

Abstract
Cell surface proteolysis is essential for communication between cells and results in the shedding of membrane-protein ectodomains. However, physiological substrates of the contributing proteases are largely unknown. We developed the secretome protein enrichment with click sugars (SPECS) method, which allows proteome-wide identification of shedding substrates and secreted proteins from primary cells, even in the presence of serum proteins. SPECS combines metabolic glycan labelling and click chemistry-mediated biotinylation and distinguishes between cellular and serum proteins. SPECS identified 34, mostly novel substrates of the Alzheimer protease BACE1 in primary neurons, making BACE1 a major sheddase in the nervous system. Selected BACE1 substrates—seizure-protein 6, L1, CHL1 and contactin-2—were validated in brains of BACE1 inhibitor-treated and BACE1 knock-out mice. For some substrates, BACE1 was the major sheddase, whereas for other substrates additional proteases contributed to total substrate shedding. The new substrates point to a central function of BACE1 in neurite outgrowth and synapse formation. SPECS is also suitable for quantitative secretome analyses of primary cells and may be used for the discovery of biomarkers secreted from tumour or stem cells.
Keywords: Alzheimer’s disease, bioorthogonal chemistry, click chemistry, proteases, quantitative proteomics
Introduction
Proteolysis is an irreversible post-translational modification mediated by over 500 different proteases in man. Proteases control the function or mediate the degradation of virtually all proteins in the cell, but the biological functions of many proteases are unknown, because no or only few physiological substrates have been identified. This is particularly true for a large class of membrane-bound proteases, referred to as sheddases, which mostly cleave single-span membrane proteins at the extracellular surface of cellular membranes. This process is termed as ectodomain shedding and is essential for the communication between cells (Reiss and Saftig, 2009; Bai and Pfaff, 2011; Lichtenthaler et al, 2011). Shedding is involved in various physiological and pathophysiological conditions, including Alzheimer’s disease.
One of the sheddases is the aspartyl protease BACE1 (β-secretase), which is a key drug target for Alzheimer’s disease, as it mediates the shedding of amyloid precursor protein (APP) and catalyses the first step in the generation of the pathogenic Aβ peptide (Vassar et al, 2009). Possible side effects of BACE1 inhibition in patients may result from a reduced cleavage of additional, largely unknown physiological BACE1 substrates. Besides APP, BACE1 also cleaves neuregulin-1 and contributes to myelination in the peripheral nervous system (Hu et al, 2006; Willem et al, 2006). Additionally, several new phenotypes of BACE1-deficient mice were reported recently, such as schizophrenic symptoms, increased mortality, epileptic seizures, hyperactivity, anxiety, impaired axon guidance and protection against diet-induced obesity (Harrison et al, 2003; Dominguez et al, 2005; Laird et al, 2005; Savonenko et al, 2008; Wang et al, 2008; Hu et al, 2010; Farah et al, 2011; Meakin et al, 2011; Rajapaksha et al, 2011). These phenotypes mostly affect brain and pancreas, where BACE1 expression is highest (Vassar et al, 1999), but it remains unclear which substrates are affected in these tissues.
The secretome of a cell comprises soluble, secreted proteins and the membrane protein ectodomains proteolytically released by sheddases (sheddome). Proteomic identification of secretome proteins from the conditioned medium of cells is in principle possible by mass spectrometry, but has been difficult due to three fundamental limitations (Makridakis and Vlahou, 2010). First, secretome proteins have low concentrations in conditioned media. Second, the use of media supplements such as fetal calf serum or the neuronal supplement B27 introduces albumin and other serum proteins at concentrations (up to 5 g/l) much higher than the secretome proteins (Price and Brewer, 2001). Third, secretome proteins can be masked by highly abundant cytosolic proteins released from broken or apoptotic cells. Thus, the mass spectrometer used for protein identification predominantly identifies albumin, other serum proteins and cytosolic proteins, but not the cell-derived secretome proteins. To circumvent these limitations, previous studies used serum- or protein-free cell culture conditions. However, cellular stress and incompatibility with the culture of many cell types are major drawbacks of this approach, making identification and quantification of secreted proteins in primary cells, such as neurons, impossible. Additionally, many sheddases are less active in the absence of serum. As a consequence, the desired protease is frequently overexpressed or added exogenously in vitro, as carried out for example for BACE1, meprin β and MT1-MMP (Tam et al, 2004; Hemming et al, 2009; Jefferson et al, 2011). While this type of approach can demonstrate which substrates may in principle be cleaved by a protease, false positive substrate identification is a major risk of protease overexpression, for example because of mislocalization of the protease (Huse et al, 2002).
Here, we developed a novel technique for quantitative proteomics of cell culture supernatants containing serum or albumin, called secretome protein enrichment with click sugars (SPECS), which solves the challenges mentioned above. SPECS distinguishes between secretome proteins and exogenous serum proteins within the conditioned medium. We used SPECS to determine the secretome of human embryonic kidney 293 (HEK293T) cells and of primary, murine neurons in the presence of serum proteins. Additionally, SPECS was used to identify novel, physiological BACE1 substrates in primary neurons. Selected BACE1 substrates—seizure-protein 6, L1, CHL1 and contactin-2—were validated in brains of BACE1 inhibitor-treated and BACE1 knock-out mice.
Results
Development of the SPECS method
SPECS exploits the fact that the majority of secreted proteins (66%) and potential shedding substrates (87% of type I and type II transmembrane proteins) is glycosylated as annotated in Uniprot. SPECS consists of metabolic labelling of cellular glycoproteins with azido sugars followed by copper-free click chemistry-mediated biotinylation of cellular, but not of serum glycoproteins (Figure 1A). The click-chemistry reaction consists of the bioorthogonal, chemical [3+2] cycloaddition of an azide moiety with a strained cycloalkyne (Jewett and Bertozzi, 2010). We used the biotinylated, strained cycloalkyne dibenzylcyclooctyne (DBCO-PEG12-biotin) (Figure 1B) and tetraacetyl-N-azidoacetyl-mannosamine (ManNAz), which is metabolically converted to N-azidoacetyl-sialic acid and incorporated into terminal positions in N- and O-linked glycans (Sletten and Bertozzi, 2011). Due to the lack of an active transport of N-acetylamino sugars across the plasma membrane of mammalian cells, the hydroxy-groups of the sugar are peracetylated to permit passive diffusion of the sugars into the cytosol, where the acetyl groups are cleaved off by cellular esterases.
Figure 1.

Overview and validation of SPECS method. (A) Detailed description of the work flow of the SPECS method including a timeline. (B) Schematic representation of the click reaction between the azide group (blue) of 1,3,4,6-acetyl-N-acetyl-azido-mannosamine (ManNAz) and the strained alkyne (red) of dibenzylcycloocytne-PEG12-Biotin. (C) APP and APLP2 shedding were analysed in the presence of the BACE1 inhibitor C3 and ManNAz or DMSO as a control. Subsequently, click-chemistry reaction was performed in the conditioned medium. Full-length APP (APPfl) and APLP2 (APLP2fl) in the lysates as well as secreted APP and APLP2 (sAPPtotal, sAPPβ, sAPLP2) were detected by immunoblot. The analysis shows no significant changes of the shedding of APP and APLP2 upon addition of ManNAz. Biotinylated proteins (specific bands and broad smear) were detected with Streptavidin-HRP only when ManNAz was present. Acetylated (Ac) tubulin serves as a loading control. (D) Purification of glycoproteins by streptavidin pull down after click-chemistry reaction. Left: aliquot of conditioned medium is directly loaded (input). Right: upon streptavidin pull down, sAPPtotal is enriched by about two-fold, whereas serum albumin in the coomassie gel is reduced over 50-fold, leading to a specific enrichment of the glycosylated sAPPtotal by over 100-fold. (E) Distribution of glycoprotein types among all glycoproteins identified in the HEK293T (293T) secretome and the neuronal secretome. Only such proteins were included, which were detected by at least two peptides. Detailed listing of identified proteins and their topology is in Supplementary Tables 1, 2, 5 and 6.
After ManNAz labelling the conditioned medium contained the labelled ectodomains released by shedding together with cell-derived secreted proteins (Figure 1A). Free ManNAz was removed by ultrafiltration followed by in-vitro click reaction, resulting in biotinylation of secretome proteins (Figure 1A). Subsequent ultrafiltration removed excessive biotinylated cyclooctyne. After streptavidin pull down, the protein samples were separated by SDS–PAGE, followed by in-gel trypsinization. The resulting peptides were analysed by nanoLC coupled to high-resolution mass spectrometry. Peptide identification was done by Andromeda. The SPECS method can be carried out with little hands-on time within 6 days, which includes 2 days of metabolic sugar labelling and 2 days of mass spectrometric analysis.
To exclude any interference of metabolic glycan labelling with cellular physiology, we investigated in primary neurons the known shedding of the APP and its homologue, the amyloid precursor-like protein 2 (APLP2), by the protease BACE1, which is a main drug target in Alzheimer’s disease (Vassar et al, 2009) and is also involved in myelination (Hu et al, 2006; Willem et al, 2006). The presence of ManNAz neither altered shedding of APP and APLP2 or its response to the specific BACE1 inhibitor C3 (Stachel et al, 2004; Figure 1C) nor did it affect neuronal viability, in agreement with a previous study (Almaraz et al, 2012). Secreted APP (sAPPtotal) was enriched by over 100-fold relative to albumin (Figure 1D). These results demonstrate that SPECS works successfully and is compatible with primary cells, such as neurons.
Determination of cellular secretomes and sheddomes
Next, we used SPECS to determine the secretome of neurons and of HEK293T cells, an immortalized cell line. HEK293T cells were grown in the presence of serum with or without ManNAz. In all, 254 proteins were specifically identified in the presence but not in the absence of ManNAz and constitute the secretome of HEK293T cells. The secretome comprised 142 membrane protein ectodomains, which constitute the sheddome, and 112 secreted proteins (Figure 1E; Supplementary Tables 1, 2, 3 and 4). To determine the neuronal secretome, primary E15/E16 wild-type neurons were cultured in the presence of ManNAz. Additionally, the neurons were incubated with or without the BACE1 inhibitor C3 to determine which of the secretome proteins are BACE1 substrates (next paragraph). Relative label-free protein quantification of identified unique protein groups was performed using the MaxQuant software suite. In all, 427 glycoproteins were identified in total, with 283 of them being identified in at least four out of five experiments without inhibitor. These 283 proteins comprise 97 secreted proteins and 186 shed membrane proteins (sheddome) and constitute the secretome of primary neurons (Figure 1E; Supplementary Tables 5 and 6, raw data and peptides are given in Supplementary Tables 7 and 8). While a previous study identified 34 proteins as the neuronal secretome using serum-free conditions (Thouvenot et al, 2008), SPECS identified 283 proteins with high sequence coverage, demonstrating the advantage of the SPECS method. It is possible that neurons and HEK293T cells secrete additional proteins, in particular low-abundant proteins below the detection limit of the SPECS method or proteins with a molecular mass <30 kDa, which would be lost during the filtration steps of the SPECS method. However, smaller molecular mass cut-off columns may also be used, which would allow detection of small-sized glycosylated secretome proteins. In all, 112 proteins of the neuronal secretome (47%) were shared with the HEK293T cell secretome, whereas the remaining proteins include many proteins with neuronal functions, such as neuroligins and LINGO-1 (Supplementary Table 5).
Identification of neuronal BACE1 sheddome
While the majority of the neuronal secretome was not affected by C3 treatment, 40 proteins showed a reduced shedding or secretion (Table I, top part). Proteins were included into this hit list, if their levels were quantifiable in at least four out of the five biological replicates and had a variance score ≤0.35 in the C3-treated samples. The 40 proteins include 34 membrane proteins (Table I), which we refer to as the neuronal BACE1 substrates (BACE1 sheddome), even though it remains possible that the shedding of some substrates may have been reduced indirectly. In all, 8 of the 34 proteins had previously been identified as candidate substrates in BACE1 overexpressing cells (Hemming et al, 2009). The 23 remaining proteins besides APP and its homologues are novel BACE1 substrates.
Table 1. Changes in protein secretion and shedding upon BACE1 inhibitor C3 treatment.
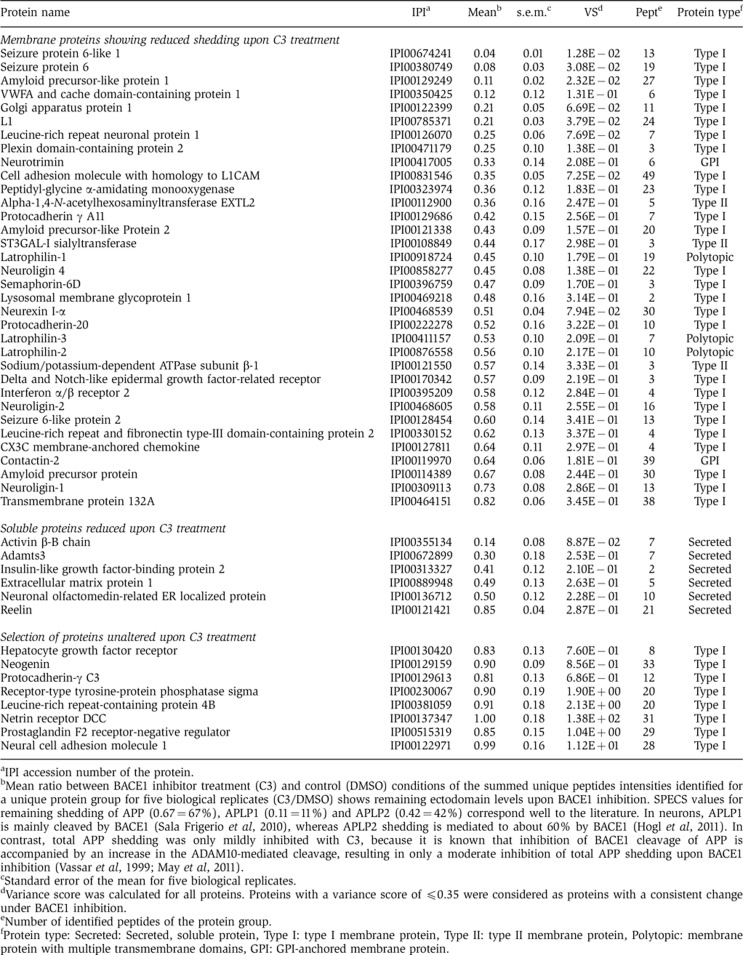
aIPI accession number of the protein.
bMean ratio between BACE1 inhibitor treatment (C3) and control (DMSO) conditions of the summed unique peptides intensities identified for a unique protein group for five biological replicates (C3/DMSO) shows remaining ectodomain levels upon BACE1 inhibition. SPECS values for remaining shedding of APP (0.67=67%), APLP1 (0.11=11%) and APLP2 (0.42=42%) correspond well to the literature. In neurons, APLP1 is mainly cleaved by BACE1 (Sala Frigerio et al, 2010), whereas APLP2 shedding is mediated to about 60% by BACE1 (Hogl et al, 2011). In contrast, total APP shedding was only mildly inhibited with C3, because it is known that inhibition of BACE1 cleavage of APP is accompanied by an increase in the ADAM10-mediated cleavage, resulting in only a moderate inhibition of total APP shedding upon BACE1 inhibition (Vassar et al, 1999; May et al, 2011).
cStandard error of the mean for five biological replicates.
dVariance score was calculated for all proteins. Proteins with a variance score of ≤0.35 were considered as proteins with a consistent change under BACE1 inhibition.
eNumber of identified peptides of the protein group.
fProtein type: Secreted: Secreted, soluble protein, Type I: type I membrane protein, Type II: type II membrane protein, Polytopic: membrane protein with multiple transmembrane domains, GPI: GPI-anchored membrane protein.
The protein list of BACE1 substrates includes APP and its homologues APLP1 and APLP2 (Table I), which are all three known BACE1 substrates and validate the SPECS analysis. Importantly, the extent to which the shedding of APP and its homologues was reduced (Table I) corresponds well to previous results obtained by quantitative immunoblots in neurons or brain (Vassar et al, 1999; Sala Frigerio et al, 2010; Hogl et al, 2011; May et al, 2011). For example, total APP shedding was only mildly inhibited with C3, because inhibition of BACE1 cleavage of APP has been shown to be accompanied by an increase in the ADAM10-mediated cleavage, resulting in only a moderate inhibition of total APP shedding upon BACE1 inhibition (Vassar et al, 1999; May et al, 2011). These results demonstrate that SPECS is well suited for the quantitative analysis of protein shedding. For other proteins on the BACE1 sheddome list shedding was reduced by about 20% (transmembrane protein 132A) to over 95% (seizure 6-like protein) (Table I, top part), suggesting that for some substrates (transmembrane protein 132A) BACE1 only contributes to a small extent to total shedding, whereas other substrates are nearly exclusively cleaved by BACE1 in neurons.
Peptides identified from individual membrane proteins were exclusively derived from their ectodomains (Supplementary Figure 1A, shown in yellow), indicating that they derive from true ectodomain shedding and not from full-length proteins released by broken cells. Additionally, for three BACE1 substrates (APP, APLP1, seizure 6-like protein 1 (SEZ6L1)) semi-tryptic peptides were identified (Supplementary Figure 1A, shown in green), which were reduced after BACE1 inhibition (Supplementary Figure 1B), suggesting that they derive from direct BACE1 cleavage. Indeed, the peptide from APP corresponds exactly to the known BACE1 cleavage site (Supplementary Figure 1B) and demonstrates that SPECS allows cleavage site determination.
Six of the forty proteins were soluble proteins and showed an inhibition of secretion by about 20–90% (Table I, middle part), including the TGFβ superfamily member activin β and insulin-like growth factor-binding protein 2. Because these are known soluble proteins, their reduced secretion is likely to be a secondary consequence of BACE1 inhibition. However, they may be useful as diagnostic markers to monitor the efficacy of BACE1 inhibitors in Alzheimer’s patients.
Validation of BACE1 substrates in primary neurons
To further validate the SPECS data, the shedding inhibition by C3 was analysed in the absence of ManNAz and quantified by immunoblot for four novel BACE1 substrates besides APP, APLP1 and APLP2. For most of the novel substrates, no antibodies are available which allow detection of the endogenous, shed ectodomain by immunoblot. We chose four proteins, where suitable antibodies were available and which showed mild, moderate and strong shedding inhibition by C3 in the SPECS measurement (Table I, top part). The quantitative comparison of SPECS and immunoblots yielded the following shedding inhibition upon C3 treatment: 12.3/7.8% (SPECS/immunoblot) for seizure protein 6 (SEZ6) (Figure 2A and quantification in Figure 2B); 23/21% for cell adhesion protein L1; 49/35% for close homologue of L1 (CHL1); 56/64% for contactin-2, a GPI-anchored protein. The values obtained by immunoblots were similar to the SPECS measurements and demonstrate the quantitative accuracy of SPECS. The reduced shedding was accompanied by increased levels of the full-length proteins in the cell lysate (Figure 2A and quantification in Figure 2B), showing that C3 reduced substrate shedding and not simply substrate expression. Interestingly, a strong reduction in shedding was not always accompanied by a strong increase in full-length protein levels in the lysate, suggesting that additional cellular mechanisms besides shedding control the full-length protein levels.
Figure 2.

Validation of BACE1 substrates in primary neurons. (A) Primary neurons were treated with DMSO or with the BACE1 inhibitor C3 overnight. Sez6, CHL1, L1 and contactin-2 showed reduced shedding into the cell supernatant (Sup) and accumulation of their membrane-tethered precursors in the lysate (Lys) after treatment with C3. The specific BACE1 cleavage product APPsβ served as internal control for BACE1 inhibition. Additionally, proteolytic processing of the APP family and of the two negative controls N-Cadherin and PTPRF was investigated. The increase in the cell lysate was different for the individual BACE1 substrates and did not linearly depend on the extent of shedding reduction in the conditioned medium. Acetylated (Ac) tubulin served as a loading control. (B) Quantification of the C3/DMSO ratio of (A) for six independent experiments for supernatant and cell lysate of Sez6, CHL1, L1 and contactin-2. Quantification of the C3/DMSO ratio of the SPECS method (taken from Table I) is also included in order to show the correlation between immunoblot and SPECS results. Quantification includes the positive controls APP, APLP1, APLP2 and the negative controls PTPRF and N-Cadherin, which are not BACE1 substrates. Given are mean and s.e.m. (n=6). Figure source data can be found with the Supplementary data.
To rule out a C3 inhibitor off-target effect, the shedding analysis was repeated in primary neurons from E15/E16 BACE1 knock-out mouse brains (Figure 3A). For all four investigated substrates, shedding reduction in the supernatant and accumulation in the cell lysate was very similar to the C3-treated neurons (quantification in Figure 3B).
Figure 3.

Validation of BACE1 substrates in BACE1 knock-out neurons and cell-surface labelling of contactin-2 upon BACE1 inhibition. (A) Analysis as in Figure 2, but using wild-type and BACE1 knock-out neurons. (B) Quantification of the BACE1−/−/Wt ratio of (A) for six independent experiments for supernatant and cell lysate of Sez6, CHL1, L1 and contactin-2. Quantification includes the positive controls APP, APLP1, APLP2 and the negative controls PTPRF and N-Cadherin. (C) Cell-surface staining of contactin-2 in primary neurons. Under control conditions (DMSO) sparse labelling of discrete puncta was visible besides background labelling of the secondary antibody of the cell soma. However, upon BACE1 inhibition strong cell-surface labelling was visible for contactin-2. Neurons were additionally stained with tau antibody and TO-PRO3-Iodine to stain for neuronal processes and the nuclei. White bar indicates 50 μM. Figure source data can be found with the Supplementary data.
BACE1 inhibition increases cell surface levels of contactin-2
Among the four tested proteins, a strong increase (nearly three-fold) in the cellular protein levels upon BACE1 inhibition and knock-out was observed for contactin-2. Contactin-2 is a cell adhesion protein with different binding partners, including APP (Ma et al, 2008), and is involved in the organization of axonal subdomains at the node of Ranvier of myelinating fibres (Karagogeos, 2003; Poliak and Peles, 2003). To localize the cellular compartment of contactin-2 accumulation, neurons were treated with or without the BACE1 inhibitor C3 and stained for cell surface contactin-2. Under control conditions, a discrete punctate staining was observed for contactin-2 (Figure 3C). In contrast, C3-treated neurons showed a prominent surface staining of contactin-2 in their neuronal processes. This result demonstrates that BACE1 negatively regulates contactin-2 levels at the plasma membrane of the neurites.
Validation of BACE1 substrates in BACE1-deficient mouse brains
Next, the proteolytic processing of the novel substrates was analysed in postnatal day 7 (P7) wild-type and BACE1-deficient mouse brains. The shedding reduction of SEZ6, CHL1 and L1 resembled the reduction observed in the C3 inhibitor-treated neurons and the BACE1 knock-out neurons (Figure 4A and quantification in Figure 4B).
Figure 4.

Validation of BACE1 substrates in brain extracts of P7 wild-type and BACE1 knock-out mice. (A) P7 brains from wild-type (wt) and BACE1 (B1−/−) knock-out mice were separated into a soluble (DEA) and a membrane fraction (Lys), containing the soluble proteins (DEA) and the membrane proteins (Lys), respectively. The soluble ectodomains of SEZ6, CHL1 and L1 were reduced in the DEA fraction, while the full-length products had increased levels in the lysate. For contactin-2, the increased levels were observed in the membrane fraction. No change was observed in the soluble fraction, because the GPI-anchored membrane form of the protein was also solubilized with the prolonged DEA extraction in this experimental series. The BACE1-specfic cleavage product APPsβ and BACE1 expression in the lysate served as internal controls. Additionally, proteolytic processing of the APP family and of the two negative controls N-Cadherin and PTPRF was investigated. (B) Quantification of data from (A). The ratio of B1−/−/Wt for all four proteins in the DEA and the lysate fraction is shown. Quantification includes the positive controls APP, APLP1, APLP2 and the negative controls PTPRF and N-Cadherin, which are not BACE1 substrates. Given are mean and s.e.m. (n=6). Figure source data can be found with the Supplementary data.
BACE1 expression in the brain is high during mouse development until about 2 weeks after birth and then drops sharply (Willem et al, 2006). This raises the possibility that some substrates are cleaved by BACE1 as long as BACE1 expression is high, but that their shedding is taken over by other proteases when BACE1 expression decreases. To test this possibility, the proteolytic processing of SEZ6, L1, CHL1 and contactin-2 was analysed in P60 BACE1 knock-out brains (Figure 5A). The reduction of shedding and accumulation of full-length proteins of SEZ6, L1 and contactin-2 was comparable to the reduction in P7 knock-out brains, demonstrating that the three proteins are still efficiently cleaved in the brain at a time when BACE1 expression has dropped. Full-length CHL1 accumulated, similarly to the P7 brains, but no reduction of total CHL1 shedding was observed (Figure 5B). However, the secreted CHL1 appeared to have a slightly lower apparent molecular weight, which may be indicative of processing by an alternative protease in the absence of BACE1. Additionally, in the adult brain, CHL1 is not only expressed in neurons but also in oligodendrocyte precursors and astrocytes (Hillenbrand et al, 1999), where BACE1 is not expressed or only at low levels. Thus, it is possible, that in the whole brain the reduction of neuronal BACE1-mediated CHL1 shedding is not visible when CHL1 shedding by other proteases still occurs normally in non-neuronal brain cells.
Figure 5.

Validation of BACE1 substrates in brain extracts of adult P60 wild-type and BACE1 knock-out mice. (A) P60 wt and BACE1 knock-out brains were separated into a soluble (DEA) and a membrane fraction (Lys), containing the soluble proteins (DEA) and the membrane proteins (Lys), respectively, and analysed for Sez6, L1, CHL1, contactin-2 and the APP family. In contrast to Figure 4, the reduction of soluble contactin-2 ectodomain could be analysed because a reduced length of the DEA extraction was used. BACE1 expression in the lysate and the BACE1-specific cleavage product APPsβ served as internal controls. (B) Quantification of (A) of Sez6, L1, CHL1 and contactin-2 (CNTN2) is shown for six independent experiments. As positive control, the APP family and as negative control N-Cadherin and PTPRF were included in the analysis. Figure source data can be found with the Supplementary data.
Taken together, the analysis of P7 and P60 BACE1-deficient mouse brains demonstrates that SEZ6, L1, CHL1 and contactin-2 are physiological BACE1 substrates not only in dissociated neurons, but also in vivo in whole brain.
BACE1 inhibitor reduces substrate cleavage in vivo in mice
A reduction in BACE1 substrate shedding can be used to demonstrate efficient BACE1 inhibition in clinical trials of Alzheimer’s disease as recently shown for APPsβ (May et al, 2011). To test whether the newly identified substrates may also be suitable to monitor BACE1 inhibition in vivo, we treated P10 mice for 16 h with a single dose of the BACE1 inhibitor LY2811376, which was recently shown to efficiently reduce Aβ generation in a phase I clinical trial (May et al, 2011). sAPPβ served as a positive control for BACE1 inhibition and was reduced to 19% (Figure 6A and quantification in Figure 6B). Likewise, shedding of SEZ6, L1, CHL1 and contactin-2 was reduced, suggesting that, besides APPsβ, a reduced secretion of novel BACE1 substrates may be tested as biomarker to monitor BACE1 inhibition.
Figure 6.

Validation of BACE1 substrates in brain extracts of BACE1 inhibitor-treated mice. (A) P10 mice were treated with BACE1 inhibitor LY2811376 for 16 h. Brains were separated into a soluble (DEA) and a membrane fraction (Lys), containing the soluble proteins (DEA) and the membrane proteins (Lys), respectively. The soluble ectodomains of SEZ6, CHL1 and L1 were reduced in the DEA fraction, while the full-length products had increased levels in the lysate. In contrast to Figure 4, the reduction of soluble contactin-2 ectodomain was detectable, because a reduced length of the DEA extraction was used. sAPPβ is shown as a positive control. The inhibitor does not fully block BACE1 in vivo (May et al, 2011). (B) Quantification of data from (A). The ratio of inhibitor/control for all four proteins in the DEA and the lysate fraction is shown. Given are mean and s.e.m. (n=6). Figure source data can be found with the Supplementary data.
Discussion
Ectodomain shedding is essential for the communication between cells and is mediated by over 20 different membrane-anchored sheddases (Freeman, 2009; Reiss and Saftig, 2009; Vassar et al, 2009; Lichtenthaler et al, 2011). However, for many sheddases, including BACE1, little is known about their substrates, and thus their function, in particular under physiological conditions. The novel method SPECS is well suited for substrate identification of sheddases. Because SPECS also identifies soluble, secreted proteins, additional applications of this method include whole secretome studies, such as secretory processes in T cells, neurons and pancreatic cells as well as discovery of biomarkers, for example, secreted from tumour and stem cells.
Compared to previous studies, SPECS is a fundamentally different approach to identify secreted proteins and shedding substrates. SPECS allows growing cells in the presence of serum, which avoids cellular stress induced by serum-free conditions. The purification and enrichment of secretome proteins yields enhanced protein sequence coverage and allows more reliable protein identification and quantification. Furthermore, primary cells, such as neurons, which require albumin-rich or serum-like culture supplements, become amenable to quantitative secretome and sheddome analysis. Thus, SPECS allows studying biological processes directly in the relevant primary cell type.
Previous studies carried out under serum-free conditions frequently required protease overexpression of the protease of interest or its recombinant addition in vitro to the cell medium (Tam et al, 2004; Hemming et al, 2009; Jefferson et al, 2011). The major risk of this approach is artificial substrate discovery, which is clearly seen from our BACE1 substrate list in comparison to a previous study, which identified potential substrates of the neuronal BACE1 protease based on BACE1 overexpression in HEK293T and HeLa cells (Hemming et al, 2009). While eight substrates are in common, several of the previously identified proteins (hepatocyte growth factor receptor, neogenin, protocadherin gamma C3, receptor-type tyrosine-protein-phosphatase sigma and NCAM) did not show a significant shedding change in our substrate list of primary neurons (see lower part of Table I) and are likely to be false positives due to BACE1 overexpression in the previous study. In contrast, SPECS enabled the study of endogenous BACE1 inhibition in neurons and additionally yielded quantitative data about the extent of substrate cleavage by BACE1.
The identification of the BACE1 sheddome provides new insights into the mechanisms that govern ectodomain shedding in general. First, BACE1 has been considered as a sheddase for a few specific proteins, including APP and neuregulin, whereas the metalloproteases ADAM10 and ADAM17 have been assumed to mediate the majority of membrane protein shedding (Reiss and Saftig, 2009). However, the 34 identified neuronal membrane proteins, which require BACE1 for their shedding, correspond to 19% of the 183 shed membrane proteins identified in neurons, making BACE1 a major contributor to the shedding process in neurons. Second, some proteins are nearly exclusive BACE1 substrates (top of Table I, SEZ6L1, SEZ6, APLP1, VWFA and cache domain-containing protein 1), whereas the other ones are also cleaved to a variable degree by additional proteases. For example, shedding of APLP2 in neurons is mediated to about 40% by ADAM10, as shown previously (Hogl et al, 2011), and to about 60% by BACE1 (Table I). Thus, cleavage of a shedding substrate by two (or even more) distinct proteases is not a rare event, but appears to be occurring frequently, at least in the brain. Future studies need to address whether the distinct proteases lead to functionally different cleavage products, as it seems to be the case for APP (Ring et al, 2007; Li et al, 2010). Third, substrate cleavage by a sheddase is a tissue-specific event, which is in agreement with high BACE1 expression in neurons and its lower expression in non-neuronal tissue (Vassar et al, 1999). For example, CHL1 in neurons is largely cleaved by BACE1 (Table I), whereas it is mainly cleaved by ADAM8 in non-neuronal tissue (Naus et al, 2004). Another example is L1, which—similarly to APP—is predominantly cleaved by ADAM10 in non-neuronal cells (Maretzky et al, 2005), but mainly requires BACE1 for its shedding in neurons (Table I).
Besides APP and its homologues, a few additional glycosylated membrane proteins were previously described as BACE1 substrates (Vassar et al, 2009; Lichtenthaler et al, 2011), but not identified by SPECS in neurons. Some of them are not expressed in neurons (e.g., PSGL-1). For other ones (e.g., neuregulin), the levels of the secreted ectodomains may have been below the detection limit of the mass spectrometric analysis or the molecular mass of the ectodomain was below 30 kDa (e.g., voltage-gated sodium channels), such that the ectodomains would be lost during the filtration steps of the SPECS method.
The BACE1 sheddome contains a larger number of proteins with unknown functions, including APP. However, several of these proteins have been implicated in neurite outgrowth (SEZ6, L1, LRRN1, neurotrimin, CHL1, brain EGF-repeat containing transmembrane protein) and synapse formation (neurexin-1a and neuroligins), pointing to a function of BACE1 in development and correct wiring of the brain, which is in agreement with the high BACE1 expression during development and in the early postnatal period (Willem et al, 2006). A role for BACE1 in neurite outgrowth is in good agreement with two recent studies demonstrating defects in axon guidance in the olfactory bulb in BACE1-deficient mice (Rajapaksha et al, 2011; Cao et al, 2012). Several of the above substrates are expressed in the olfactory bulb, such that their reduced cleavage may be responsible for the observed deficits in axonal targeting in BACE1-deficient mice.
Some proteins, for example, L1 and CHL1 (Zhang et al, 2008), contribute to myelination, raising the intriguing possibility that the peripheral hypomyelination in BACE1 knock-out mice (Hu et al, 2006; Willem et al, 2006) is not exclusively mediated by the reduced cleavage of neuregulin-1.
The two top hits in the BACE1 sheddome list are SEZ6 (Gunnersen et al, 2007), which was known to undergo shedding by an unknown protease (Osaki et al, 2011), and its homologue SEZ6L1, which are predominantly expressed in neurons (Miyazaki et al, 2006). A third homologue, seizure 6-like protein 2 (SEZ6L2), is also on the substrate list, but requires BACE1 to a much lower extent for its total shedding. For SEZ6, we can further conclude that the cleavage site must lie between the most C-terminal SEZ6 peptide identified (Supplementary Figure 1) and the transmembrane domain. A good candidate for the cleavage site is the peptide bond between leucine906 and aspartate907 (Supplementary Figure 1), which is composed of the same amino acids as the efficiently BACE1-cleaved, Swedish variant of APP (Vassar et al, 1999).
For most of the BACE1 sheddome proteins, it remains unknown whether the biological function is mediated by the soluble ectodomain, as it appears to be the case for neuregulin (Willem et al, 2006), or by the full-length protein, as recently shown for TMEM27, a substrate for BACE2, a pancreatic protease homologous to BACE1 (Esterhazy et al, 2011). Thus, future studies need to address whether loss-of-BACE1 cleavage leads to gain or loss-of-function of a given substrate and how this is phenotypically reflected in BACE1-deficient mice. Potentially, the phenotypes are not just mediated by a single substrate but by combinations of different ones. Interestingly, mutations in SEZ6, which dramatically reduce the length of SEZ6 protein, are found to a high incidence in epileptic patients (Yu et al, 2007), raising the possibility that loss of SEZ6 shedding may contribute to the higher susceptibility of epileptic seizures in BACE1 knock-out mice (Hu et al, 2010).
The identification of novel BACE1 substrates by SPECS is not only the basis for a better understanding of BACE1 function and of the phenotypes in BACE1 knock-out mice, but will also allow to better evaluate the therapeutic potential of BACE1 and to develop biomarkers for monitoring potential side effects of BACE1 inhibition in clinical trials of Alzheimer’s disease.
Materials and methods
Materials
The following antibodies were used: pAb CHL1 (AF2147), pAb Contactin-2 (AF4439; R&D Systems), L1 C-terminal (Clone 2C2; Abcam), mAb L1 antibodies 14.10 and 555 (kindly provided by Peter Altevogt), pAb Sez6 (kindly provided by Jenny Gunnersen), PTPRF (Neuromab), mAb APP C-terminal antibody 2C11, mAb sAPPβ BAWT (Kuhn et al, 2010), pAb 192wt (kindly provided by Dale Schenck), mAb 22C11 (kindly provided by Konrad Beyreuther), pAb EP2456Y clone TAU (Millipore), mAb clone C32 N-Cadherin (BD Bioscience), mAb Acetylated tubulin (Sigma-Aldrich, T7451), mAb BACE1 3D5 (kindly provided by Bob Vassar), pAb APLP1 antibody (Proteintech; 12305-2-AP), pAb APLP2 antibody (Calbiochem; 171617, 171616), HRP coupled anti-mouse and anti-secondary (DAKO), HRP coupled anti-goat and anti-sheep (Santa Cruz). The following reagents and media were used: neurobasal medium, HBSS and B27 (Invitrogen), C3 (β-secretase inhibitor IV; Calbiochem, 565788), acetonitrile (ACN), water, formic acid with LC-MS grade, iodoacetamide (Sigma-Aldrich), Trypsin (Promega), TO-PRO-3-Iodide (Invitrogen).
Isolation of primary neurons
Wild-type and BACE1 knock-out mice used for preparation of primary neurons and brain tissues were obtained from The Jackson Laboratory (B6.129-Bace1tm1Pcw/J). All animal experiments were performed according to the European community council directive (86/609/ECC). Neurons were isolated as described previously (Mitterreiter et al, 2010) at E15/E16 and cultured in neurobasal medium supplemented with 2% B27, 100 U/ml penicillin, 100 μg/ml streptomycin and 0.5 mM glutamine. Experiments were carried out after 4–7 days in vitro (DIV).
Immunofluorescence
In all, 30 000 primary neurons were seeded per well in a 96-well format. Neurons were either treated with DMSO or C3. Media were removed after 2 days of incubation and neurons were incubated in primary antibody 4D7 (1:200) in 3% BSA for 30 min on ice followed by post fixation with 4% PFA for 15 min on ice. Excessive PFA was quenched with NH4Cl and neurons were washed twice with PBS. Afterwards anti-mouse Alexa-488 (1:1000) in 3% BSA was incubated for another hour on the fixed neurons. The next step comprising a second 15 min post fixation with 4% PFA followed by permeabilization with 0.1% Triton X-100 and a second quenching of excessive PFA with NH4Cl. Afterwards, neurons were stained with anti-Tau antibody (1:1000) for 1 h followed by rabbit Alexa-555 (1:1000) and TO-PRO-3-iodine (1:1000) for 1 h to stain for neuronal processes and nuclei. Neurons were washed twice with PBS and afterwards investigated with a Zeiss LSM5 confocal microscope.
Brain fractionation
Brains were isolated from P8 mice. The brains were homogenized in 2 ml of 4°C cold diethyl amine buffer with a motorized potter directly followed by neutralization of the homogenate with TRIS buffer. Homogenate was briefly centrifuged at 14 000 r.p.m. for 10 min. Supernatant was transferred to a new vial and subject to clarifying ultracentrifugation at 55 000 r.p.m. with a TLA55 rotor for 30 min. Meanwhile, the remaining pellets were washed once more with PBS to remove the remaining soluble fraction. Pellet was lysed in STE buffer (150 mM NaCl, 50 mM TRIS, 2 mM EDTA) with 1% Triton for 30 min on ice followed by centrifugation in a cooled table-top centrifuge at 14 000 r.p.m. Resulting lysate was transferred into a fresh tube.
Metabolic labelling with azido sugar
For each condition, 40 million 4 DIV neurons were labelled with 1 μmol of tetraacetyl-N-azidoacetyl-mannosamine diluted in 20 ml neurobasal medium (50 μmol/l) supplemented with 2% B27 for 2 days. Neurons were cultured in the presence or absence of 1 μM BACE1 inhibitor C3. For HEK293T, 40 million cells were used and incubated in DMEM plus 5% FCS.
Purification of azido sugar-labelled proteins
Conditioned medium was collected and filtered through 0.45 μm PVDF filter (Millex) into a VivaSpin 20 column (30 kDa) at 4°C. To remove non-metabolized tetraacetyl-N-azidoacetyl-mannosamine VivaSpin 20 columns were centrifuged at 4600, r.p.m. at 4°C. The retentate was filled with 20 ml PBS. This procedure was repeated three times. In the last step, the PBS refill step was omitted. Instead, 250 nM of DBCO-PEG12-biotin (Click-chemistry tools) diluted in 1 ml ddH2O with 5% (v/v) DMSO was added to retentate to biotinylate metabolically azide-labelled glycoproteins. Columns were incubated overnight at 4°C. For removal of non-reacted DBCO-PEG12-Biotin, VivaSpin20 columns were subject to three times of centrifugation with subsequent PBS buffer refill. After last step of centrifugation, the retentate was diluted in 5 ml PBS with 2% SDS (v/v) and 2 mM TCEP. For purification of biotinylated proteins, the sample was loaded on a 10-ml polyprep column with a streptavidin bed of 300 μl beads. After binding of proteins, streptavidin was washed 3 × with 10 ml PBS with 1% SDS. Afterwards, streptavidin beads were boiled with urea sample buffer containing 3 mM biotin to compete for the binding of biotinylated proteins with streptavidin.
SDS–PAGE separation, trypsinization
Proteins were separated on a 10% Tris/glycine SDS gel. Afterwards, qualitatively equal gel slices were cut out from the gel with the exception of the remaining albumin band at around 60 kDa. Proteins in the gel slices were subject to trypsinization according to standard protocols (Shevchenko et al, 2006).
Mass spectrometric analysis
Mass spectrometry experiments were performed on an Easy nLC nanoflow HPLC system II (Proxeon) connected to an LTQ-Velos Orbitrap (Thermo Fisher Scientific). Peptides were separated by reverse phase chromatography using in-house made 15 cm columns (New Objective, FS360-75-8-N-S-C15) packed with C18-AQ 2,4 μm resin (Dr Maisch GmbH, Part No. r124.aq). A 90-min gradient (5–40%) at a flow rate of 400 nl/min was used. The measurement method consisted of an initial FTMS scan recorded in profile mode with 30 000 m/z resolution, a mass range from 300 to 2000, m/z and a target value of 1 000 000. Subsequently, collision-induced dissociation (CID) fragmentation was performed for the 14 most intense ions with an isolation width of 2 Da in the ion trap. A target value of 10 000, enabled charge state screening, a monoisotopic precursor selection, 35% normalized collision energy, an activation time of 10 ms, wide band activation and a dynamic exclusion list with 30 s exclusion time were applied.
Analysis of mass spectrometry data
For data analysis of the HEK293T one biological replicate and for the neuronal secretome, five biological replicates were analysed with the freely available MaxQuant suite (version 1.2.0.18) (Cox and Mann, 2008). Protein identification was performed using the integrated Andromeda search algorithm (Cox et al, 2011). Mass recalibration was done by a first search in a reduced murine protein database. Main search was performed using the murine IPI database (version 3.68) allowing for N-terminal acetylation and oxidation of methionine as variable modifications and carbamidomethylation of cysteine as fixed modifications. Two missed cleavages were allowed. Peptide as well as protein false discovery rate was set to <1%. Mass accuracy was set to 6 p.p.m. for the main search and 20 p.p.m. for the first search. Label-free quantification was performed between control (DMSO) and the treatment condition (C3) on the basis of unique peptides. A variance score (VS=absolute value of (standard error of the mean/(1−mean))) was calculated for all proteins. Proteins with a VS of ≤0.35 were considered as proteins with a consistent change upon BACE1 inhibition.
In-vivo BACE1 inhibitor experiment
Ten-day-old BALB/c mouse pups were purchased from Charles River Laboratories (Wilmington, MA, USA). All animal experiments were performed in accordance with the European Communities Council Directive #86/609 and the directives of the Danish National Committee on Animal Research Ethics. The specific BACE1 inhibitor LY-2811376 [(S)-4-(2,4-difluoro-5-pyrimidin-5-yl-phenyl)-4-methyl-5,6-dihydro-4H-[1,3]thiazin-2-ylamine] (May et al, 2011) was synthesized following the schemes provided by Lilly Research Labs (Indianapolis, IN, USA; patent WO2009134617) and dissolved in 100% polyethylene glycol 400 for systemic administration (subcutaneously, s.c.) as dictated by compound solubility. Animals were treated every 12 h with 100 mg/kg and 16 h after the first application mice were sacrificed by decapitation. Snap frozen brains were homogenized in 10 volumes (w/v) of 0.2% diethylamine (DEA) containing 50 mM NaCl (pH 10) and protease inhibitors. DEA fraction was ultracentrifuged at 100 000 g for 30 min. The resulting supernatant was retained as the soluble fraction and neutralized by addition of 10% 0.5 M Tris/HCl, pH 6.8. The DEA insoluble material was homogenized with RIPA buffer and cleared by ultracentrifugation for 30 min at 100 000 g.
Synthesis of 1,3,4,6-tetraacetyl-N-acetylazido-D-mannosamine
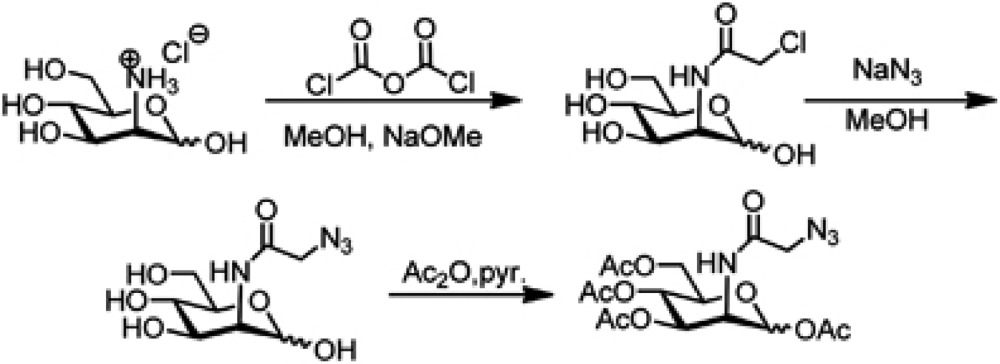
To a solution of D-mannosamine hydrochloride (1 g; 4.64 mmol) in methanol (50 ml), sodium methylate (30%w/w NaOMe in MeOH, 1.66 ml, 4.64 mmol, 1 equiv.) was added and the mixture was stirred at room temperature for 30 min until complete dissolution. Then, NEt3 (0.47 g; 4.64 mmol, 1 equiv.) and chloroacetic anhydride (871 mg; 5.1 mmol, 1.1 equiv.) were added to the solution, and stirred at room temperature overnight. After finishing, the solvent was evaporated and the crude product was used in subsequent reaction without further purification (if necessary sodium bicarbonate was added to neutralize the solution!)
To a solution of N-chloroacetyl-mannosamine in MeOH/H2O (10:1 (20/2 ml)), sodium azide NaN3 (1.06 g; 16.24 mmol, 3.5 equiv.) was added. The mixture was stirred for 5 h at 65°C. Subsequently, the reaction mixture was concentrated and dried in vacuo. The residue was then suspended in pyridine (20 ml) and acetic anhydride (20 m) was added to the solution. The reaction was stirred at room temperature overnight. After concentration in vacuo, the residue was dissolved in EtOAc (50 ml) and washed with 1 N HCl, NaHCO3 and brine (each 50 ml) (CAUTION: extraction with sodium bicarbonate causes gas formation and excess pressure in the separating funnel). After drying over MgSO4 the crude product was purified by column chromatography (CH/ EE 1: 1) to afford the desired product (mixture of anomers α/β—54/46) as a white-yellowish oil that eventually can solidify upon lyophilization.
Yield Y=55% (over 3 steps).
1,3,4,6-Tetraacetyl-N-azidoacetyl-D-mannosamine
[α]D20=+6.5 (c=0.53; CHCl3).
1H-NMR: (400 MHz, CDCl3): δ (p.p.m.)=6.67 (d, 3J=9.0 Hz, 1H), 6.63 (d, 3J=9.3 Hz, 1H); 6.03 (d, 3J=1.8 Hz, 1 H); 5.88 (d, 3J=1.6 Hz, 1H), 5.33 (dd, 3J=10.2 Hz, 3J=4.3 Hz, 1H); 5.21 (t, 3J=10.0 Hz, 1H); 5.15 (t, 3J=9.8 Hz, 1H); 5.05 (dd, 3J=9.9 Hz, 3J=3.9 Hz, 1H); 4.72 (ddd, 3J=9.0 Hz, 3J=3.8 Hz, 3J=1.6 Hz, 1H); 4.61 (ddd, 3J=9.3 Hz, 3J=4.2 Hz, 3J=1.9 Hz, 1H); 4.27–4.19 (m, 2H); 4.16–3.99 (m, 7H); 3.81 (ddd, 3J=9.6 Hz, 3J=4.6 Hz, 3J=2.5 Hz, 1H); 2.17 (s, 3H); 2.10 (s, 3H); 2.10 (s, 6H); 2.05 (s, 6H); 1.99 (s, 3H); 1.99 (s, 3H).
13C-NMR (100 MHz, CDCl3): δ (p.p.m.)=170.5; 170.1; 170.1; 169.6; 168.3; 168.1; 167.4; 166.8; 91.3; 90.3; 73.4; 71.4; 70.3; 68.8; 65.1; 65.0; 61.8; 61.7; 52.6; 52.4; 49.7; 49.3; 20.8; 20.7; 20.7; 20.6; 20.6.
IR (ATR Platinum Diamond): (cm−1)=3339 (vw), 1739 (w), 1208 (m), 1036 (m).
MS (FAB, 3-NBA), m/z (%): 371 [C14H19N4O8+] (50), 139 (100).
HR-MS (FAB, 3-NBA), C16H22N4O10: cald. 431.1414; found 431.1417.
Supplementary Material
Acknowledgments
We thank Bernhard Küster for critically reading the manuscript and Katrin Moschke, Nina Scheithauer and Heike Hampel for excellent technical help. We thank the following institutions for financial support: the Deutsche Forschungsgemeinschaft for SFB596 to SFL, CH, MW and AI, for project RO 2226/10-1 to SR, the competence network degenerative dementias (BMBF) to SFL and CH, the Boehringer Ingelheim Foundation for a Scholarship to SH, the Friedrich Baur Foundation to SFL, the AD-IG NGFNplus to CH and MW and the HELMA to CH and MW.
Author contributions: PHK, AI, SB and SFL conceived or designed the experiments. PHK, KK, SH, AC, UZ and CV performed the experiments. PHK, SH, SR, AI, SB, CV and SFL analysed the data. PHK and SFL wrote the manuscript. MW, US, AH, CH and SR contributed essential material.
Footnotes
CV is an employee of H. Lundbeck, Denmark.
References
- Almaraz RT, Aich U, Khanna HS, Tan E, Bhattacharya R, Shah S, Yarema KJ (2012) Metabolic oligosaccharide engineering with N-Acyl functionalized ManNAc analogs: cytotoxicity, metabolic flux, and glycan-display considerations. Biotechnol Bioeng 109: 992–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai G, Pfaff SL (2011) Protease regulation: the Yin and Yang of neural development and disease. Neuron 72: 9–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Rickenbacher GT, Rodriguez S, Moulia TW, Albers MW (2012) The precision of axon targeting of mouse olfactory sensory neurons requires the BACE1 protease. Sci Rep 2: 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805 [DOI] [PubMed] [Google Scholar]
- Dominguez D, Tournoy J, Hartmann D, Huth T, Cryns K, Deforce S, Serneels L, Camacho IE, Marjaux E, Craessaerts K, Roebroek AJ, Schwake M, D’Hooge R, Bach P, Kalinke U, Moechars D, Alzheimer C, Reiss K, Saftig P, De Strooper B (2005) Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J Biol Chem 280: 30797–30806 [DOI] [PubMed] [Google Scholar]
- Esterhazy D, Stutzer I, Wang H, Rechsteiner MP, Beauchamp J, Dobeli H, Hilpert H, Matile H, Prummer M, Schmidt A, Lieske N, Boehm B, Marselli L, Bosco D, Kerr-Conte J, Aebersold R, Spinas GA, Moch H, Migliorini C, Stoffel M (2011) Bace2 is a beta cell-enriched protease that regulates pancreatic beta cell function and mass. Cell Metab 14: 365–377 [DOI] [PubMed] [Google Scholar]
- Farah MH, Pan BH, Hoffman PN, Ferraris D, Tsukamoto T, Nguyen T, Wong PC, Price DL, Slusher BS, Griffin JW (2011) Reduced BACE1 activity enhances clearance of myelin debris and regeneration of axons in the injured peripheral nervous system. J Neurosci 31: 5744–5754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman M (2009) Rhomboids: 7 years of a new protease family. Semin Cell Dev Biol 20: 231–239 [DOI] [PubMed] [Google Scholar]
- Gunnersen JM, Kim MH, Fuller SJ, De Silva M, Britto JM, Hammond VE, Davies PJ, Petrou S, Faber ES, Sah P, Tan SS (2007) Sez-6 proteins affect dendritic arborization patterns and excitability of cortical pyramidal neurons. Neuron 56: 621–639 [DOI] [PubMed] [Google Scholar]
- Harrison SM, Harper AJ, Hawkins J, Duddy G, Grau E, Pugh PL, Winter PH, Shilliam CS, Hughes ZA, Dawson LA, Gonzalez MI, Upton N, Pangalos MN, Dingwall C (2003) BACE1 (beta-secretase) transgenic and knockout mice: identification of neurochemical deficits and behavioral changes. Mol Cell Neurosci 24: 646–655 [DOI] [PubMed] [Google Scholar]
- Hemming ML, Elias JE, Gygi SP, Selkoe DJ (2009) Identification of beta-secretase (BACE1) substrates using quantitative proteomics. PLoS ONE 4: e8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillenbrand R, Molthagen M, Montag D, Schachner M (1999) The close homologue of the neural adhesion molecule L1 (CHL1): patterns of expression and promotion of neurite outgrowth by heterophilic interactions. Eur JNeurosci 11: 813–826 [DOI] [PubMed] [Google Scholar]
- Hogl S, Kuhn PH, Colombo A, Lichtenthaler SF (2011) Determination of the proteolytic cleavage sites of the amyloid precursor-like protein 2 by the proteases ADAM10, BACE1 and gamma-secretase. PLoS ONE 6: e21337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R (2006) Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci 9: 1520–1525 [DOI] [PubMed] [Google Scholar]
- Hu X, Zhou X, He W, Yang J, Xiong W, Wong P, Wilson CG, Yan R (2010) BACE1 deficiency causes altered neuronal activity and neurodegeneration. J Neurosci 30: 8819–8829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse JT, Liu K, Pijak DS, Carlin D, Lee VM, Doms RW (2002) Beta-secretase processing in the trans-Golgi network preferentially generates truncated amyloid species that accumulate in Alzheimer’s disease brain. J Biol Chem 14: 14. [DOI] [PubMed] [Google Scholar]
- Jefferson T, Causevic M, auf dem Keller U, Schilling O, Isbert S, Geyer R, Maier W, Tschickardt S, Jumpertz T, Weggen S, Bond JS, Overall CM, Pietrzik CU, Becker-Pauly C (2011) Metalloprotease meprin beta generates nontoxic N-terminal amyloid precursor protein fragments in vivo. J Biol Chem 286: 27741–27750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett JC, Bertozzi CR (2010) Cu-free click cycloaddition reactions in chemical biology. Chem Soc Rev 39: 1272–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagogeos D (2003) Neural GPI-anchored cell adhesion molecules. Front Biosci 8: s1304–s1320 [DOI] [PubMed] [Google Scholar]
- Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, Kremmer E, Rossner S, Lichtenthaler SF (2010) ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J 29: 3020–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC (2005) BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci 25: 11693–11709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wang B, Wang Z, Guo Q, Tabuchi K, Hammer RE, Sudhof TC, Zheng H (2010) Soluble amyloid precursor protein (APP) regulates transthyretin and Klotho gene expression without rescuing the essential function of APP. Proc Natl Acad Sci USA 107: 17362–17367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenthaler SF, Haass C, Steiner H (2011) Regulated intramembrane proteolysis – lessons from amyloid precursor protein processing. J Neurochem 117: 779–796 [DOI] [PubMed] [Google Scholar]
- Ma QH, Futagawa T, Yang WL, Jiang XD, Zeng L, Takeda Y, Xu RX, Bagnard D, Schachner M, Furley AJ, Karagogeos D, Watanabe K, Dawe GS, Xiao ZC (2008) A TAG1-APP signalling pathway through Fe65 negatively modulates neurogenesis. Nat Cell Biol 10: 283–294 [DOI] [PubMed] [Google Scholar]
- Makridakis M, Vlahou A (2010) Secretome proteomics for discovery of cancer biomarkers. J Proteomics 73: 2291–2305 [DOI] [PubMed] [Google Scholar]
- Maretzky T, Schulte M, Ludwig A, Rose-John S, Blobel C, Hartmann D, Altevogt P, Saftig P, Reiss K (2005) L1 is sequentially processed by two differently activated metalloproteases and presenilin/gamma-secretase and regulates neural cell adhesion, cell migration, and neurite outgrowth. Mol Cell Biol 25: 9040–9053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PC, Dean RA, Lowe SL, Martenyi F, Sheehan SM, Boggs LN, Monk SA, Mathes BM, Mergott DJ, Watson BM, Stout SL, Timm DE, Smith Labell E, Gonzales CR, Nakano M, Jhee SS, Yen M, Ereshefsky L, Lindstrom TD, Calligaro DO et al. (2011) Robust central reduction of amyloid-beta in humans with an orally available, non-peptidic beta-secretase inhibitor. J Neurosci 31: 16507–16516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meakin PJ, Harper AJ, Hamilton DL, Gallagher J, McNeilly AD, Burgess LA, Vaanholt LM, Bannon KA, Latcham J, Hussain I, Speakman JR, Howlett DR, Ashford ML (2011) Reduction in BACE1 decreases body weight, protects against diet-induced obesity and enhances insulin sensitivity in mice. Biochem J 441: 281–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitterreiter S, Page RM, Kamp F, Hopson J, Winkler E, Ha HR, Hamid R, Herms J, Mayer TU, Nelson DJ, Steiner H, Stahl T, Zeitschel U, Rossner S, Haass C, Lichtenthaler SF (2010) Bepridil and amiodarone simultaneously target the Alzheimer’s disease beta- and gamma-secretase via distinct mechanisms. J Neurosci 30: 8974–8983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T, Hashimoto K, Uda A, Sakagami H, Nakamura Y, Saito SY, Nishi M, Kume H, Tohgo A, Kaneko I, Kondo H, Fukunaga K, Kano M, Watanabe M, Takeshima H (2006) Disturbance of cerebellar synaptic maturation in mutant mice lacking BSRPs, a novel brain-specific receptor-like protein family. FEBS Lett 580: 4057–4064 [DOI] [PubMed] [Google Scholar]
- Naus S, Richter M, Wildeboer D, Moss M, Schachner M, Bartsch JW (2004) Ectodomain shedding of the neural recognition molecule CHL1 by the metalloprotease-disintegrin ADAM8 promotes neurite outgrowth and suppresses neuronal cell death. J Biol Chem 279: 16083–16090 [DOI] [PubMed] [Google Scholar]
- Osaki G, Mitsui S, Yuri K (2011) The distribution of the seizure-related gene 6 (Sez-6) protein during postnatal development of the mouse forebrain suggests multiple functions for this protein: an analysis using a new antibody. Brain Res 1386: 58–69 [DOI] [PubMed] [Google Scholar]
- Poliak S, Peles E (2003) The local differentiation of myelinated axons at nodes of Ranvier. Nat Rev Neurosci 4: 968–980 [DOI] [PubMed] [Google Scholar]
- Price PJ, Brewer GJ (2001) Serum-free media for neural cell cultures. InProtocols for Neural Cell Culture Fedoroff S, Richardson A (eds)Vol. 19: pp255–264Springer: Totowa, NJ [Google Scholar]
- Rajapaksha TW, Eimer WA, Bozza TC, Vassar R (2011) The Alzheimer’s beta-secretase enzyme BACE1 is required for accurate axon guidance of olfactory sensory neurons and normal glomerulus formation in the olfactory bulb. Mol Neurodegener 6: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss K, Saftig P (2009) The ‘A Disintegrin And Metalloprotease’ (ADAM) family of sheddases: Physiological and cellular functions. Semin Cell Dev Biol 20: 126–137 [DOI] [PubMed] [Google Scholar]
- Ring S, Weyer SW, Kilian SB, Waldron E, Pietrzik CU, Filippov MA, Herms J, Buchholz C, Eckman CB, Korte M, Wolfer DP, Muller UC (2007) The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J Neurosci 27: 7817–7826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala Frigerio C, Fadeeva JV, Minogue AM, Citron M, Van Leuven F, Staufenbiel M, Paganetti P, Selkoe DJ, Walsh DM (2010) beta-Secretase cleavage is not required for generation of the intracellular C-terminal domain of the amyloid precursor family of proteins. FEBS J 277: 1503–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savonenko AV, Melnikova T, Laird FM, Stewart KA, Price DL, Wong PC (2008) Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc Natl Acad Sci USA 105: 5585–5590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1: 2856–2860 [DOI] [PubMed] [Google Scholar]
- Sletten EM, Bertozzi CR (2011) From mechanism to mouse: a tale of two bioorthogonal reactions. Acc Chem Res 44: 666–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachel SJ, Coburn CA, Steele TG, Jones KG, Loutzenhiser EF, Gregro AR, Rajapakse HA, Lai MT, Crouthamel MC, Xu M, Tugusheva K, Lineberger JE, Pietrak BL, Espeseth AS, Shi XP, Chen-Dodson E, Holloway MK, Munshi S, Simon AJ, Kuo L et al. (2004) Structure-based design of potent and selective cell-permeable inhibitors of human beta-secretase (BACE-1). J Med Chem 47: 6447–6450 [DOI] [PubMed] [Google Scholar]
- Tam EM, Morrison CJ, Wu YI, Stack MS, Overall CM (2004) Membrane protease proteomics: Isotope-coded affinity tag MS identification of undescribed MT1-matrix metalloproteinase substrates. Proc Natl Acad Sci USA 101: 6917–6922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thouvenot E, Urbach S, Dantec C, Poncet J, Seveno M, Demettre E, Jouin P, Touchon J, Bockaert J, Marin P (2008) Enhanced detection of CNS cell secretome in plasma protein-depleted cerebrospinal fluid. J Proteome Res 7: 4409–4421 [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC et al. (1999) Beta-secretase cleavage of Alzheimer‘s amyloid precursor protein by the transmembrane aspartic protease BACE. Science (New York, NY) 286: 735–741 [DOI] [PubMed] [Google Scholar]
- Vassar R, Kovacs DM, Yan R, Wong PC (2009) The beta-secretase enzyme BACE in health and Alzheimer’s disease: regulation, cell biology, function, and therapeutic potential. J Neurosci 29: 12787–12794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Song L, Laird F, Wong PC, Lee HK (2008) BACE1 knock-outs display deficits in activity-dependent potentiation of synaptic transmission at mossy fiber to CA3 synapses in the hippocampus. J Neurosci 28: 8677–8681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C (2006) Control of peripheral nerve myelination by the beta-secretase BACE1. Science (New York, NY) 314: 664–666 [DOI] [PubMed] [Google Scholar]
- Yu ZL, Jiang JM, Wu DH, Xie HJ, Jiang JJ, Zhou L, Peng L, Bao GS (2007) Febrile seizures are associated with mutation of seizure-related (SEZ) 6, a brain-specific gene. J Neurosci Res 85: 166–172 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yeh J, Richardson PM, Bo X (2008) Cell adhesion molecules of the immunoglobulin superfamily in axonal regeneration and neural repair. Restor Neurol Neurosci 26: 81–96 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.