an impairment in the final stages of erythroid maturation. Hemotoxylin and eosin staining of the same sample (Figure 4B) showed a clear prevalence of primitive erythroid cells (identified on the basis of their morphology) with the frequent presence of binucleated cells (about 4%). As discussed below, we think that this could reflect an underlying variable penetrance/expressivity of the gelsolin defect. Of note, in both cases, the cell population representing the transitional stages between R2 and R3 was missing, suggesting impairment of late stages of differentiation.
Figure 4.
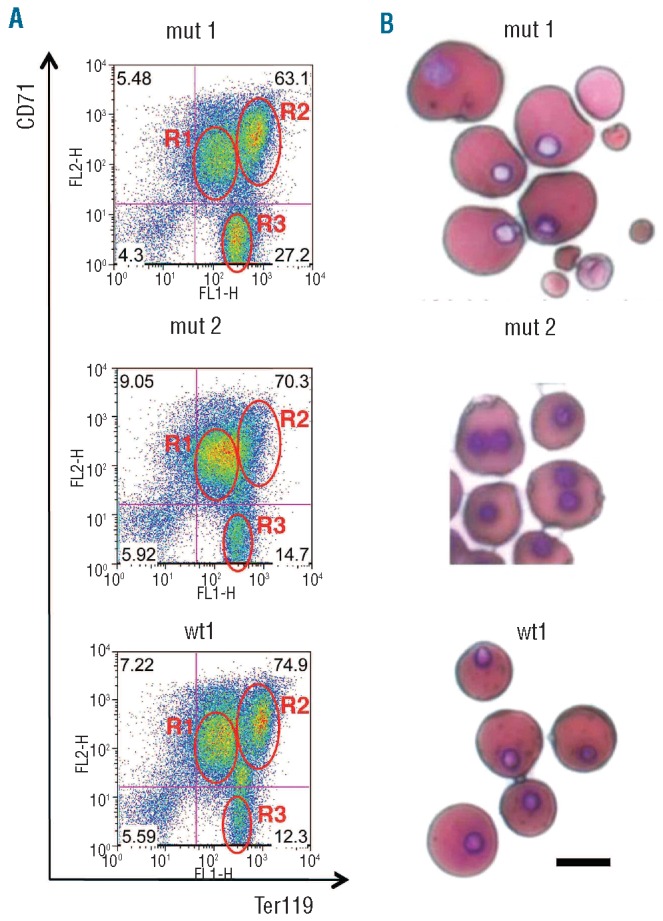
(A) FACS analysis on blood cells from E13.5 wt and Gsn−/−embryos with anti-CD71 and anti-Ter119 antibodies. Three major distinct populations of progressively differentiating cells are present in the circulation: R1: CD71+Ter119+; R2: CD71+Ter119++; R3: CD71− Ter119++. (B) Hematoxylin/eosin staining of the same samples as in (A) Bar: 10 μm.
Adult gelsolin null mice are unable to recover from phenylhydrazine-induced anemia
Very few Gsn−/− mice were born (20% of the expected Mendelian ratio, 5% instead of 25% of total mice; Online Supplementary Table S1), but grew to adulthood in apparently healthy conditions. These mice, however, showed decreased exercise endurance in the treadmill test and, strikingly, the running times of the Gsn−/− mice were decreased by 60% (total count of activity per night: 2459±435 runs/night Gsn−/− mice versus 8760±917 runs/night wt mice, P<0.006, n= 3). No significant differences in muscle mass were observed (unpublished data). These results suggest an impairment in the physical performance of the Gsn−/− mice.
Although the hematologic parameters of the Gsn−/− mutants were grossly normal (data not shown), morphological analysis of blood showed the presence of RBC with irregular shape, frequently with reduced central pallor and more than two concavities, thus resembling knizocytes (Figure 5A), which have been described in hemolytic anemia, hereditary stomatocytosis, and thalassemia.12 Methylene blue staining of blood smears revealed a higher number of reticulocytes in mutant mice than in wt mice (12.5% versus 4.4%, respectively) (data not shown).
Figure 5.

Adult Gsn−/− mice are unable to recover from PHZ-induced anemia: (A) Morphological analysis of blood from adult Gsn−/− mice showed the presence of knizocytes. Bar: 5 μm. (B) Osmotic lysis test. X axis: NaCl concentration; Y axis: amount of proteins released upon osmotic lysis, measured as absorbance at 595 nm (Bradford method). (C) The mean weight of the spleen is increased in Gsn−/− mice under PHZ-induced erythropoietic stress. (D) Morphological analysis of spleen sections from wt and Gsn−/− mice. Bar: 200 μm. (E) Spleen cells isolated from wt and Gsn−/− mice and stained with anti CD71-FITC and antiTer119-PE antibodies for FACS analysis. The panel shows dot plots of wt and Gsn−/− mice. (F) Percentage of positive cells within the four major distinct populations of progressively differentiating cells (CD71++Ter119-, CD71++ Ter119++, CD71+Ter119++ and CD71− Ter119++ cells) were evaluated on total spleen cells. Histograms represent the mean of three wt and three Gsn−/− mice; significance levels were calculated by a t test for unpaired data.
To characterize the functional properties of RBC in Gsn−/− mice we performed an osmotic fragility test, comparing the resistance of mutant and wt cells when incubated with decreasing concentrations of NaCl, ranging from 160 mM NaCl to H2O. The amount of released proteins at the different NaCl concentrations was slightly higher in mutant RBC, suggesting a mild but significant intrinsic fragility of these cells under both normal and hypotonic conditions (Figure 5B).
To better define the mechanism of erythroid compensation in the few adult Gsn−/− escaping embryonic death, we treated the same mutant animals analyzed above with phenylhydrazine (PHZ) to induce acute anemia. PHZ (15 mg/Kg body weight) was injected intraperitoneally for two consecutive days; on the second day after the second PHZ administration, Gsn−/− mice were sacrificed and hematologic parameters and spleen morphology were analyzed.
Gsn−/− mice presented accentuated pallor after PHZ treatment, when compared to wt controls (not shown) and hematologic analysis revealed a reduced number of RBC and low hematocrit (RBC ×106/μL: wt 9.19±3 versus KO 4.94±1.2; hematocrit %: wt 43.8±9.1 versus KO 29.9±5.5; mean corpuscular volume μm3: wt 47.3±4.3 versus KO 46.3±0.9) together with a reduction in platelet counts (platelets × 106/μL: wt 1932±1125 versus KO 909±313).
The mean weight of the spleen was increased in Gsn−/− mice under PHZ stress (Figure 5C). Morphological analysis of spleen sections confirms the presence of a higher number of red cells with respect to wt mice treated with PHZ (Figure 5D). Flow cytometric analysis on spleen cells stained with antibodies against CD71 and Ter119 (Figure 5E, F) revealed an increased percentage of immature erythroid cells (CD71++Ter119−) in Gsn−/− mice when compared to the percentage in wt mice(P=0.029). Afterwards, we detected no difference between wt and mutants at the intermediate stages and a trend towards a decrease in more mature cells (CD71−Ter119++, P=0.27 for n=3). It is thus possible that compensatory mechanisms operate during maturation. Taken together, these data indicate that Gsn−/− mice are able to respond to stress erythropoiesis, but nevertheless fail to fully recover from induced anemia.
Ex vivo gelsolin null erythroblasts fail to differentiate properly in hanging drop cultures
To get insight into the defect of erythroid terminal differentiation in Gsn−/− cells, we set up hanging drop cultures starting from E13.5 ex vivo fetal livers. This protocol enables a quantitative differentiation of primary definitive erythroid cells to mature enucleated erythrocytes within 2 days of culture.24
No significant difference was observed when fetal liver cells isolated from wt and Gsn−/− mice were disaggregated, stained with O-dianosidine to mark hemoglobinized cells and counterstained with hematoxylin/eosin (Figure 6A,B): in both samples the distribution of cells at the different stages of differentiation (from pro-erythroblast, dividing pro-erythroblasts, basophilic, polychromatic and orthochromatic cells to reticulocytes) was very similar. Twenty-four hours after cell seeding, a significant proportion of hemoglobinized cells (brown staining) undergoing enucleation (black arrows) or already enucleated (green arrows) was present in wt cultures (Figure 6C). In contrast, cells from Gsn−/− fetal livers showed a substantial delay in erythroid differentiation, with a marked prevalence of immature cells (Figure 6D). At 48 h, massive enucleation took place in wt cultures (Figure 6E,G), whereas the majority of Gsn−/− cells became hemoglobinized but failed to undergo proper enucleation (Figure 6F,H). These data are summarized in Figure 6I. Moreover, many Gsn−/− cells presented two or more distinct nuclei (reminiscent of binucleated cells observed in the circulation in Figure 4B), suggesting that the lack of gelsolin function and thus the inability to sever actin filaments, results in an impairment of the process of cytodieresis and nuclear extrusion required for terminal erythroid maturation.
Figure 6.
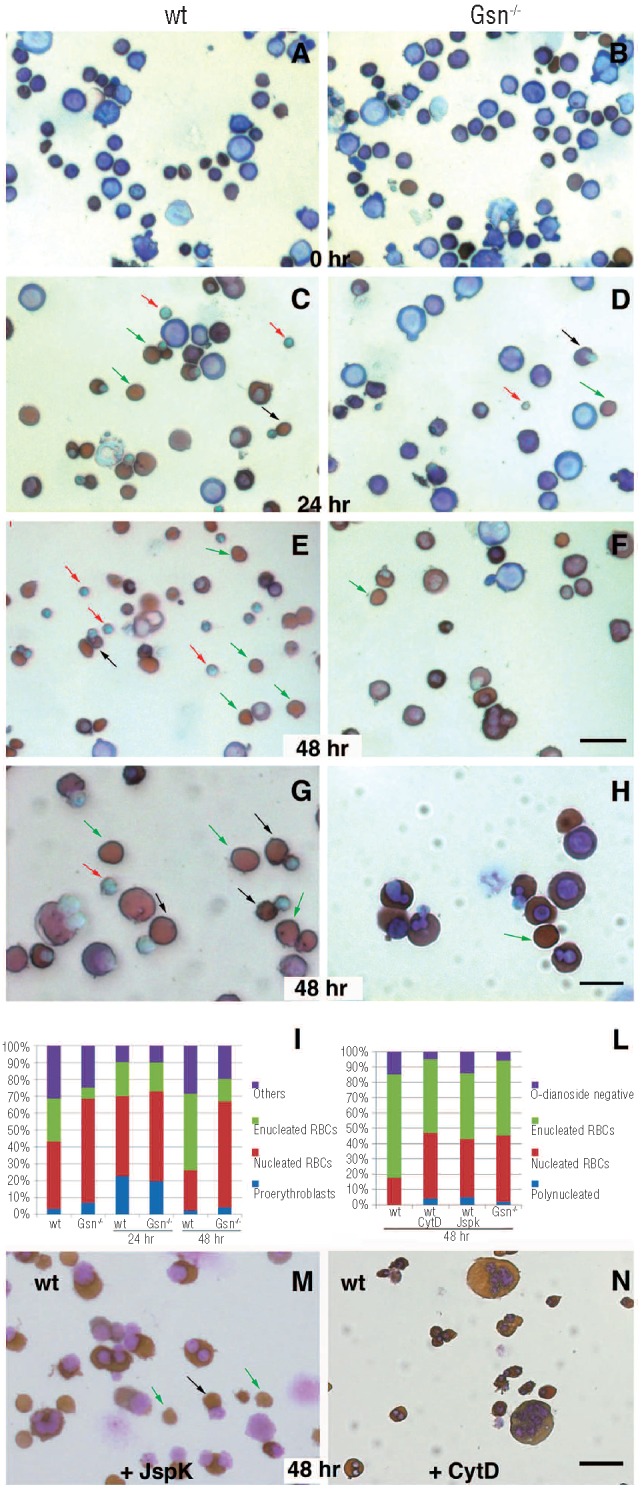
Ex vivo Gsn−/− erythroblasts fail to differentiate properly in hanging drop cultures. (A, B) Fetal liver cells isolated from wt and Gsn−/− - mice are disaggregated, stained with O-dianosidine (brown) and counter-stained with hematoxylin/eosin. (C, D) At 24 h after cell seeding, a significant proportion of hemoglobinized cells (brown staining) undergoing enucleation (black arrows) or already enucleated (green arrows) was present in wt cultures (C), in contrast with the sharp prevalence of immature cells present in the Gsn−/− cells (D). (E-H) At 48 h, massive enucleation took place in wt cultures (E, G), whereas the majority of Gsn−/− derived cells (F, H) became hemoglobinized but failed to undergo proper enucleation. Pyrenocytes are indicated by red arrows (F, H). (I) Relative proportion of cells at different stages of differentiation (> 100 cells scored in 3 independent fields). (L) The addition of Jasplakinolide (JspK) or Cytochalasin D (CytD) to wt cells mimics the accumulation of polynucleated cells and the delay in maturation observed in Gsn−/− samples. (M, N) Representative fields of wt cells treated with JspK (left) or CytD (right). Bars: 15 μm (A-F and M-N); Bar: 7 μm (G-H).
On this basis, we reasoned that the perturbation of actin metabolism produced by the addition of Cytochalasin D, which blocks polymerization and elongation of actin filaments, and of Jasplakinolide, which stabilizes actin filaments, might mimic the absence of gelsolin in wt-derived cultures (and eventually exacerbate the enucleation failure in mice lacking gelsolin). Addition of the drugs shows that our prediction was respected (Figure 6L-N): the addition of Jasplakinolide (Figure 6M) or Cytochalasin D (Figure 6N) to the wt cells decreased the proportion of enucleated cells and induced the accumulation of polynucleated cells in a proportion similar to that observed in mutant cultures.
Discussion
Gelsolin is a Ca++-dependent protein that regulates actin filament length by severing preassembled filaments. After severing, gelsolin remains attached to the fast-growing end of the filament and caps it, thus preventing its growth by addition of new actin monomers. Gsn−/− mice generated in a C57BL/6 mixed genetic background are viable but show impaired cellular functions requiring motile responses (recruitment of leukocytes in inflammation) and shape changes (platelet activation), as expected on the basis of gelsolin’s role in cytoskeleton remodeling.1
Since the genetic background is known to cause considerable variation in the phenotype of genetically engineered mice, possibly due to the presence of multiple functional interactions of different alleles at modifier loci,28 the Gsn−/− mutation was transferred into the inbred BALB/c background. On this genetic background the majority of null mice died in utero, with only very few mice (Figure 2) surviving to adulthood in apparently good health (although they performed poorly under physical exercise conditions, see treadmill test).
The cause of embryonic death is likely rather complex, resulting from the impairment of various cell functions and developmental processes requiring proficient actin remodeling; the defect in erythropoiesis described here is probably one component of the observed embryonic failure.
Although a previous paper reported gelsolin expression in chicken erythroid progenitors,22 no further evidence on the role of gelsolin in erythropoiesis was available. In this paper we report that the BALB/c genetic background uncovers a non-redundant gelsolin requirement for proper terminal erythroid maturation.
Gelsolin is normally already expressed at low levels in c-Kit+Ter119− progenitors isolated from E13.5 fetal livers and accumulates along with erythroid differentiation, in parallel with the emergence of Ter119 positivity (Figure 1).
The persistence of primitive nucleated erythroid cells in Gsn−/− mice, until E17.5 (Figure 2), suggests either a delay in maturation (and thus enucleation) possibly due to the inefficient actin remodeling and/or the presence of some compensatory mechanism to sustain a defective definitive erythropoiesis. In this regard, Figure 4A shows that primitive circulating cells from mut1 and mut2 present morphological abnormalities, confirming that this cell population is affected by the lack of gelsolin.
Blood cells from these two mutant phenotypes are characterized by the presence of either binucleated or fragmented cells, respectively. Small differences in the time of pregnancy and/or in embryo development within the same litter could account for some intrinsic phenotypic variation. However, we speculate that the observed heterogeneity might reflect, at least in part, a underlying variable expressivity of the Gsn−/− defect null mutation. Embryos with a mild phenotype at E13.5 (as mut1) will probably survive to later stages due to some compensatory effect, whereas embryos with the most severe phenotype (as mut2) will die around E13.5. Analysis of embryonic circulating RBC at E17.5 (Figures 2 and 3) revealed, together with the persistence of primitive cells expressing embryonic βH1 globin, the presence of abnormal cells, some of which were nucleated or hypochromic and/or misshaped. These latter abnormal cells suggest defective cellular maturation of definitive erythroid cells, possibly accompanied by intrinsic fragility of defective RBC, as suggested by the moderate anemia observed in Gsn−/− embryos.
Impaired actin remodeling could likely result in either impaired terminal maturation/enucleation and/or increased fragility of RBC.29 In late erythropoiesis actin filaments assemble the contractile ring that marks the border between the incipient reticulocyte and the condensed nucleus prior to its expulsion, and Rac GTPases and their downstream target mDia2, a formin required for nucleation of unbranched actin filaments, are required for nuclear extrusion of in vitro-cultured mouse fetal liver cells. Once mature enucleated RBC are released into the circulation, short actin filaments ensure, by securing the hexagonal spectrin scaffold, the mechanical property (flexibility and tensile strength) of the plasma membrane, together with a network of associated proteins. In both cases, the length of the actin filaments is tightly regulated by a variety of proteins promoting: (i) the polymerization of actin monomers, (ii) the maintenance of filament length by capping their ends thus preventing further association or dissociation and (iii) the disassembly of preexisting filaments, by severing them. Mutations in some of these proteins, such as adducins,14 and E-Tmod,16 in mice are associated with erythroid defects resembling human hereditary hemolytic anemias, as a consequence of increased RBC fragility.
Our results suggest that the absence of gelsolin influences both processes: data from the hanging drops cultures from E13.5 mouse fetal liver cells (Figure 6) confirm the inability of the mutant cells to undergo proper cytodieresis and enucleation, with a consistent accumulation of binucleated cells, a feature often observed in congenital dyserythropoietic anemia.30 This defect is mimicked by the addition of Jasplakinolide to wt cells, which has the effect of stabilizing actin filaments, or of Cytochalasin D, which blocks polymerization and elongation (Figure 6), suggesting that the failure of proper actin turnover might be the direct cause of the observed phenotype in gelsolin null mutants.
On the other hand, the few Gsn−/− animals reaching adulthood had almost normal hematologic parameters although their RBC had an altered morphology and slightly increased fragility. On this point, it is worth considering that the analysis on adult mice inevitably refers only to the small fraction of animals that escape intrauterine death and might not, therefore, fully reproduce the effect of the lack of gelsolin in adult cells. Finally, when challenged with PHZ, Gsn−/− mice failed to recover from induced acute anemia, although they did respond to stress erythropoiesis with massive production of red cells by the spleen (Figure 5).
Together, these data suggest that the impairment of erythroid differentiation/maturation might be a significant determinant of the embryonic lethality observed in BALB/c Gsn−/− mice. Definitive erythroblasts proliferate, differentiate and finally enucleate within the erythroblastic islands, consisting in highly specialized niches composed of erythroblasts surrounding a central macrophage.5 A still largely unknown network of cell-cell adhesions, together with the secretion of regulatory cytokines, modulate the crosstalk between these two types of cell.
Since gelsolin is also expressed in macrophages, it will be of great interest to investigate whether, in analogy with other actin cytoskeleton regulatory proteins,31,32 it is also required for the establishment of the erythropoietic microenvironment in erythroblastic islands.5
Acknowledgments
We thank James Palis, Alberto Zanella and Andrea Brendolan for precious advice and discussion; Cristina Vercellati, Anna Paola Marcello, Elisa Fermo, Federica Colleoni and Giorgio Scarì for technical support; and Ilaria Alborelli, Luciana Petti and Alessandro Farinato for helping with experiments and discussion.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding: this work was supported by grants from PRIN to AR and from the Italian Ministry of Health RC-2010 to LS.
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Witke W, Sharpe AH, Hartwig JH, Azuma T, Stossel TP, Kwiatkowski DJ. Hemostatic, inflammatory, and fibroblast responses are blunted in mice lacking gelsolin. Cell. 1995;81(1):41–51. doi: 10.1016/0092-8674(95)90369-0. [DOI] [PubMed] [Google Scholar]
- 2.Koury ST, Koury MJ, Bondurant MC. Cytoskeletal distribution and function during the maturation and enucleation of mammalian erythroblasts. J Cell Biol. 1989;109(6):3005–13. doi: 10.1083/jcb.109.6.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ney PA. Normal and disordered reticulocyte maturation. Curr Opin Hematol. 2011;18(3):152–7. doi: 10.1097/MOH.0b013e328345213e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krauss SW, Lo AJ, Short SA, Koury MJ, Mohandas N, Chasis JA. Nuclear substructure reorganization during late-stage erythropoiesis is selective and does not involve caspase cleavage of major nuclear substructural proteins. Blood. 2005;106(6):2200–5. doi: 10.1182/blood-2005-04-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chasis JA, Mohandas N. Erythroblastic islands: niches for erythropoiesis. Blood. 2008;112(3):470–8. doi: 10.1182/blood-2008-03-077883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ji P, Jayapal SR, Lodish HF. Enucleation of cultured mouse fetal erythroblasts requires Rac GTPases and mDia2. Nat Cell Biol. 2008;10(3):314–21. doi: 10.1038/ncb1693. [DOI] [PubMed] [Google Scholar]
- 7.Bennett V, Gilligan DM. The spectrin-based membrane skeleton and micron-scale organization of the plasma membrane. Annual review of cell biology. 1993:927–66. doi: 10.1146/annurev.cb.09.110193.000331. [DOI] [PubMed] [Google Scholar]
- 8.Salomao M, Zhang X, Yang Y, Lee S, Hartwig JH, Chasis JA, et al. Protein 4.1R-dependent multiprotein complex: new insights into the structural organization of the red blood cell membrane. Proc Natl Acad Sci USA. 2008;105(23):8026–31. doi: 10.1073/pnas.0803225105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohandas N, Gallagher PG. Red cell membrane: past, present, and future. Blood. 2008;112(10):3939–48. doi: 10.1182/blood-2008-07-161166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Birkenmeier CS, Barker JE. Hereditary haemolytic anaemias: unexpected sequelae of mutations in the genes for erythroid membrane skeletal proteins. J Pathol. 2004;204(4):450–9. doi: 10.1002/path.1636. [DOI] [PubMed] [Google Scholar]
- 11.Mohandas N, Evans E. Mechanical properties of the red cell membrane in relation to molecular structure and genetic defects. Annual review of biophysics and biomolecular structure. 1994:23787–818. doi: 10.1146/annurev.bb.23.060194.004035. [DOI] [PubMed] [Google Scholar]
- 12.Delaunay J. The molecular basis of hereditary red cell membrane disorders. Blood Rev. 2007;21(1):1–20. doi: 10.1016/j.blre.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 13.Gilligan DM, Lozovatsky L, Gwynn B, Brugnara C, Mohandas N, Peters LL. Targeted disruption of the beta adducin gene (Add2) causes red blood cell spherocytosis in mice. Proc Natl Acad Sci USA. 1999;96(19):10717–22. doi: 10.1073/pnas.96.19.10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muro AF, Marro ML, Gajović S, Porro F, Luzzatto L, Baralle FE. Mild spherocytic hereditary elliptocytosis and altered levels of alpha- and gamma-adducins in beta-adducin-deficient mice. Blood. 2000;95(12):3978–85. [PubMed] [Google Scholar]
- 15.Chen H, Khan AA, Liu F, Gilligan DM, Peters LL, Messick J, et al. Combined deletion of mouse dematin-headpiece and beta-adducin exerts a novel effect on the spectrinactin junctions leading to erythrocyte fragility and hemolytic anemia. J Biol Chem. 2007;282(6):4124–35. doi: 10.1074/jbc.M610231200. [DOI] [PubMed] [Google Scholar]
- 16.Chu X, Chen J, Reedy MC, Vera C, Sung K-LP, Sung LA. E-Tmod capping of actin filaments at the slow-growing end is required to establish mouse embryonic circulation. Am J Physiol Heart Circ Physiol. 2003;284(5):H1827–38. doi: 10.1152/ajpheart.00947.2002. [DOI] [PubMed] [Google Scholar]
- 17.Arai M, Kwiatkowski DJ. Differential developmentally regulated expression of gelsolin family members in the mouse. Dev Dyn. 1999;215(4):297–307. doi: 10.1002/(SICI)1097-0177(199908)215:4<297::AID-AJA2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 18.Teubner A, Sobek-Klocke I, Hinssen H, Eichenlaub-Ritter U. Distribution of gelsolin in mouse ovary. Cell Tissue Res. 1994;276(3):535–44. doi: 10.1007/BF00343950. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka J, Kira M, Sobue K. Gelsolin is localized in neuronal growth cones. 1993;76(2):268–71. doi: 10.1016/0165-3806(93)90217-x. [DOI] [PubMed] [Google Scholar]
- 20.Lueck A, Yin HL, Kwiatkowski DJ, Allen PG. Calcium regulation of gelsolin and adseverin: a natural test of the helix latch hypothesis. Biochemistry. 2000;39(18):5274–9. doi: 10.1021/bi992871v. [DOI] [PubMed] [Google Scholar]
- 21.Barkalow K, Witke W, Kwiatkowski DJ, Hartwig JH. Coordinated regulation of platelet actin filament barbed ends by gelsolin and capping protein. J Cell Biol. 1996;134(2):389–99. doi: 10.1083/jcb.134.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hinssen H, Vandekerckhove J, Lazarides E. Gelsolin is expressed in early erythroid progenitor cells and negatively regulated during erythropoiesis. J Cell Biol. 1987;105(3):1425–33. doi: 10.1083/jcb.105.3.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kingsley PD, Malik J, Fantauzzo KA, Palis J. Yolk sac-derived primitive erythroblasts enucleate during mammalian embryogenesis. Blood. 2004;1;104(1):19–25. doi: 10.1182/blood-2003-12-4162. [DOI] [PubMed] [Google Scholar]
- 24.Gutiérrez L, Lindeboom F, Ferreira R, Drissen R, Grosveld F, Whyatt D, et al. A hanging drop culture method to study terminal erythroid differentiation. Exp Hematol. 2005;33(10):1083–91. doi: 10.1016/j.exphem.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 25.Cantù C, Ierardi R, Alborelli I, Fugazza C, Cassinelli L, Piconese S, et al. Sox6 enhances erythroid differentiation in human erythroid progenitors. Blood. 2011;117(13):3669–79. doi: 10.1182/blood-2010-04-282350. [DOI] [PubMed] [Google Scholar]
- 26.Cantu’ C, Grande V, Alborelli I, Cassinelli L, Cantu’ I, Colzani MT, et al. A highly conserved SOX6 double binding site mediates SOX6 gene downregulation in erythroid cells. Nucleic Acids Res. 2011;39(2):486–501. doi: 10.1093/nar/gkq819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen K, Liu J, Heck S, Chasis JA, An X, Mohandas N. Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc Natl Acad Sci USA. 2009;106(41):17413–8. doi: 10.1073/pnas.0909296106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doetschman T. Inflluence of genetic background on genetically engineered mouse phenotypes. Methods Mol Biol. 2009;530:423–33. doi: 10.1007/978-1-59745-471-1_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalfa TA, Pushkaran S, Mohandas N, Hartwig JH, Fowler VM, Johnson JF, et al. Rac GTPases regulate the morphology and deformability of the erythrocyte cytoskeleton. Blood. 2006;108(12):3637–45. doi: 10.1182/blood-2006-03-005942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Queisser W, Spiertz E, Jost E, Heimpel H. Proliferation disturbances of erythroblasts in congenital dyserythropoietic anemia type I and II. Acta Haematol. 1971;45(2):65–76. doi: 10.1159/000208608. [DOI] [PubMed] [Google Scholar]
- 31.Liu X-S, Li X-H, Wang Y, Shu R-Z, Wang L, Lu S-Y, et al. Disruption of palladin leads to defects in definitive erythropoiesis by interfering with erythroblastic island formation in mouse fetal liver. Blood. 2007;110(3):870–6. doi: 10.1182/blood-2007-01-068528. [DOI] [PubMed] [Google Scholar]
- 32.Soni S, Bala S, Kumar A, Hanspal M. Changing pattern of the subcellular distribution of erythroblast macrophage protein (Emp) during macrophage differentiation. Blood Cells Mol Dis. 2007;38(1):25–31. doi: 10.1016/j.bcmd.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]