

Background: FPR2/ALX is activated by many ligands, including annexin A1 (AnxA1), which activates resolution circuits in inflammation.
Results: Cells transfected with FPR2/ALX and clones with domains swapped with FPR1 afforded identification of N-terminal and extracellular loop II domains as transducers of AnxA1 signaling.
Conclusion: We identified AnxA1 distinct domains of FPR2/ALX and unveiled potential specific signaling.
Significance: FPR2/ALX domain identification permits development of anti-inflammatory AnxA1 mimetics.
Keywords: 7-Helix Receptor, Annexin, Cell Biology, Inflammation, Leukocyte, Anti-inflammatory Targets, Lipoxin A4 Receptor, Resolution of Inflammation
Abstract
Understanding how proresolving agonists selectively activate FPR2/ALX is a crucial step in the clarification of proresolution molecular networks that can be harnessed for the design of novel therapeutics for inflammatory disease. FPR2/ALX, a G protein-coupled receptor belonging to the formyl peptide receptor (FPR) family, conveys the biological functions of a variety of ligands, including the proresolution mediators annexin A1 (AnxA1) and lipoxin A4, as well as the activating and proinflammatory protein serum amyloid A. FPR2/ALX is the focus of intense screening for novel anti-inflammatory therapeutics, and the small molecule compound 43 was identified as a receptor ligand. Here, we used chimeric FPR1 and FPR2/ALX clones (stably transfected in HEK293 cells) to identify the N-terminal region and extracellular loop II as the FPR2/ALX domain required for AnxA1-mediated signaling. Genomic responses were also assessed with domain-specific effects emerging, so the N-terminal region is required for AnxA1 induction of JAG1 and JAM3, whereas it is dispensable for modulation of SGPP2. By comparison, serum amyloid A non-genomic responses were reliant on extracellular loops I and II, whereas the small molecule compound 43 activated extracellular loop I with downstream signaling dependent on transmembrane region II. In desensitization experiments, the N-terminal region was dispensable for AnxA1-induced FPR2/ALX down-regulation in both the homologous and heterologous desensitization modes.
Introduction
The biological properties of G protein-coupled receptors are fundamental to the control of the host inflammatory reaction mounted against an insult, in terms of both promoting the response (e.g. chemokine-mediated leukocyte recruitment) and limiting its duration and intensity. This second side of the innate immune response is currently referred to as the resolution of inflammation phase. Within this context, an important role is played by the duo annexin 1 (AnxA1)3 and its receptor, formyl peptide receptor (FPR) type 2, whose acronym is FPR2/ALX because it also conveys the inhibitory signals induced by lipoxin A4 and resolvin D1 (1). FPR2/ALX is an intriguing receptor not only because it was the first one shown to mediate actions elicited by lipids, peptides, and proteins but also because it can mediate activating proinflammatory responses, such as those elicited by serum amyloid A (SAA) (2). A recent study by Ye and co-workers (3) chiefly demonstrated the ability of FPR2/ALX to modulate cell responsiveness in a ligand-biased fashion. Based on analogies with G protein-coupled receptors, several mechanistic hypotheses can be put forward to explain the versatility of this receptor, including homologous and/or heterologous dimerization, as well as the recruitment of ligand-specific signaling pathways (2, 4). Another plausible option is that ligands might activate specific receptor domains, thus promoting at least in part not overlapping downstream responses. As an example, the small lipid lipoxin A4 has been shown to activate FPR2/ALX by interacting with extracellular loop III and the associated transmembrane region (5).
This study was undertaken with a particular focus on the domains required for AnxA1-dependent receptor activation. We took advantage of recently characterized stable clones expressing chimeric receptors with different regions of FPR2/ALX and the cognate receptor FPR1 (6). Activation of these chimeric receptors by AnxA1 was analyzed and compared, in some experiments, with responses evoked by SAA and, for completion and to assess the potential impact these finding might have on drug discovery, by the small molecule agonist compound 43 (C43) (7).
EXPERIMENTAL PROCEDURES
Cell Culture
HEK293 cells stably transfected with FPR1 and FPR2/ALX were obtained as described (8) and maintained in culture with DMEM/F-12 supplemented with 10% FCS, gentamycin (50 μg/ml), nonessential amino acids (500 μg/ml), and Geneticin (200 μg/ml) solution. Fig. 1A shows the schematics for FPR2/ALX. HEK293 cells transfected with the FPR1-FPR2 chimera were obtained as described (6) and are listed in Fig. 1B. Cells were cultured in DMEM supplemented with 10% FCS, l-glutamine (500 μg/ml), penicillin/streptomycin (500 μg/ml), HEPES (1 m), and Geneticin (800 μg/ml) solution.
FIGURE 1.

Schematic representation of native and chimeric human FPR1 and FPR2/ALX clones used in the study. A, FPR2/ALX sequence with amino acids different from human FPR1 highlighted in red. B, FPR1-FPR2/ALX clones used in this study. Each clone is named alphabetically and has distinct domains of FPR1 replaced with the indicated FPR2/ALX amino acid sequences. C, typical Ca2+ flux responses evoked in FPR2/ALX cells upon treatment with AnxA1 or C43 (left panel) and with SAA (right panel). Data (means ± S.E. of five distinct experiments done in triplicate) are reported as percent ionomycin (1 μm) response.
Ligands and Inhibitors
AnxA1 was produced as described previously (9). SAA was purchased from PeproTech Laboratories (London, United Kingdom), and C43 was a generous gift from Amgen (Thousands Oaks, CA). The MEK1 inhibitor PD98059 and the calcium inhibitor BAPTA-AM were obtained from Cell Signaling Technologies (Hertfordshire, United Kingdom) and Calbiochem, respectively.
Human Polymorphonuclear Leukocyte Isolation
Peripheral blood was collected from healthy volunteers by intravenous withdrawal in 3.2% sodium citrate solution (1:10). All healthy volunteers gave oral and written consent, and cell separation was covered by Ethical Approval 05/Q0603/34 (East London and The City Research Ethics). Granulocytes (polymorphonuclear leukocytes) were isolated from blood via density centrifugation on a Histopaque 1119/1077 gradient (Sigma-Aldrich) according to the manufacturer's instructions and suspended in PBS containing 0.5% BSA. Contaminating erythrocytes were removed by lysis with cold Milli-Q water.
Calcium Mobilization Assay
HEK293 cells or freshly prepared polymorphonuclear leukocytes were incubated with 2 μm Fura 2-AM (Molecular Probes, Paisley, United Kingdom) and 1 μm Pluronic acid F-127 (Molecular Probes) in extracellular solution (13 mm glucose, 10 mm HEPES, 147 mm NaCl, 2 mm KCl, 1 mm MgCl2, and 2 mm CaCl2 at pH 7.3) at 37 °C for 1 h in the dark. Subsequently, cells were washed three times with the extracellular solution and added to 96-well plates prior to stimulation with agonists at the indicated concentrations. Ionomycin (1 μm) was used as a positive control. Mobilization of intracellular calcium was measured by recording the ratio of fluorescence emission at 510 nm after sequential excitation at 340 and 380 nm using the NOVOstar microplate reader (BMG LABTECH Ltd., Aylesbury, United Kingdom).
Western Blot Analysis
The protein content of the lysate was determined via Bradford protein assay (Bio-Rad). Cells lysates were boiled in 6× Laemmli buffer, analyzed by standard SDS-PAGE (12%), and electrophoretically transferred to PVDF (Millipore, Watford, United Kingdom). Membranes were incubated with rabbit anti-phospho-p44/42 MAPK (1:1000 dilution) and anti-total p44/42 (clone 137F5; 1:1000 dilution) antibodies (Cell Signaling Technologies) diluted in Tris-buffered saline containing 0.1% Tween 20 (TBST) and 1% BSA overnight at 4 °C. Membranes were washed for 30 min with TBST and incubated with HRP-conjugated goat anti-rabbit secondary antibody (1:2000 dilution; Dako, Cambridge, United Kingdom) for 2 h at room temperature. After stripping, membranes were also incubated with anti-β-actin antibody (1:10,000 dilution; Sigma-Aldrich) in TBST and 5% nonfat dry milk. Human actin was detected with HRP-conjugated goat anti-mouse antibody (1:5000 dilution). Proteins were then detected using an ECL detection kit and visualized on Hyperfilm (GE Healthcare).
Flow Cytometry Analysis
FPR1 and FPR2/ALX cell surface expression was measured by incubation with anti-FPR1 (R&D Systems, Abingdon, United Kingdom) or anti-FPR2 (Genovac, Brussels, Belgium) monoclonal antibody (mAb; 1:100 dilution in both cases); a final staining with a rabbit anti-mouse IgG (clone STAR9B; 1:200 dilution; Serotec) was then conducted. Flow cytometry analysis was performed by analyzing ≥10,000 events using a FACSCalibur flow cytometer (BD Biosciences) equipped with CellQuest software (BD Biosciences), followed by analysis using FlowJo (Version 9.2, TreeStar Inc., Stanford, CA). Results are reported as median fluorescence intensity units.
Real-time PCR Using SYBR Green I Dye
The assay was performed as shown previously (10). Briefly, RNA was extracted using an RNeasy kit (Qiagen 74104) according to the manufacturer's instructions. Total RNA was normalized at 5 μg/reaction. cDNA was produced using SuperScript III (Invitrogen). Samples were normalized to 100 ng of cDNA/well and loaded in duplicate for each gene using Power SYBR Green PCR Master Mix (AB 4367659). QuantiTect primer assays (Qiagen) were used: human GADPH (QT01192646), human SGPP2 (QT00041832), human JAG1 (QT00031948), and mouse JAM3 (QT00024997). GAPDH was used as an endogenous control. PCR ramping protocols were standardized for QuantiTect primer assay sets at 95 °C for 15 min, followed by 40 cycles at 94 °C for 15 s, 55 °C for 30 s, and 72 °C for 30 s.
Statistical Analysis
Data are expressed as means ± S.E. of experiments conducted at least three times. Data were analyzed using Student's t test (two groups) or one-way analysis of variance, followed by Dunnett's post hoc tests (more than two groups). In all cases, a p value of <0.05 was taken as significant.
RESULTS
FPR1, FPR2/ALX, and Chimeric Receptors
Human FPR1 and FPR2/ALX display ∼69% homology (2) as illustrated in Fig. 1A. To evaluate the contribution of single domains in FPR2/ALX activation, we used eight different chimeric receptors with domains swapped between FPR1 and FPR2/ALX, generated as described previously (6) and detailed in Fig. 1B. The degree of receptor expression, as demonstrated in Ref. 6, was assessed using specific mAbs against FPR1 or FPR2. Supplemental Fig. 1 reports these results and shows that chimeras C, D, and G were recognized by the anti-FPR2/ALX mAb, whereas all others clones were detected with the anti-FPR1 mAb (supplemental Fig. 1).
Calcium Mobilization Induced by FPR1 and FPR2/ALX Agonists
AnxA1 and its bioactive peptides are able to mobilize intracellular Ca2+ in primary polymorphonuclear leukocytes (11, 12) and cells transfected with FPR2/ALX and/or FPR1, respectively (12, 13). Using native FPR2/ALX, AnxA1 afforded concentration-dependent responses, with an EC50 value of ∼6 nm (Fig. 1C). SAA and C43 also produced canonical concentration-dependent responses, with EC50 values of ∼30 and ∼100 nm, respectively (Fig. 1C).
To compare the ligand-binding domains activated by the different agonists, Ca2+ mobilization in chimeric HEK293 cells was studied. AnxA1 (selected concentration, 10 nm) promoted Ca2+ flux in native FPR2/ALX, with similar responses in clones C, G, and H (all ranging from 40 to 60% of the maximal response), with little or no effect on FPR1-expressing cells and the other clones (Fig. 2A). The addition of SAA (100 nm) afforded ∼50% of the ionomycin response with both native FPR2/ALX cells and clones E, F, G, and H, whereas no responses were detected with the other clones, including clone B, which bears extracellular domain III of FPR2/ALX (Fig. 2B). The small molecule C43 (1 μm) produced a similar degree of response in FPR2/ALX cells and was active only in clones E and F (Fig. 2C). For completion, we also tested formyl-methionyl-leucyl-phenylalanine (300 nm; as selected from preliminary analyses (not shown)), which, as expected, was active only in native FPR1 (but not FPR2/ALX) cells; with respect to the clones analyzed, formyl-methionyl-leucyl-phenylalanine was particularly active in clones A and B (supplemental Fig. 2).
FIGURE 2.

Clone-specific calcium mobilization induced by AnxA1, SAA, and C43. HEK293 cells expressing native and chimeric clones were treated with AnxA1 (10 nm; A), SAA (0.1 μm; B), or C43 (1 μm; C) (concentrations selected from Fig. 1). Dashed lines indicate the degree of response produced with control CMV empty plasmid-transfected HEK293 cells in the presence of the respective ligand. Data (means ± S.E. of more than three distinct experiments done in triplicate) are reported as percent ionomycin (1 μm) response. Asterisks indicate similarity in responses between each clone (as indicated) and native FPR2/ALX-transfected cells. FPR1-FPR2/ALX clones are represented, with the part of the FPR2/ALX receptor insert (red) in the FPR1 structure (black) (D).
ERK Activation Induced by AnxA1 and the Two FPR2/ALX Agonists
Next, we determined whether the clone specificity that emerged so far could be confirmed with another readout for FPR2/ALX activation, such as phosphorylation of ERK (8, 12). As sometimes evident with transfected cells, the overexpressed receptor was, in some instances, already (partially) activated, yet the addition of AnxA1 (10 nm) provoked clear ERK phosphorylation in both FPR2/ALX and clones C, G, and H as monitored at 5 and 10 min post-addition (Fig. 3A). Clone E was also tested because it was unresponsive to AnxA1 for transient Ca2+ flux, and the same inactivity held true for phospho-ERK. For completion, we also tested SAA and C43. The positive association between Ca2+ flux and phospho-ERK was also observed for C43 (1 μm) because it activated both the native receptor and clones E and F (Fig. 3C). SAA induced ERK phosphorylation in FPR2/ALX cells and in clones C, G, and H (Fig. 3B).
FIGURE 3.

Clone-specific ERK1/2 phosphorylation induced by AnxA1, SAA, and C43. HEK293 cells expressing native and chimeric clones were treated with AnxA1 (10 nm; A), SAA (0.1 μm; B), or C43 (1 μm; C) (concentrations selected from Fig. 1) for 5 or 10 min as indicated, and the phosphorylation of ERK1/2 was monitored by Western blot analysis. Representative blots from three or more experiments with distinct cell preparations are shown. tERK, total ERK.
Genomic Modulation Induced by AnxA1
Next, we tested whether the rapid responses elicited by the FPR2/ALX domains expressed in clones C, G, and H upon application of AnxA1 could also lead to genomic responses. We have recently reported the specific profiles of gene expression post-AnxA1/FPR2 interaction, with a particular validation for the markedly up-regulated JAG1 (Jagged1) and SGPP2 (sphingosine-1-phosphate phosphatase type 2) and down-regulated JAM3 (junctional adhesion molecule-3) (10). The results obtained here with FPR2/ALX-HEK cells confirmed this profile, though JAG1 induction was relatively modest in this set of experiments (Fig. 4). Analysis of the clones indicated disparate outcomes: clone H afforded SGGP2 induction similar to the native receptor (Fig. 4A), whereas clone C was the most faithful for JAG1 induction (Fig. 4B). Inhibition of JAM3 (strongly evident for native FPR2/ALX) was replicated in all three clones (C, G, and H) (Fig. 4C). Using pharmacological inhibitors, we demonstrated that SGPP2 induction in clone H does not require calcium mobilization or ERK phosphorylation, pathways that, in contrast, are needed for JAM3 inhibition using clones C and H (supplemental Fig. 4).
FIGURE 4.
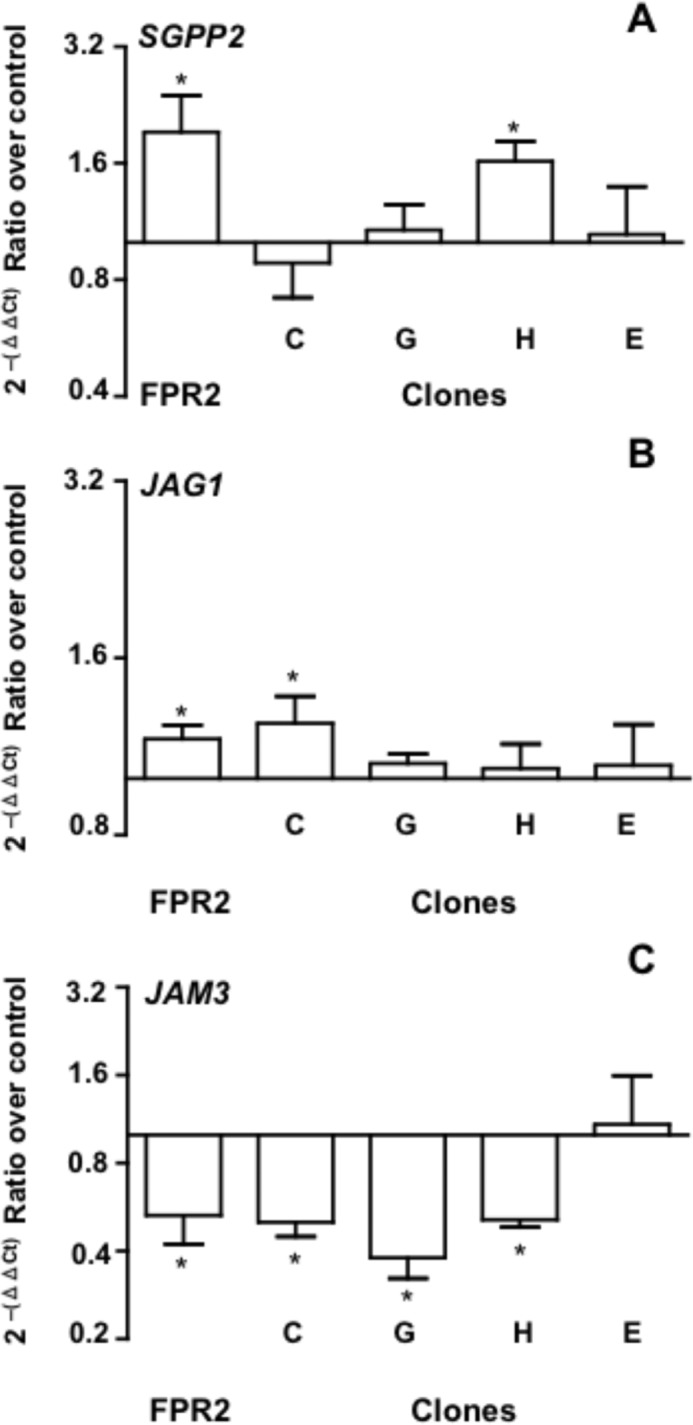
Clone-specific genomic responses evoked by AnxA1. Real-time PCR data for SGPP2 (A), JAG1 (B), and JAM3 (C) gene product expression in HEK293 cells expressing native and the indicated chimeric clones following 4 h of incubation with AnxA1 (0.5 μm). The response produced with control CMV empty plasmid-transfected HEK293 cells was taken as 1. Data (means ± S.E.) are from three distinct experiments performed in duplicate. *, p < 0.05 versus control CMV-transfected HEK293 cells.
AnxA1 and SAA Cross-receptor Desensitization
AnxA1 and SAA can inhibit each other's effects even when added to the same cell (3, 15). We attempted to identify the receptor domain required for this biological response. The data in Fig. 5A confirm the homologous and heterologous desensitization against AnxA1-induced Ca2+ flux in FPR2/ALX cells as exerted by preincubation with AnxA1 or SAA, respectively. The same effect was quantified with clones G and H, but not with clone C (Fig. 5B), indicating that the FPR2/ALX N-terminal domain is insufficient to afford desensitization of the AnxA1 response. Finally, to apply translation potential to these findings, which were produced with artificial receptors and transfected cells, we studied the cross-desensitization between AnxA1 and SAA in primary neutrophils. SAA (100 nm) desensitized AnxA1-induced Ca2+ flux (Fig. 6A), and conversely, AnxA1 attenuated the SAA response to a similar degree as the homologous desensitization (Fig. 6B). Of interest, when tested at 1 μm, C43 also provided modest desensitization of the responses in primary neutrophils (Fig. 6), whereas it was much more efficient in FPR2/ALX-transfected HEK cells (supplemental Fig. 3).
FIGURE 5.

N-terminal domain of FPR2/ALX is dispensable for homologous and heterologous desensitization. A, HEK293 cells expressing native and chimeric clones C, G, and H were treated with vehicle, AnxA1 (10 nm), and SAA (0.1 μm) for 5 min, followed by a second treatment with AnxA1 (10 nm). Data (means ± S.E. of more than three distinct experiments done in triplicate) are reported as percent ionomycin (1 μm) response. *, p < 0.05 versus vehicle pretreatment. B, selected clones (see Fig. 1B for details).
FIGURE 6.
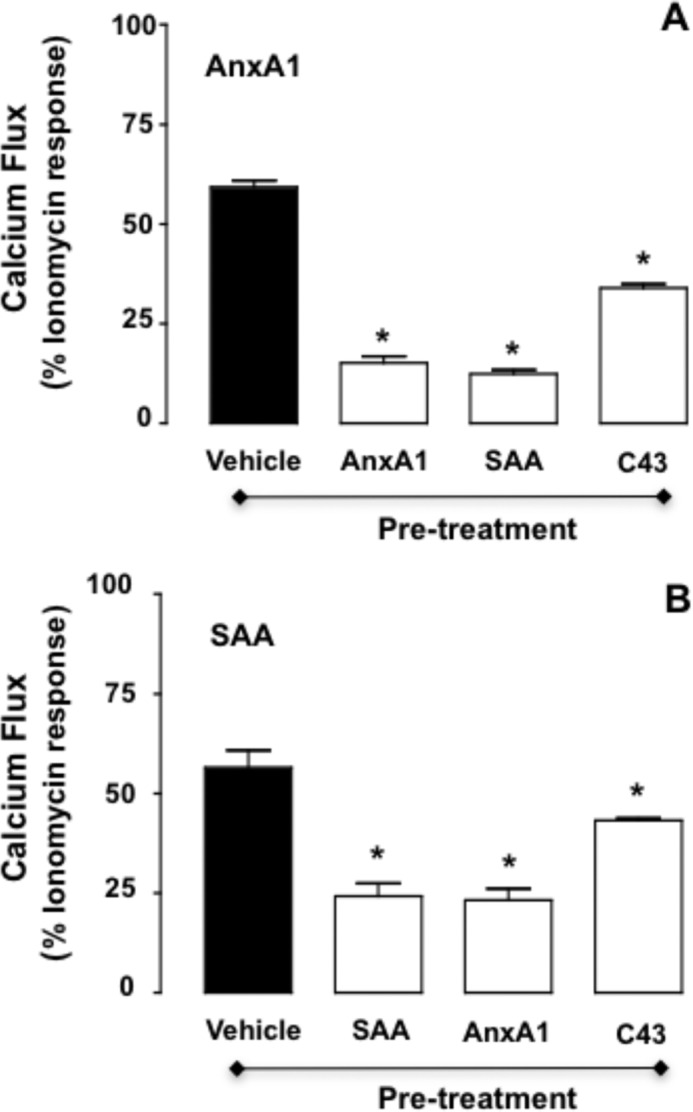
FPR2/ALX desensitization in human neutrophils upon agonist stimulations. Freshly isolated human neutrophils were treated with vehicle, AnxA1 (10 nm), SAA (0.1 μm), or C43 (1 μm) for 5 min prior to the addition of AnxA1 (10 nm; A) or SAA (0.1 μm; B). Data (means ± S.E. of more than three distinct experiments done in triplicate) are reported as percent ionomycin (1 μm) response. *, p < 0.05 versus native vehicle pretreatment.
DISCUSSION
FPR2/ALX is a G protein-coupled receptor that is able to recognize many ligands, often of a very different chemical nature (2, 5). Following binding with their agonists, FPR2/ALX undergoes a conformational change that allows functional interaction with Gαi1, Gαi2, and Gαi3 and association with G0, Gz, and Gα16 (16). This event triggers activation of a variety of signaling pathways, including fluxes in intracellular calcium and activation of phospholipases A2, C, and D, PI3K, and MAPK (17). Receptor activation would also cause rapid phosphorylation, leading to phospholipase C-mediated receptor desensitization and internalization (18). FPR2/ALX downstream activation of the MAPK pathway, in particular ERK1/2, regulates cell chemokinesis (19). However, a major conundrum in the biology of this receptor is, on one hand, the large numbers of putative ligands and, on the other hand, the apparent ability to elicit contrasting cellular responses in a ligand-biased fashion. The latter point has been chiefly demonstrated in a recent study in macrophages, where both putative “proinflammatory” (or rather activating) FPR2/ALX agonists (such as SAA) and inhibitory agonists (such as AnxA1 and lipoxin A4) were able to convey their message through this receptor and afford functional antagonism to each other (3). This study aimed to shed light on this heavily ligand-biased FPR2/ALX pharmacology by assessing the possibility that distinct receptor domains might interact with distinct ligands and potentially elicit different receptor downstream responses. We took advantage of well characterized stably transfected cells with varied FPR1-FPR2/ALX chimeric receptors (6) and began by assessing robust readouts, such as Ca2+ fluxes and ERK phosphorylation. An important control useful also for identifying the epitope recognized by commercially available mAbs was the overall amount of receptor expression. It appears that the anti-FPR2/ALX mAb likely binds to a tridimensional epitope formed between the receptor N-terminal domain and extracellular loops II and III. When the signal was positive, this mAb revealed similar receptor density on the cell surface as in native FPR2/ALX-transfected cells. All other chimeras were detected solely by anti-FPR1 mAb. It is evident that the mAbs are able somehow to penetrate the transmembrane domains because the anti-FPR2/ALX antibody was active in clone D (containing sequence 40–86 of the native receptor), whereas the anti-FPR1 mAb was not active in clone F (containing sequence 106–145 of the FPR2/ALX receptor).
The combined data produced with the clones for AnxA1 indicate that the N-terminal domain of FPR2/ALX is sufficient for this ligand to elicit rapid downstream responses, whereas the second extracellular loop is required to provoke more sustained changes, such as those leading to modulation of gene expression. Here, we selected three genes as readouts from our previous study (10) and confirmed modulation upon FPR2/ALX engagement; intriguingly, clone C, which could transduce Ca2+ and phospho-ERK, modulated JAG1 and JAM3, but clone H, with both extracellular loops II and III, was nearly as active as the native receptor in SGPP2 induction. This dichotomy of responses led us to propose the model discussed below and shown in Fig. 7. It is worth noting that, in a previous study using a similar approach but different chimeric strategy, the FPR2/ALX agonist lipoxin A4 was shown to interact with extracellular loop III (5).
FIGURE 7.

Schematic summary for agonist binding of FPR2/ALX. A, AnxA1 requires the N terminus and/or extracellular loop II to induce receptor activation. SAA interacts with extracellular loops I and II for FPR2/ALX, whereas the small molecule C43 penetrates into the extracellular loop I and transmembrane regions for full agonism. The reported binding site for lipoxin A4 (LXA4) is also indicated as reported (5). B, summary of AnxA1-evoked post-FPR2/ALX signaling. The FPR2/ALX N-terminal domain conveys AnxA1 signaling through calcium mobilization and ERK phosphorylation, leading to JAG1 up-regulation and JAM3 down-regulation. Extracellular loop II is more complex in its downstream AnxA1-evoked events. It participates in AnxA1-mediated JAM3 down-regulation through calcium flux and ERK phosphorylation, and it may involve unknown pathways for SGGP2 induction.
SAA is an acute-phase protein that is able to activate a variety of receptors, including FPR2/ALX (20, 21). Our data confirm the genuine agonistic activity of SAA on FPR2/ALX without any activation of FPR1. In terms of receptor domains, analysis of Ca2+ fluxes and ERK phosphorylation indicates an interaction with extracellular loops I and II, whereas the N-terminal and extracellular loop III domains are dispensable for the Ca2+ response. Concerning C43, in line with the ability of small molecules to “penetrate” inside G protein-coupled receptor loops (22), this FPR2/ALX agonist (7, 23) did not elicit Ca2+ flux responses in FPR1-transfected cells and was active in native FPR2/ALX, as well as in clones E and F; therefore, extracellular loop I, transmembrane region III, and intracellular loop II contain the binding site for C43. It should be noted here that C43 produces anti-inflammatory effects when given in vivo (7), which are lost in Fpr2/Fpr3 null mice (15); however, other studies have claimed an involvement of Fpr1 (the mouse counterpart of human FPR1) in its anti-inflammatory profile (24, 25), yet activation of human FPR1 could not be detected in these experimental settings with the stably transfected cells. Next, studies should establish whether C43 permits FPR1 and FPR2/ALX dimerization.
We completed this investigation by determining FPR2/ALX homologous and heterologous desensitization because they had already been reported for AnxA1 and SAA (3) and also because of the functional consequences these processes may have in modulating cell responsiveness in inflammatory settings (12, 15). It emerged that AnxA1 engagement of FPR2/ALX extracellular loop II is required for desensitization to occur, as determined by Ca2+ readout. As shown recently (26), the C terminus (present in clone H) may also have an important role in FPR2/ALX desensitization. Analyses conducted with clone C, which contained solely the FPR2/ALX N-terminal region linked to the nearly full FPR1 sequence, corroborated the specificity of full-length AnxA1 for FPR2/ALX and its inability to engage FPR1. This is in contrast to the ability of short AnxA1-derived peptides to activate all members of the human FPR family (12, 27). We have recently discussed how this dichotomy of protein/peptide behavior may be of biological significance in the context of resolution of inflammation (28).
In summary, we studied the domains of FPR2/ALX engaged by AnxA1 in comparison with SAA, which elicits opposite cellular responses (3), and the small molecule C43 in view of the potential exploitation of the pharmacology of this receptor for the development of novel anti-inflammatory therapeutics. The picture that emerges is illustrated in Fig. 7, which shows that AnxA1 can interact with the N-terminal domain and extracellular loop II. Because of the large structure of this protein (29) and the fact that, in the presence of calcium, it undergoes a conformational change with exposure of its N-terminal domain (30, 31), we hypothesize that at least two FPR2/ALX sites are required to accommodate this agonist (Fig. 7), a hypothesis proposed quite a few years ago when structural data were scarce (32). A recent study proposed a three-point binding model for small molecule ligand interaction with FPRs, for FPR2/ALX (33), confirming original studies with FPR (14). These binding sites are within the non-conserved amino acid residues 84, 85 (point 1), 163 (point 2), and 284 (point 3) (14, 33). Also in our model, AnxA1 binds the FPR2/ALX receptor in the binding pocket that comprises residue 163 (present in clone G). Residues 84, 85, and 163 are in the FPR2/ALX-binding region required by C43 and SAA to induce receptor activation.
In conclusion, information has been obtained regarding the ligand-specific domains required for classical FPR2/ALX readouts, such as Ca2+ fluxes and ERK phosphorylation. Because of the emerging importance of FPR2/ALX in the regulation of the inflammatory process, we reasoned that a better appreciation of the binding domains and the ensuing signaling characteristic of the interaction with the endogenous agonist AnxA1 is not only of biological importance but will also be significant in the development of AnxA1 mimetics able to engage this effector of resolution.
Supplementary Material
This work was supported by Wellcome Trust Programme 086867/Z/08. This work forms part of the research themes contributing to the translational research portfolio of Barts and The London Cardiovascular Biomedical Research Unit, which is supported and funded by the National Institute for Health Research.

This article contains supplemental Figs. 1–4.
- AnxA1
- annexin 1
- FPR
- formyl peptide receptor
- SAA
- serum amyloid A
- C43
- compound 43
- BAPTA-AM
- 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid acetoxymethyl ester
- mAb
- monoclonal antibody.
REFERENCES
- 1. Serhan C. N., Savill J. (2005) Resolution of inflammation: the beginning programs the end. Nat. Immunol. 6, 1191–1197 [DOI] [PubMed] [Google Scholar]
- 2. Ye R. D., Boulay F., Wang J. M., Dahlgren C., Gerard C., Parmentier M., Serhan C. N., Murphy P. M. (2009) International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol. Rev. 61, 119–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li Y., Cai L., Wang H., Wu P., Gu W., Chen Y., Hao H., Tang K., Yi P., Liu M., Miao S., Ye D. (2011) Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor 2. Oncogene 30, 3887–3899 [DOI] [PubMed] [Google Scholar]
- 4. Dufton N., Perretti M. (2010) Therapeutic anti-inflammatory potential of formyl peptide receptor agonists. Pharmacol. Ther. 127, 175–188 [DOI] [PubMed] [Google Scholar]
- 5. Chiang N., Fierro I. M., Gronert K., Serhan C. N. (2000) Activation of lipoxin A4 receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation. J. Exp. Med. 191, 1197–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Le Y., Ye R. D., Gong W., Li J., Iribarren P., Wang J. M. (2005) Identification of functional domains in the formyl peptide receptor-like 1 for agonist-induced cell chemotaxis. FEBS J. 272, 769–778 [DOI] [PubMed] [Google Scholar]
- 7. Bürli R. W., Xu H., Zou X., Muller K., Golden J., Frohn M., Adlam M., Plant M. H., Wong M., McElvain M., Regal K., Viswanadhan V. N., Tagari P., Hungate R. (2006) Potent hFPRL1 (ALXR) agonists as potential anti-inflammatory agents. Bioorg. Med. Chem. Lett. 16, 3713–3718 [DOI] [PubMed] [Google Scholar]
- 8. Hayhoe R. P., Kamal A. M., Solito E., Flower R. J., Cooper D., Perretti M. (2006) Annexin 1 and its bioactive peptide inhibit neutrophil-endothelium interactions under flow: indication of distinct receptor involvement. Blood 107, 2123–2130 [DOI] [PubMed] [Google Scholar]
- 9. Pederzoli-Ribeil M., Maione F., Cooper D., Al-Kashi A., Dalli J., Perretti M., D'Acquisto F. (2010) Design and characterization of a cleavage-resistant annexin A1 mutant to control inflammation in the microvasculature. Blood 116, 4288–4296 [DOI] [PubMed] [Google Scholar]
- 10. Renshaw D., Montero-Melendez T., Dalli J., Kamal A., Brancaleone V., D'Acquisto F., Cirino G., Perretti M. (2010) Downstream gene activation of the receptor ALX by the agonist annexin A1. PLoS ONE 5, e12771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Solito E., Kamal A., Russo-Marie F., Buckingham J. C., Marullo S., Perretti M. (2003) A novel calcium-dependent pro-apoptotic effect of annexin 1 on human neutrophils. FASEB J. 17, 1544–1546 [DOI] [PubMed] [Google Scholar]
- 12. Walther A., Riehemann K., Gerke V. (2000) A novel ligand of the formyl peptide receptor: annexin 1 regulates neutrophil extravasation by interacting with the FPR. Mol. Cell 5, 831–840 [DOI] [PubMed] [Google Scholar]
- 13. Ernst S., Lange C., Wilbers A., Goebeler V., Gerke V., Rescher U. (2004) An annexin 1 N-terminal peptide activates leukocytes by triggering different members of the formyl peptide receptor family. J. Immunol. 172, 7669–7676 [DOI] [PubMed] [Google Scholar]
- 14. Lala A., Gwinn M., De Nardin E. (1999) Human formyl peptide receptor function role of conserved and non-conserved charged residues. Eur. J. Biochem. 264, 495–499 [DOI] [PubMed] [Google Scholar]
- 15. Dufton N., Hannon R., Brancaleone V., Dalli J., Patel H. B., Gray M., D'Acquisto F., Buckingham J. C., Perretti M., Flower R. J. (2010) Anti-inflammatory role of the murine formyl peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J. Immunol. 184, 2611–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Migeotte I., Communi D., Parmentier M. (2006) Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 17, 501–519 [DOI] [PubMed] [Google Scholar]
- 17. Selvatici R., Falzarano S., Mollica A., Spisani S. (2006) Signal transduction pathways triggered by selective formyl peptide analogues in human neutrophils. Eur. J. Pharmacol. 534, 1–11 [DOI] [PubMed] [Google Scholar]
- 18. Le Y., Wetzel M. A., Shen W., Gong W., Rogers T. J., Henderson E. E., Wang J. M. (2001) Desensitization of chemokine receptor CCR5 in dendritic cells at the early stage of differentiation by activation of formyl peptide receptors. Clin. Immunol. 99, 365–372 [DOI] [PubMed] [Google Scholar]
- 19. Wenzel-Seifert K., Hurt C. M., Seifert R. (1998) High constitutive activity of the human formyl peptide receptor. J. Biol. Chem. 273, 24181–24189 [DOI] [PubMed] [Google Scholar]
- 20. Liang T. S., Wang J. M., Murphy P. M., Gao J. L. (2000) Serum amyloid A is a chemotactic agonist at FPR2, a low-affinity N-formyl peptide receptor on mouse neutrophils. Biochem. Biophys. Res. Commun. 270, 331–335 [DOI] [PubMed] [Google Scholar]
- 21. He R., Sang H., Ye R. D. (2003) Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood 101, 1572–1581 [DOI] [PubMed] [Google Scholar]
- 22. Baker J. G., Hill S. J. (2007) Multiple GPCR conformations and signaling pathways: implications for antagonist affinity estimates. Trends Pharmacol. Sci. 28, 374–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krishnamoorthy S., Recchiuti A., Chiang N., Yacoubian S., Lee C. H., Yang R., Petasis N. A., Serhan C. N. (2010) Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. U.S.A. 107, 1660–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sogawa Y., Shimizugawa A., Ohyama T., Maeda H., Hirahara K. (2009) The pyrazolone originally reported to be a formyl peptide receptor (FPR) 2/ALX-selective agonist is instead an FPR1 and FPR2/ALX dual agonist. J. Pharmacol. Sci. 111, 317–321 [DOI] [PubMed] [Google Scholar]
- 25. Sogawa Y., Ohyama T., Maeda H., Hirahara K. (2011) Formyl peptide receptor 1 and 2 dual agonist inhibits human neutrophil chemotaxis by the induction of chemoattractant receptor cross-desensitization. J. Pharmacol. Sci. 115, 63–68 [DOI] [PubMed] [Google Scholar]
- 26. Rabiet M. J., Macari L., Dahlgren C., Boulay F. (2011) N-Formyl peptide receptor 3 (FPR3) departs from the homologous FPR2/ALX receptor with regard to the major processes governing chemoattractant receptor regulation, expression at the cell surface, and phosphorylation. J. Biol. Chem. 286, 26718–26731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dalli J., Montero-Melendez T., McArthur S., Perretti M. (2012) Annexin A1 N-terminal derived peptide Ac2-26 exerts chemokinetic effects on human neutrophils. Front. Pharmacol. 3, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brancaleone V., Dalli J., Bena S., Flower R. J., Cirino G., Perretti M. (2011) Evidence for an anti-inflammatory loop centered on polymorphonuclear leukocyte formyl peptide receptor 2/lipoxin A4 receptor and operative in the inflamed microvasculature. J. Immunol. 186, 4905–4914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rosengarth A., Gerke V., Luecke H. (2001) X-ray structure of full-length annexin 1 and implications for membrane aggregation. J. Mol. Biol. 306, 489–498 [DOI] [PubMed] [Google Scholar]
- 30. Rosengarth A., Luecke H. (2003) A calcium-driven conformational switch of the N-terminal and core domains of annexin A1. J. Mol. Biol. 326, 1317–1325 [DOI] [PubMed] [Google Scholar]
- 31. Gerke V., Creutz C. E., Moss S. E. (2005) Annexins: linking Ca2+ signaling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 6, 449–461 [DOI] [PubMed] [Google Scholar]
- 32. Gao J. L., Murphy P. M. (1993) Species and subtype variants of the N-formyl peptide chemotactic receptor reveal multiple important functional domains. J. Biol. Chem. 268, 25395–25401 [PubMed] [Google Scholar]
- 33. Kirpotina L. N., Khlebnikov A. I., Schepetkin I. A., Ye R. D., Rabiet M. J., Jutila M. A., Quinn M. T. (2010) Identification of novel small molecule agonists for human formyl peptide receptors and pharmacophore models of their recognition. Mol. Pharmacol. 77, 159–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.