

Abstract
HIV-associated neurocognitive disorders (HAND) continue to be a consequence of HIV-1 infection among clade B-infected individuals. In contrast, the incidence of severe neurological impairment is lower among clade C-infected patients in regions of Sub-Saharan Africa and India. Biological aspects such as replication, cytopathicity, inflammatory response, and neurotoxicity unique to each clade influence neuropathogenicity and ultimately affect the clinical outcome of the disease. We hypothesize that productive infection by clade C isolates leads to macrophage-mediated neurotoxicity, although to a lesser extent than clade B isolates. Using a panel of primary isolates of clades B and C we demonstrated that clade B has higher replication efficiency in monocyte-derived macrophages (MDM) through reverse transcriptase activity assay and HIV-1 p24 antigen ELISA. To test the neurotoxicity of clades B and C, we used an in vitro neurotoxicity model. Conditioned medium from clade B-infected MDM was neurotoxic to rat and human neuron cultures. In contrast, clade C isolates mediated neurotoxicity when a higher initial viral titer was used for MDM infection. Furthermore, neurotoxicity mediated by isolates of both clades correlated with virus replication in MDM. Together, these results suggest that in comparison to clade B, primary isolates of clade C have slower replication kinetics in primary MDM, leading to lower levels of macrophage-mediated neurotoxicity. Elucidating the differences in replication and macrophage-mediated neurotoxicity between isolates of HIV-1 clades B and C will provide important insights needed to clarify the disparity seen in HAND incidence.
Keywords: HIV-1 clade B, HIV-1 clade C, Neurotoxicity, Macrophages, HIV-associated neurocognitive disorders
Introduction
HIV-1-associated neurocognitive disorders (HAND) continue to be of significant clinical consequence among HIV-1-infected individuals. HAND includes asymptomatic neurocognitive impairment, mild neurocognitive disorder (MND), and the most severe manifestation, HIV-1-associated dementia—HAD (reviewed by Boisse et al. 2008). Following the introduction of antiretroviral therapy, 11% of HIV-1 clade B-infected individuals develop HAD (Kolson and Gonzalez-Scarano 2000), which is characterized by motor, behavioral, and cognitive dysfunction. However, neuropsychological profiling using the International Dementia Scale, a standardized test, found lower incidences of HAD in developing regions (~1–2% in India) where clade C predominates (Clifford et al. 2007; Riedel et al. 2006). Less severe forms of neurological impairment, such as MND, have become more prevalent among HIV-sero-positive individuals in developed countries. Cross-sectional studies of infected individuals in India reported a 56–60% prevalence of mild cognitive deficits, not qualifying as motor/cognitive impairment or dementia (Gupta et al. 2007; Yepthomi et al. 2006). The mechanisms underlying the disparity between HIV-1 clades B and C, in regard to HAND severity, are unclear.
HIV-infected macrophages enter the central nervous system, mediating neuronal injury and dysfunction through the secretion of viral proteins, inflammatory factors, excess glutamate, and other neurotoxins (Jiang et al. 2001; Zheng and Gendelman 1997). Low incidence of severe neurological manifestations in clade C-infected individuals does not correlate with the preferential tropism for CCR5 (Abebe et al. 1999; Zhang et al. 2006). HIV-infected macrophages drive the pathogenesis of HAND; thus, preferential usage of CCR5 coreceptor by clade C would presumably cause a higher rather than lower incidence of severe neurological deficits. Neuropathological studies in India revealed the presence of HIV-1-infected macrophages in post mortem brains, but an absence of multinucleated giant cells (MNGC) and multifocal microglial nodules, both of which are frequently found in clade B-infected patients (Mahadevan et al. 2007). Pro-inflammatory molecules and markers known to be increased among clade B-infected individuals, such as interleukin (IL)-1α, IL-6, tumor necrosis factor-α (TNF-α), β2-microglobulin, and neopterin have been identified in the serum and plasma of clade C-infected individuals from India and Ethiopia (Ayehunie et al. 1993; Kamat et al. 2009). Additionally, clade C viral proteins, such as Tat, exhibit distinct clade-specific modulation of neuropathogenic molecules from their clade B counterparts (Campbell et al. 2007; Li et al. 2008; Mishra et al. 2008). Biological aspects unique to each clade, including genetic diversity of viral proteins, viral replication capacity, cytopathicity, inflammatory response, and neurotoxicity may all contribute to different neuropathogenic properties and ultimately affect the clinical outcome of the neurological disease.
Clade C, the most commonly transmitted subtype, accounts for more than 50% of HIV-1 infections world-wide. However, the majority of HIV-1 research focuses on infection models using clade B strains. We undertook this study to describe the replication efficiency and neurotoxicity of a panel of primary isolates of clades B and C using established in vitro models of human monocyte-derived macrophages (MDM) and primary rat cortical neurons (RCN). We found that clade B primary isolates have higher replication efficiency in MDM than clade C isolates. Productive infection by clade B isolates was associated with increased neurotoxicity. Clade C-mediated neurotoxicity was observed when a higher initial viral titer was used for MDM infection. The observed neurotoxicity mediated by isolates of both clades correlated with virus replication in MDM. Our findings show that in comparison to clade B, primary isolates of clade C studied here have lower replication efficiency in primary MDM, leading to lower levels of macrophage-mediated neurotoxicity.
Materials and Methods
Isolation and Culture of Primary MDM, RCN, and Human Neurons
Human monocytes were isolated from peripheral blood mononuclear cells (PBMCs) of HIV-1, -2, and hepatitis B seronegative donors after leukopheresis, and counter-current centrifugal elutriation (Gendelman et al. 1988). Primary monocytes were cultured as adherent monolayers at a density of 1.1 × 106 cells/well in 24-well plates and differentiated for 7 days in Dulbecco’s modified Eagle’s medium (DMEM, GIBCO Invitrogen Corp., Carlsbad, CA) supplemented with 10% heat-inactivated pooled human serum (Cambrex Bio Science, Walkersville, MD), 50 μg/ml gentamicin (GIBCO Invitrogen Corp.), 10 μg/ml ciprofloxacin (Fisher Scientific, Dubuque, IA), and 1,000 U/ml highly purified recombinant human macrophage-colony stimulating factor (M-CSF, a generous gift from Wyeth Pharmaceutical, Cambridge, MA). PBMCs were cultured in RPMI-1640 (GIBCO Invitrogen Corp.) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen), 10 IU/ml IL-2 (R&D Systems, Minneapolis, MN), and 5 μg/ml penicillin–streptomycin (GIBCO, Invitrogen, Corp.). Primary cortical neurons were prepared from cortices of embryonic day 17–18 (E17-18) Sasco Sprague–Dawley rat fetuses as previously described (Zheng et al. 2001). Briefly, the cortex was dissected and individual cells were mechanically dissociated in Neurobasal™ medium (Invitrogen Corp.) and differentiated in Neurobasal™ medium containing B27 supplement (GIBCO Invitrogen Corp.), 0.5 mM glutamine (Sigma–Aldrich), and 25 μg/ml penicillin–streptomycin. For neurotoxicity assays, neurons were plated onto poly-d-lysine-coated 96-well plates at a density of 5 × 104 cells/well. Cultured neurons were assumed to be mature 7–12 days after plating. All experiments were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center. Human fetal brain tissue (gestational age 13–16 weeks) was obtained from elective abortions in full compliance with the University of Nebraska Medical Center (UNMC) and NIH ethical guidelines. Human neuronal cultures were prepared as previously described (Zheng et al. 1999). Human neurons were plated on poly-d-lysine-coated 15-mm coverslips in 24-well plates at a density of 2 × 105 cells/well. Cells were cultured in neurobasal media supplemented with 1% B27, 1% penicillin/streptomycin, and 0.5 mM glutamine. Ten days after culture, neurons were deemed mature.
Titration of HIV-1 Isolates, MDM Infection and Collection of MDM-Conditioned Medium (MCM)
HIV-1JR-FL was obtained from the AIDS Research and Reference Reagents Program at the NIH National Institute of Allergy and Infectious Diseases (Koyanagi et al. 1987). HIV-1ADA was isolated from the PBMCs of an infected patient with Kaposi’s sarcoma (Gendelman et al. 1988). Clade B primary viruses (D02-2562 BG, A00-086 CPx, D02-2562 Sp, and G0048 CPx) were isolated from the spleen (Sp), basal ganglia (BG), or choroid plexus (CPx) in the trigone of the lateral ventricle of infected patients with HIV Encephalitis (HIVE) (Burkala et al. 2005). Clade C primary isolates (2873M, 2669M, 1084M, and 1157I) were isolated from PBMCs of HIV-1-infected mother and infant pairs in Africa (Zhang et al. 2005, 2006). All HIV-1 isolates (listed in Table 1) use CCR5 as a co-receptor (Burkala et al. 2005; Gendelman et al. 1988; Koyanagi et al. 1987; Zhang et al. 2005, 2006). Primary viruses were cultured in phytohemagglutinin (PHA, 5 μg/ml, Sigma–Aldrich) stimulated PBMCs for 14 days before titration. Stock viruses were screened for mycoplasma and endotoxin using hybridization and Limulus amebocyte lysate assays, respectively. The 50% tissue culture infective dose (TCID50) of each viral stock was determined by monitoring infection of TZM-bl cells obtained through NIH AIDS Research and Reference Reagent Program as described previously (Derdeyn et al. 2000; Marozsan et al. 2004; Platt et al. 1998; Wei et al. 2002). The stock viral titers of each isolate are listed in Table 1. Seven days after plating, MDM were infected with the aforementioned viral isolates using TCID50/ml indicated in Table 1. On the second day after infection, medium was removed and substituted with MDM culture medium (DMEM with 10% heat-inactivated pooled human serum, 50 μg/ml gentamicin, and 10 μg/ml ciprofloxacin) (Gendelman et al. 1988). On 4, 7, 14, or 21 days after infection, cells were changed to 0.5 ml/well fresh serum-free Neurobasal™ medium supplemented with 25 ug/ml penicillin–streptomycin and 5 mM glutamine for 24 h. Culture supernatants were collected as MCM and subsequently stored at −80°C.
Table 1.
HIV-1 isolates
| HIV-1 isolate | Classification | Source | Reference | TCID50/mla |
|---|---|---|---|---|
| ADA | Lab-adapted B | PBMCs | Gendelman et al. (1988) | 6.31 × 104 |
| JR-FL | Lab-adapted B | Brain Tissue | Koyanagi et al. (1987) | 2.51 × 104 |
| D02-2562 BG | Primary B | Basal ganglia | Burkala et al. (2005) | 2.51 × 104 |
| A00-086 CPx | Primary B | Choroid plexus | Burkala et al. (2005) | 1.26 × 104 |
| D02-2562 Sp | Primary B | Spleen | Burkala et al. (2005) | 1.58 × 105 |
| G0048 CPx | Primary B | Choroid plexus | Burkala et al. (2005) | 1.58 × 105 |
| 2873M | Primary C | PBMCs | Zhang et al. (2006) | 1.00 × 104 |
| 2669M | Primary C | PBMCs | Zhang et al. (2006) | 2.00 × 104 |
| 1084I | Primary C | PBMCs | Zhang et al. (2006) | 1.00 × 105 |
| 1157I | Primary C | PBMCs | Zhang et al. (2005) | 2.51 × 104 |
TCID50/ml determined by titration in TZM-bl cells as detailed in the Sect. Materials and Methods
Monitoring of Viral Replication and Cytopathicity
HIV-1 reverse transcriptase (RT) activity was determined from triplicate samples of cell culture supernatants as previously described (Koenig et al. 1986). Briefly, 10 μl of supernatant was incubated in a reaction mixture of 0.05% Nonidet P-40, 10 μg of poly(A)/ml, 0.25 μg of oligo(dT)/ml, 5 mM dithiothreitol, 150 mM KCl, 15 mM MgCl2, and [3H]TTP in Tris–HCl buffer (pH 7.9) for 24 h at 37°C. Radiolabeled nucleotides were precipitated with cold 10% trichloroacetic acid on filter paper plates in an automatic cell harvester and washed with 95% ethanol. Radioactivity was estimated by liquid scintillation spectroscopy. HIV-1 p24 antigen levels were determined in culture supernatants by enzyme-linked immunosorbent assay—ELISA (Beckman Coulter, Miami, FL) according to the manufacturer’s instructions.
The efficiency of infection was analyzed after 14 and 21 days using flow cytometry (FACS Caliber, BD Biosciences) by evaluating the number of p24-expressing cells. Following culture in 50 ml Teflon flasks, cells were washed with Flow Cytometry Stain Buffer (BD Pharmingen, BD Biosciences) and incubated at room temperature in the dark for 30 min with APC mouse antihuman CD14-conjugated antibody or APC mouse IgG isotype control (BD Pharmingen). Cells were washed with stain buffer and fixed in the dark at room temperature for 20 min using fixation buffer (eBiosciences, San Diego, California). Cells were then washed twice using permeabilization buffer (eBiosciences) and incubated at room temperature for 20 min in the dark with HIV-1 p24 FITC-conjugated antibody (Santa Cruz Biotechnology) or FITC mouse IgG isotype control (BD Pharmingen). Cells were washed twice and resuspended in stain buffer, and HIV-1 p24 cells were then determined by flow cytometry and analyzed with BD CellQuest Software. At least 10,000 cells were analyzed per sample. HIV-1 p24-positive cells were also detected by immunocytochemistry. Human MDM were plated on 15-mm cover slips in 24-well plates and fixed with 4% paraformaldehyde at room temperature for 20 min after 7, 14, or 21 days of infection. Fixed cells were blocked with 3% BSA in PBS and incubated at 4°C overnight with primary antibodies to HIV-1 p24 (monoclonal mouse, DAKO Corp, Carpinteria, CA). Cultures were washed and incubated for 1 h at room temperature with fluorophore-labeled secondary antibody Alexa Fluor 488 goat antimouse IgG (Molecular Probes, Eugene, OR). Cover slips were then mounted on glass slides with VECTASHIELD Hard Set Mounting Medium with DAPI (VECTOR Laboratories, Inc, Burlingame, CA). Double immunostaining with CD68 and p24 and morphological changes were visualized with a Nikon Eclipse E800 fluorescent microscope equipped with a digital imaging program. Photographs have a magnification of ×200.
MAP-2 ELISAs
MAP-2 neuronal antigen was determined in RCN treated with MCM using colorimetric ELISA as described previously (Zheng et al. 2001). Briefly, fixed RCN were blocked with 3% normal goat serum in PBS and incubated for 1 h with antibodies against MAP-2 (monoclonal mouse, Millipore-Chemicon International, Billerica, MA), followed by antimouse biotinylated antibody (VECTOR Laboratories) for 30 min. Avidin/biotin complex solution (VECTOR Laboratories) was added for 30 min, then color was developed using TMB substrate (Sigma–Aldrich) and terminated with 1 M H2SO4 (Sigma–Aldrich). The absorbance was read at 450 nm using a microplate reader (Bio-Rad Laboratories, Hercules, CA). About 50 μM glutamate (Sigma–Aldrich) was used as a positive neurotoxicity control.
MTT Reduction Assay
Cell viability of MDM and RCN was assessed by MTT reduction as described previously (Jiang et al. 2001; Zhao et al. 2004). Briefly, cells were incubated with 10% MTT (Sigma–Aldrich) solution in Neurobasal media for 30 min at 37°C. The extent of MTT conversion to formazan by mitochondrial dehydrogenase, indicating cell viability, was determined by measuring absorbance at 490 nm using a microplate reader (Bio-Rad Laboratories). About 50 μM glutamate (Sigma–Aldrich) and the NMDA receptor antagonist MK801 (Invitrogen Co., Carlsbad, CA) alone and in combination were used as controls.
In Situ TUNEL Assay
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was used to determine apoptosis of human neurons treated with 25% MCM. About 3 days after treatment, apoptotic neurons were detected using the In Situ Cell Death Detection kit (Roche, Manheim, Germany), following the manufacturer’s instructions. Results are expressed as a percentage of TUNEL-positive cells over the total amount of neurons (nuclei stained by DAPI). Random images were acquired from immunostained fields using a Nikon Eclipse E800 microscope. A minimum of ten fields in duplicate wells was counted for each treatment condition and photographs have a magnification of ×200.
Statistical Analyses
The data were evaluated statistically by the analysis of variance (ANOVA), followed by a Bonferroni posttest or Tukey’s test for multiple comparisons. Data were analyzed as ± standard error of the mean (SEM), and significance was determined as p < 0.05. Spearman’s correlation was used to examine associations. To account for any donor-specific differences, all experiments were performed in triplicate from at least three donors.
Results
Replication Differences in MDM Among a Panel of Primary HIV-1 Isolates
Investigations on HIV-1 infection in MDM using primary clade C isolates have been sparse. We obtained a panel of CCR5-tropic HIV-1 primary isolates, which included four clade B isolates D02-2562 BG, A00-086 CPx, A00-275 Sp, and G0048 CPx and four clade C isolates 2873M, 2669M, 1084I, and 1157I (Table 1). All primary isolates were initially cultured and expanded in PBMCs. Viral replication in PBMCs was monitored by measuring RT activity at 3, 5, 10, and 14 days postinfection (Supplemental Fig. 1). Isolates from both clades generated a comparable exponential increase of RT activity in the culture medium. Intraclade differences were evident as isolate D02-2562 Sp (clade B) had higher RT activity than all other isolates. Similarly, clade C isolates 1084I and 1157I had lower RT activity at all time points. The primary isolates were titrated in TZM-bl cells and comparable titers were found (Table 1). To characterize the infection course in primary human MDM, two clade B laboratory-adapted strains (HIV-1ADA and HIV-1JR-FL) were used as infection controls, whereas uninfected MDM were used as negative controls in each experiment. We compared the replication kinetics of viral isolates by performing parallel infections in MDM with equivalent titers (400 TCID50/ml) and measured the HIV-1 RT activity at different time points during the infection course. Primary isolates of clade B generated an exponential increase of virus in the medium and reached a maximum, comparable to laboratory-adapted strains, at 7 and 14 days postinfection (Fig. 1a and b). The clade C isolates exhibited a delayed increase in replication, as measured by RT activity, and reached a peak approximately 21 days postinfection (Fig. 1c). Through 14 days of infection, RT activity of clade C isolates in the culture medium was higher than the uninfected control, but significantly lower than day-matched clade B culture medium (p < 0.001). Replication capacity of clade B and C isolates was comparable (p > 0.05) at 21 days postinfection. However, RT activity of clade C isolates at 21 days postinfection was lower than clade B RT activity at 14 days postinfection (p < 0.001). RT activity at 28 days postinfection revealed clade C isolates, with the exception of 2669M, reached maximum replication at 21 days postinfection (data not shown). Maintenance of MDM cultures beyond 21 days becomes difficult because of the cell death that results from extensive periods of in vitro culture.
Fig. 1.
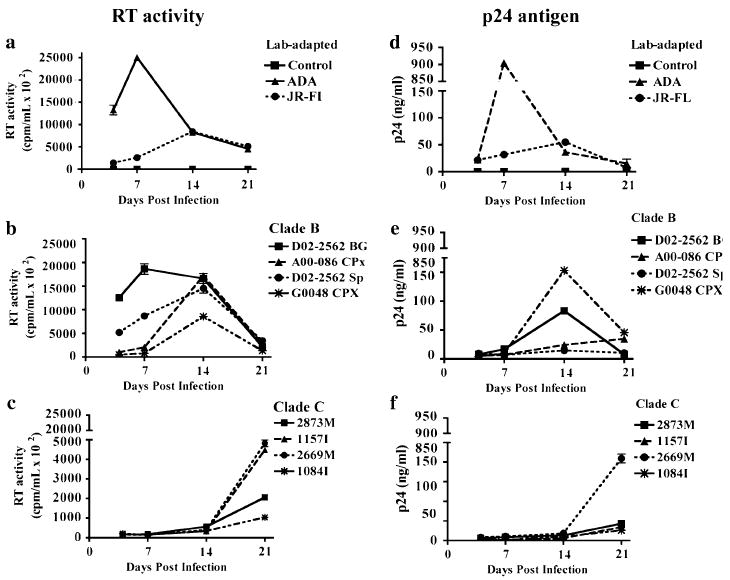
Viral replication of HIV-1 clade B and C isolates in human MDM. Macrophage cultures were infected with 400 TCID50/ml of HIV-1 laboratory-adapted strains, clade B, or clade C primary isolates. Supernatants were collected at days 4, 7, 14, and 21 postinfection. Viral replication was monitored through RT activity (a–c) and p24 antigen production (d–f). Values represent SEM from three independent experiments
To further characterize the infection course, we measured HIV-1 p24 antigen levels in cell culture supernatants using ELISA. Consistent with the RT activity, p24 production for laboratory-adapted strains (Fig. 1d) and for clade B primary isolates (Fig. 1e) peaked from 7 to 14 days postinfection. In contrast, p24 levels in clade C-infected cultures revealed a maximum average production 21 days postinfection (Fig. 1f). Taken together, these experiments suggest the four HIV-1 clade C primary isolates studied here, in comparison to clade B, replicate with delayed kinetics leading to a less robust infection in primary MDM.
Limited Cytopathicity in MDM Infected by Clade C Isolates
We have previously reported that productive infection by HIV-1 laboratory-adapted strains induces cell death (Cui et al. 2008). To determine the extent of cytopathicity induced by HIV-1 primary isolates, we infected MDM with 400 TCID50/ml of each of the viral isolates (Table 1) and evaluated MDM viability by MTT reduction assay at 4, 7, 14, and 21 days postinfection. The viability of infected MDM cultures was compared with day-matched uninfected MDM. Consistent with previous findings from our laboratory, productive infection with laboratory-adapted strains significantly reduced MDM viability at 14 and 21 days postinfection (Fig. 2a). Similarly, infection by clade B primary isolates significantly decreased cellular viability by 14 days postinfection (Fig. 2b). On the contrary, clade C-infected MDM retained higher viability than clade B throughout a period of 21 days (Fig. 2c). These findings confirm that clade B primary isolates exhibit higher levels of cytopathicity when compared with the clade C primary isolates under study.
Fig. 2.
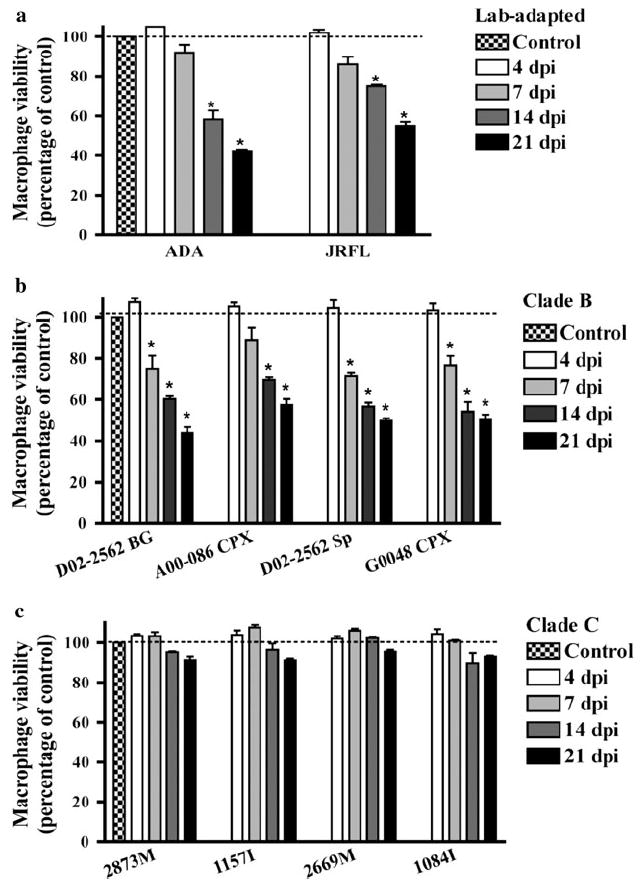
Cellular viability of macrophages infected by clade B or C isolates. MDM cultures were infected with 400 TCID50/ml of HIV-1 laboratory-adapted strains (a), clade B (b), or clade C (c) isolates. Supernatants were collected at 4, 7, 14, and 21 days postinfection (dpi). Cell viability was determined by MTT reduction assay and results correspond to the percentage of viable cells normalized to day-matched uninfected control MDM. Values represent SEM from three independent experiments and * denotes p < 0.01 in comparison to control
Neurotoxicity Mediated by MDM Infection With Clade B Isolates, but Not Clade C
Both HIV-1-infected and immune-activated macrophages secrete viral proteins, inflammatory factors, glutamate, and other molecules that contribute to neurotoxicity, causing neuronal injury, dysfunction, and death (Kaul et al. 2005). We evaluated indirect macrophage-mediated neurotoxicity through a well-defined RCN culture system. MCM from MDM cultures infected at 400 TCID50/ml was added to RCNs and cell viability was determined by MTT assay. Glutamate treatment significantly reduced RCN viability and was used as a positive control for neurotoxicity (data not shown). MCM, collected 14 and 21 days postinfection with clade B isolates, significantly reduced RCN viability (Fig. 3a and b, gray bars). In contrast, treatment with MCM collected at 14 and 21 days postinfection from clade C-infected cultures did not significantly reduce RCN viability (Fig. 3, closed bars). The data suggest conditioned medium from MDM infected with equal initial viral titers of clade B isolates, but not the four clade C isolates used, mediate significant indirect neurotoxicity in RCNs.
Fig. 3.
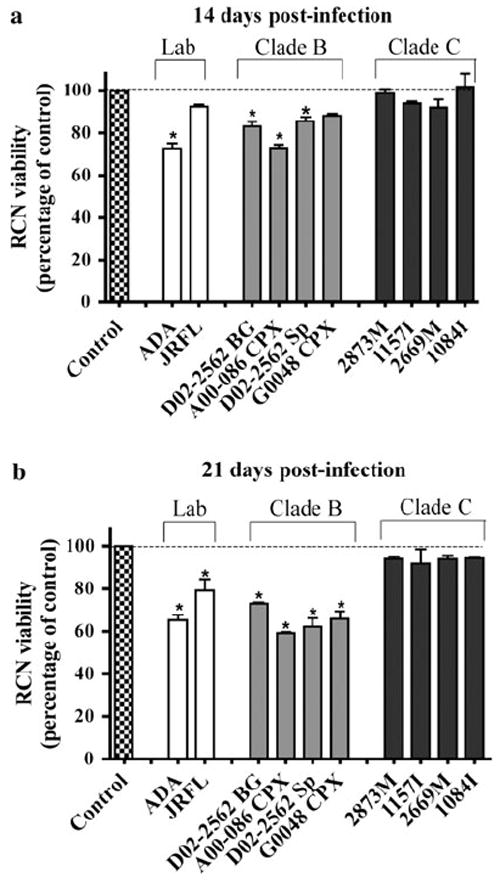
Neurotoxicity of HIV-1 clade B or C-infected MDM in RCN. RCN were treated for 4 days using conditioned media collected at 14 (a) and 21 (b) days after infection by a panel of HIV-1 laboratory-adapted strains and primary isolates of clades B and C. Cell viability was determined by MTT assay and neurotoxicity corresponds to the percentage of viable cells normalized to day-matched uninfected control conditioned media. Values represent SEM from three independent experiments and * denotes p < 0.01 in comparison to control
Increase in Replication Capacity for Clade C Isolate 2669M
As it has been suggested that differences in macrophage-mediated neurotoxicity between primary isolates of clades B and C may be attributable to unequal levels of viral replication in MDM, we performed parallel infections of MDM with increasing viral titers for clade C isolates. We measured RT activity at days 7, 14, and 21 to monitor the progress of infection and to identify which viral titers produced comparable replication levels between isolates of both clades (data not shown). Viral replication, as measured by RT activity, was comparable in cultures infected by 200 TCID50/ml of D02-2562 (clade B) and 800 TCID50/ml of 2669M (clade C) (Fig. 4a). In parallel with viral replication assessment, we evaluated the cellular viability of infected MDM (Fig. 4b). Cellular viability was significantly decreased at 14 and 21 days following infection with D02-2562. The reduction in MDM viability was consistent with the decrease in viability observed following infection with 400 TCID50/ml of clade B isolates (Fig. 2). MDM infected with 800 TCID50/ml of 2669M remained viable throughout the course of infection.
Fig. 4.
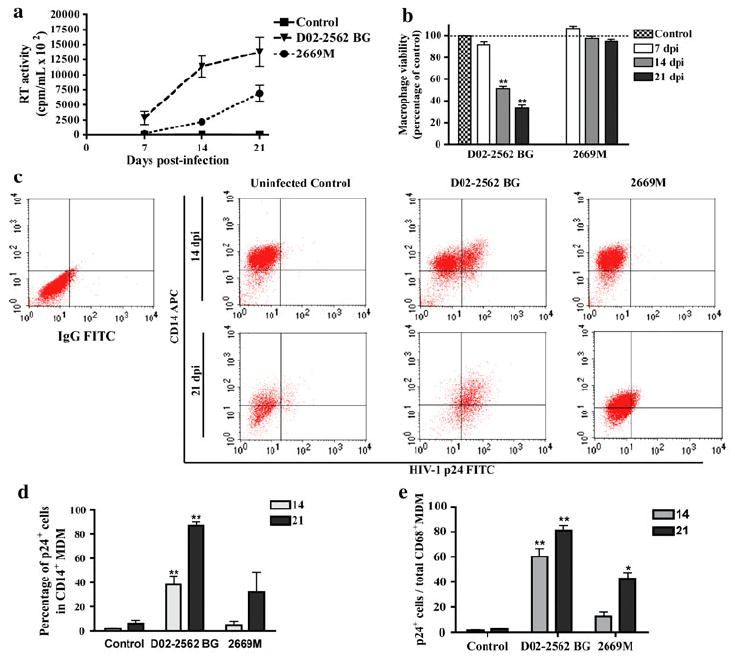
Replication capacity of isolates D02-2562 BG (clade B) and 2669M (clade C) following viral titer adjustment. MDM cultures were infected with 200 TCID50/ml of D02-2562 (clade B primary isolate) or 800 TCID50/ml 2669M (clade C primary isolate) for 7, 14, and 21 days. Viral replication was monitored in cell culture supernatants through RT activity (a). MDM viability was determined by MTT reduction assay (b). MTT results correspond to the percentage of viable cells normalized to day-matched controls. Efficiency of infection was analyzed after 14 and 21 days postinfection (dpi) using flow cytometry by evaluating the number of p24-expressing MDM (c). Percentage of p24-positive cells in CD14+ MDM with mean values shown for independent experiments (n = 6 for 14 dpi and n = 4 for 21 dpi) using MDM from different donors (d). Quantification of MDM stained with HIV-1 p24 and presented as percentage of total CD68-positive MDM (e). Values represent SEM from independent experiments and * denotes p < 0.01 in comparison to control
To directly compare the infection course in MDM, we analyzed the amount of HIV-1 p24-positive cells in the infected cultures. At 14 and 21 days postinfection, FACS analysis revealed that the number of MDM infected by clade C isolate 2669M was minimal compared to isolate D02-2562 BG (Fig. 4c). In cultures infected by D02-2562 BG, 38 and 87% of MDM were p24-positive by 14 and 21 days postinfection, respectively (Fig. 4d). The number of p24-positive MDM in 2669M cultures increased as infection progressed over time, as typically 5% of the MDM were p24-positive and the amount continued to increase to 32% by 21 days postinfection. These findings were corroborated by manual quantification of p24-positive MDMs using immunostaining, where isolate D02-2562 BG infected a significantly higher number of cells (Fig. 4e). Additionally, MDM cultures were observed for MNGC formation using HIV-1 p24 antigen and nuclear DAPI staining (Supplemental Fig. 2). MNGCs are formed by the fusion of macrophages and are a characteristic histopathologic feature of HIV-associated neuropathologies (Boisse et al. 2008; Navia et al. 1986). By day 21 postinfection, approximately 60% of cells in D02-2562BG-infected cultures were MNGCs. Although infection with 2669M (clade C) revealed p24-positive cells, MNGC formation was not evident (0% of infected cells were MNGCs) throughout the course of infection. Because a small number MDM are infected by 2669M-clade C isolate, these findings offer a possible explanation for the observation of negligible neurotoxicity.
Productive Infection Leads to Neurotoxicity
We assessed the indirect neurotoxic effects of HIV infection on RCN and found that neuronal viability was significantly reduced following treatment of RCN with MCM collected at 7, 14, and 21 days postinfection with 200 TCID50/ml of D02-2562 (Fig. 5a). Similar to previous results (Fig. 3), treatment with MCM collected at 7 and 14 days from cultures infected with 2669M had no significant effect on RCN, as viability was comparable to cultures treated with MCM from uninfected MDM. In contrast, treatment with MCM collected at 21 days from 2669M-infected cultures had a significant impact on RCN viability (Fig. 5a). MAP-2 ELISAs were used to quantify the extent of the damage in RCN following a 3-day treatment with conditioned medium from infected MDM cultures (Fig. 5b). MAP-2 is found on neuronal dendrites and cell body; thus, loss of MAP-2 immunoreactivity is more sensitive for determining the extent of neuronal injury and demise. MCM collected from D02-2562-infected cultures for 21 days caused more than 20% reduction of MAP-2 in treated RCN when compared to MCM from uninfected controls. Similarly, MCM collected 21 days after 2669M infection decreased neuronal MAP-2 expression, albeit this reduction was closer to 10–15%. The data suggest conditioned medium from MDM, infected by clade B or C primary isolates, causes neuronal damage. Additionally, viral replication as determined by RT activity in the culture medium correlates with macrophage-mediated indirect neurotoxicity (Fig. 5c), suggesting that at high levels of productive infection, HIV isolates may induce neurotoxicity of RCN.
Fig. 5.
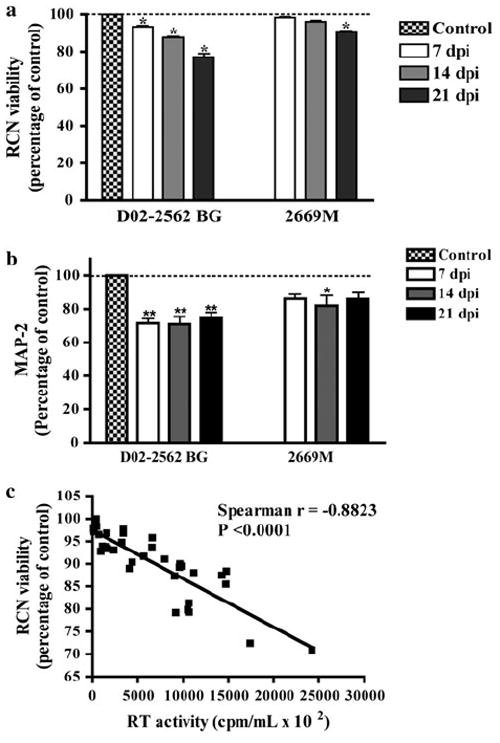
Assessment of neurotoxicity in RCN following treatment with conditioned medium from infected cultures. RCN were treated with MCM collected from cultures infected for 7, 14, and 21 days postinfection (dpi) with 200 TCID50/ml of D02-2562 (clade B primary isolate) or 800 TCID50/ml 2669M (clade C primary isolate). RCN viability was assessed by MTT assay (a). MTT results correspond to the percentage of viable cells normalized to day-matched controls. MAP-2 percentage was determined using ELISA for RCN cultures treated with clade B or C MCM (b). MAP-2 is expressed as a percentage in comparison to control MCM. The association between RT activity levels and RCN viability following treatment with MCM collected at 14 and 21 dpi was determined by Spearman’s correlation (c). Values represent SEM from three independent experiments and * and ** denote p < 0.05 and p < 0.01, respectively in comparison to control
To test whether conditioned medium also had neurotoxic effects on human neurons, cultures were treated for 3 days with MCM collected at 7, 14, and 21 days postinfection with a clade B or C isolate, and cell death was determined using TUNEL assays. Apoptosis induced by MCM from infected macrophages was evident, as cultures had visible nuclear condensation and cell shrinkage (Fig. 6a). Quantification of TUNEL assays showed treatment with D02-2562 MCM caused more apoptosis than treatment with 2669M MCM (Fig. 6b). Taken together, these results suggested that increased viral replication by primary isolates of clades B and C leads to neurotoxicity, with clade B primary isolates being the most neurotoxic to both rat and human neurons.
Fig. 6.
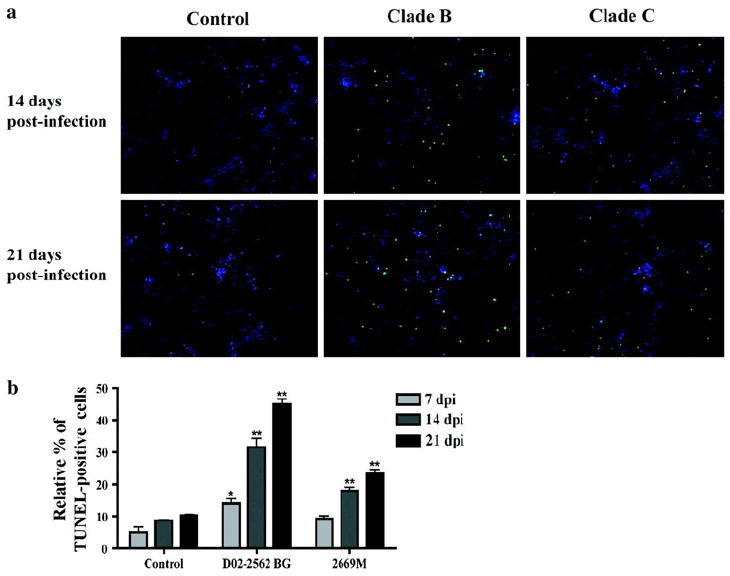
Apoptosis of human neurons following treatment with conditioned medium from infected cultures. Human neurons were treated for 3 days with MCM collected from cultures infected for 7, 14, and 21 days with 200 TCID50/ml of D02-2562 (clade B primary isolate) or 800 TCID50/ml 2669M (clade C primary isolate). Apoptotic cells were labeled with TUNEL, and nuclei of total cells were stained with DAPI (a). Photographs have a magnification of ×200. Neuronal apoptosis is expressed as a percentage of TUNEL-positive cells over the total amount of neurons (nuclei staining by DAPI, b). Data values represent SEM and * denotes p < 0.01 in comparison to control
Discussion
HIV-1 clade B-infected macrophages drive the pathogenesis of HAND seen in infected individuals. The low incidence of severe neurological manifestations associated with HIV-1 clade C infection does not correlate with the preferential CCR5 tropism of clade C viruses. In this study, we have characterized the replication efficiency and macrophage-mediated neurotoxicity of a panel of eight primary isolates from HIV-1 clades B and C. When performing parallel infections of MDM using similar initial viral titers, clade B primary isolates replicated more efficiently. Viral replication of all four clade C isolates in MDM was slow in comparison to clade B isolates. However, by increasing the viral titer of clade C isolates, we were able to obtain replication levels comparable to clade B. MDM infected with clade B isolates had lower viability than cells infected with clade C isolates, even when a lower initial viral titer was used for infection. Conditioned medium, obtained from MDM infected with clade B primary isolates, displayed neurotoxic properties. MCM from macrophages infected with clade C isolates induced neurotoxicity when higher initial viral titers were used to achieve a productive infection, as determined by replication capacity (RT activity). Lower levels of neurotoxicity by clade C isolates in this report are likely the result of an efficient, albeit slow in vitro viral replication in MDM.
Recently, understanding the effects of HIV-1 clade diversity on the induction of neuropathological disorders has obtained great interest. Neuropsychological testing revealed a lower incidence of severe neurological impairment, such as HAD in regions where clade C predominates (Clifford et al. 2007; Riedel et al. 2006). Interclade differences in macrophage-mediated neurotoxicity may account for the observed discrepancies in neurological dysfunction between HIV-1 clades B and C. Our results demonstrate that in vitro replication of clade C isolates in primary human MDM is delayed compared to clade B isolates. The replication kinetics of clade C primary isolates used in this investigation (Zhang et al. 2006) and their cloned proviruses (Zhang et al. 2010) has been studied previously along with NL4-3, a laboratory-adapted virus. For this investigation, we wanted to compare a panel of primary isolates using the same TCID50/ml to reflect the difference in replication efficiency in MDM. We then adjusted the macrophage infection by using 200 TCID50/ml for clade B isolate D02-2562 and 800 TCID50/ml for clade C isolate 2669M to achieve similar levels of viral replication (Figs. 4-6).
Clade C primary isolates have equal transmission fitness but reduced pathogenic fitness relative to other group M HIV-1 isolates (Abraha et al. 2009). Additionally, clade C R5 viruses exhibit slower replication profiles on CD4+ lymphocytes in comparison to clade B viruses, whereas clade C X4 viruses replicated with comparable efficiency to clade B X4 viruses (Pollakis et al. 2004). Nevertheless, delayed replication kinetics of clade C isolates in MDM cultures does not imply that poor replication is expected in vivo or in other cell populations. Increased in vitro fitness of clade B over clade C has been previously observed in human PBMCs, CD4+ T cells, and blood-derived macrophages, but not in skin-derived Langerhans cells (Ball et al. 2003). However, the cytokine network of the gut-associated lymphoid tissue supports HIV-1 clade C viral replication via preferential activation of the clade C promoter (Centlivre et al. 2006). Accordingly, replication kinetics and infection progress of clade C isolates may be affected or even restricted by delayed viral entry, reverse transcription, integration to the host-cell genome, or induction of the viral promoter.
It is well known that clade C is preferentially a CCR5-tropic virus with an in vitro nonsyncytium-inducing (NSI) phenotype (Abebe et al. 1999; Thompson et al. 2002). The NSI phenotype is traditionally evaluated in vitro by observing syncytia formation in MT-2 T lymphoblasts. Within the brain, mononuclear phagocyte-derived MNGCs occur as a response to viral infection and are considered a hallmark of severe HIV-1 clade B-associated neuropathology. We previously demonstrated that productive HIV-1ADA infection induces the formation of MNGCs and decreases cellular viability via apoptosis in MDM cultures (Cui et al. 2008; Huang et al. 2006). Although clade C-infected macrophages are found in HIV-infected brain tissue during autopsy, MNGC are absent (Mahadevan et al. 2007). Our current results reinforce these findings by demonstrating a lack of MNGC formation in primary MDM cultures infected with primary clade C isolates. Furthermore, this outcome provides evidence of remarkable macrophage cytopathicity, including formation of MNGC and cell death in MDM infection with clade B, but not with clade C isolates.
To the best of our knowledge, this is the first report comparing neurotoxicity of RCN and human neurons mediated by primary human macrophages infected by primary isolates of HIV-1 clades B and C. This approach allowed us to obtain physiologically relevant results regarding effects on infection progression in MDM and resulting neuronal toxicity. Previous studies have compared the in vitro neurotoxicity of HIV viral proteins and demonstrated that clade C Tat has an attenuated phenotype that renders it less neurotoxic than its clade B counterpart (Campbell et al. 2007; Li et al. 2008; Mishra et al. 2008). Diminished clade C neuropathogenesis has been confirmed in vivo using infectious molecular clones of clades B (HIV-1ADA) and C (HIV-1Indie-C1) in the HIVE SCID mouse model (Rao et al. 2008). These in vivo studies demonstrated that exposure to clade C resulted in mild and not severe cognitive dysfunction, whereas mice exposed to HIV-1ADA exhibited increased astrogliosis and neuronal loss. Moreover, commonly used laboratory-adapted strains are cultured in vitro through multiple rounds of infection and may not be representative of primary isolates. Replication efficiency increases over time in culture and possibly creates a more infectious strain of HIV than primary isolates (Lawson et al. 2002; Simon et al. 2003). Indeed, HIV-1ADA exhibited faster and higher infection efficiency than the primary isolates in our macrophage system. Hence, this investigation is unique in that we infected primary MDM and assessed neurotoxicity using primary isolates that had similar culturing history and were obtained from infected individuals in the USA and Africa.
A possible limitation to our study is that we used clade C blood isolates that may have a skewed pathogenic potential to a less neurotoxic phenotype when compared to clade B brain isolates. Brain-derived viruses are more neurotoxic than blood isolates, potentially the result of variations in the viral envelope (Power et al. 1998). The gp120 variant Asn 283 (N283) is more frequent in isolates from brain tissues of HAD patients, such as HIV-1ADA, compared to isolates from lymphoid and non-HAD brain tissues (Dunfee et al. 2006). N283 enhances macrophage tropism by enhancing viral fusion and entry when low levels of CD4 are expressed. Notably, we analyzed 528 clade C envelope sequences from the Los Alamos HIV Sequence Database and found that only one isolate, 05ZAFV7, (GenBank: DQ382365; Michler et al. 2008) contained N283. Clade C isolates used in this investigation lack the N283 mutation and may be at a disadvantage for replication in macrophages by having a limited capacity to use low-CD4 expression levels. However, it is important to note that clade C isolates in this study mediated neurotoxicity at high viral titers in our MDM culture system; suggesting neurotoxicity is a possible result of increased viral replication. Certainly, it would be interesting to assess the neurotoxicity of clade C brain isolates when they become available.
In conclusion, clade C isolates investigated herein possess slow replication kinetics in MDM and induce neurotoxicity at high viral replication levels. These clade C isolates have a neurotoxic potential that may be of consequence if viral infection progresses and immunosuppression is established. Multiple factors, including host and viral genetic factors, clinical characteristics, and opportunistic infections, may provide the environment for development of milder forms of HAND. As a consequence, it is possible that clade C-infected patients may be at risk of developing more severe cognitive impairment as disease progresses and more infected macrophages infiltrate the brain. Future studies on the mechanisms leading to variations of replication in macrophages and subsequent inflammatory responses among isolates of clades B and C and the relationship between these pathways and differences in neurovirulence can clarify the disparity of HAND incidence.
Supplementary Material
Acknowledgments
We thank Dr. Nicholas P. Whitney for assistance with immunocytochemistry, microscopy, and revision of the manuscript; Dr. Hui Peng, Dr. Changhai Tian, and Dr. Min Cui for helpful discussions regarding experiments and the manuscript. Myhanh Che, Na Ly, and Emilie Scoggins provided exceptional administrative support. This work was supported in part by research grants by the National Institutes of Health, Grant numbers: R01NS41858, R01NS61642, R21MH83525, P20RR15635, and P01NS43985; National Natural Science Foundation of China, Grant number: 81028007 to Jialin C. Zheng; CA75903 and NCRR COBRE grant RR15635 to Charles Wood; and University of Nebraska Medical Center graduate student fellowship to Agnes A. Constantino.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s12640-011-9241-3) contains supplementary material, which is available to authorized users.
Contributor Information
Agnes A. Constantino, Laboratory of Neuroimmunology and Regenerative Therapy, Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, 985930 Nebraska Medical Center, Omaha, NE 68198-5930, USA
Yunlong Huang, Laboratory of Neuroimmunology and Regenerative Therapy, Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, 985930 Nebraska Medical Center, Omaha, NE 68198-5930, USA.
Hong Zhang, Nebraska Center for Virology and The School of Biological Sciences, University of Nebraska, Lincoln, NE 68583-0900, USA.
Charles Wood, Nebraska Center for Virology and The School of Biological Sciences, University of Nebraska, Lincoln, NE 68583-0900, USA.
Jialin C. Zheng, Laboratory of Neuroimmunology and Regenerative Therapy, Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, 985930 Nebraska Medical Center, Omaha, NE 68198-5930, USA jzheng@unmc.edu Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, NE 68198-5930, USA.
References
- Abebe A, Demissie D, Goudsmit J, Brouwer M, Kuiken CL, Pollakis G, Schuitemaker H, Fontanet AL, Rinke de Wit TF. HIV-1 subtype C syncytium- and non-syncytium-inducing phenotypes and coreceptor usage among Ethiopian patients with AIDS. AIDS. 1999;13:1305–1311. doi: 10.1097/00002030-199907300-00006. [DOI] [PubMed] [Google Scholar]
- Abraha A, Nankya IL, Gibson R, Demers K, Tebit DM, Johnston E, Katzenstein D, Siddiqui A, Herrera C, Fischetti L, et al. CCR5- and CXCR4-tropic subtype C human immunodeficiency virus type 1 isolates have a lower level of pathogenic fitness than other dominant group M subtypes: implications for the epidemic. J Virol. 2009;83:5592–5605. doi: 10.1128/JVI.02051-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayehunie S, Sonnerborg A, Yemane-Berhan T, Zewdie DW, Britton S, Strannegard O. Raised levels of tumour necrosis factor-alpha and neopterin, but not interferon-alpha, in serum of HIV-1-infected patients from Ethiopia. Clin Exp Immunol. 1993;91:37–42. doi: 10.1111/j.1365-2249.1993.tb03350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball SC, Abraha A, Collins KR, Marozsan AJ, Baird H, Quinones-Mateu ME, Penn-Nicholson A, Murray M, Richard N, Lobritz M, et al. Comparing the ex vivo fitness of CCR5-tropic human immunodeficiency virus type 1 isolates of subtypes B and C. J Virol. 2003;77:1021–1038. doi: 10.1128/JVI.77.2.1021-1038.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisse L, Gill MJ, Power C. HIV infection of the central nervous system: clinical features and neuropathogenesis. Neurol Clin. 2008;26:799–819. x. doi: 10.1016/j.ncl.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Burkala EJ, He J, West JT, Wood C, Petito CK. Compartmentalization of HIV-1 in the central nervous system: role of the choroid plexus. AIDS. 2005;19:675–684. doi: 10.1097/01.aids.0000166090.31693.aa. [DOI] [PubMed] [Google Scholar]
- Campbell GR, Watkins JD, Singh KK, Loret EP, Spector SA. Human immunodeficiency virus type 1 subtype C Tat fails to induce intracellular calcium flux and induces reduced tumor necrosis factor production from monocytes. J Virol. 2007;81:5919–5928. doi: 10.1128/JVI.01938-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centlivre M, Sommer P, Michel M, Ho Tsong Fang R, Gofflo S, Valladeau J, Schmitt N, Wain-Hobson S, Sala M. The HIV-1 clade C promoter is particularly well adapted to replication in the gut in primary infection. AIDS. 2006;20:657–666. doi: 10.1097/01.aids.0000216365.38572.2f. [DOI] [PubMed] [Google Scholar]
- Clifford DB, Mitike MT, Mekonnen Y, Zhang J, Zenebe G, Melaku Z, Zewde A, Gessesse N, Wolday D, Messele T, et al. Neurological evaluation of untreated human immunodeficiency virus infected adults in Ethiopia. J Neurovirol. 2007;13:67–72. doi: 10.1080/13550280601169837. [DOI] [PubMed] [Google Scholar]
- Cui M, Huang Y, Zhao Y, Zheng J. Transcription factor FOXO3a mediates apoptosis in HIV-1-infected macrophages. J Immunol. 2008;180:898–906. doi: 10.4049/jimmunol.180.2.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O’Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol. 2000;74:8358–8367. doi: 10.1128/jvi.74.18.8358-8367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunfee RL, Thomas ER, Gorry PR, Wang J, Taylor J, Kunstman K, Wolinsky SM, Gabuzda D. The HIV Env variant N283 enhances macrophage tropism and is associated with brain infection and dementia. Proc Natl Acad Sci USA. 2006;103:15160–15165. doi: 10.1073/pnas.0605513103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendelman HE, Orenstein JM, Martin MA, Ferrua C, Mitra R, Phipps T, Wahl LA, Lane HC, Fauci AS, Burke DS. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J Exp Med. 1988;167:1428–1441. doi: 10.1084/jem.167.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta JD, Satishchandra P, Gopukumar K, Wilkie F, Waldrop-Valverde D, Ellis R, Ownby R, Subbakrishna DK, Desai A, Kamat A, et al. Neuropsychological deficits in human immunodeficiency virus type 1 clade C-seropositive adults from South India. J Neurovirol. 2007;13:195–202. doi: 10.1080/13550280701258407. [DOI] [PubMed] [Google Scholar]
- Huang Y, Erdmann N, Peng H, Herek S, Davis JS, Luo X, Ikezu T, Zheng J. TRAIL-mediated apoptosis in HIV-1-infected macrophages is dependent on the inhibition of Akt-1 phosphorylation. J Immunol. 2006;177:2304–2313. doi: 10.4049/jimmunol.177.4.2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Piggee C, Heyes MP, Murphy C, Quearry B, Bauer M, Zheng J, Gendelman HE, Markey SP. Glutamate is a mediator of neurotoxicity in secretions of activated HIV-1-infected macrophages. J Neuroimmunol. 2001;117:97–107. doi: 10.1016/s0165-5728(01)00315-0. [DOI] [PubMed] [Google Scholar]
- Kamat A, Ravi V, Desai A, Satishchandra P, Satish KS, Kumar M. Estimation of virological and immunological parameters in subjects from South India infected with human immunodeficiency virus type 1 clade C and correlation of findings with occurrence of neurological disease. J Neurovirol. 2009;15:25–35. doi: 10.1080/13550280802338652. [DOI] [PubMed] [Google Scholar]
- Kaul M, Zheng J, Okamoto S, Gendelman HE, Lipton SA. HIV-1 infection and AIDS: consequences for the central nervous system. Cell Death Differ. 2005;12(Suppl 1):878–892. doi: 10.1038/sj.cdd.4401623. [DOI] [PubMed] [Google Scholar]
- Koenig S, Gendelman HE, Orenstein JM, Canto MCD, Pezeshkpour GH, Yungbluth M, Janotta F, Aksamit A, Martin MA, Fauci AS. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. 1986;233:1089–1093. doi: 10.1126/science.3016903. [DOI] [PubMed] [Google Scholar]
- Kolson DL, Gonzalez-Scarano F. HIV and HIV dementia. J Clin Invest. 2000;106:11–13. doi: 10.1172/JCI10553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyanagi Y, Miles S, Mitsuyasu RT, Merrill JE, Vinters HV, Chen IS. Dual infection of the central nervous system by AIDS viruses with distinct cellular tropisms. Science. 1987;236:819–822. doi: 10.1126/science.3646751. [DOI] [PubMed] [Google Scholar]
- Lawson VA, Oelrichs R, Guillon C, Imrie AA, Cooper DA, Deacon NJ, McPhee DA. Adaptive changes after human immunodeficiency virus type 1 transmission. AIDS Res Hum Retroviruses. 2002;18:545–556. doi: 10.1089/088922202753747897. [DOI] [PubMed] [Google Scholar]
- Li W, Huang Y, Reid R, Steiner J, Malpica-Llanos T, Darden TA, Shankar SK, Mahadevan A, Satishchandra P, Nath A. NMDA receptor activation by HIV-Tat protein is clade dependent. J Neurosci. 2008;28:12190–12198. doi: 10.1523/JNEUROSCI.3019-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan A, Shankar SK, Satishchandra P, Ranga U, Chickabasaviah YT, Santosh V, Vasanthapuram R, Pardo CA, Nath A, Zink MC. Characterization of human immunodeficiency virus (HIV)-infected cells in infiltrates associated with CNS opportunistic infections in patients with HIV clade C infection. J Neuropathol Exp Neurol. 2007;66:799–808. doi: 10.1097/NEN.0b013e3181461d3e. [DOI] [PubMed] [Google Scholar]
- Marozsan AJ, Fraundorf E, Abraha A, Baird H, Moore D, Troyer R, Nankja I, Arts EJ. Relationships between infectious titer, capsid protein levels, and reverse transcriptase activities of diverse human immunodeficiency virus type 1 isolates. J Virol. 2004;78:11130–11141. doi: 10.1128/JVI.78.20.11130-11141.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michler K, Connell BJ, Venter WD, Stevens WS, Capovilla A, Papathanasopoulos MA. Genotypic characterization and comparison of full-length envelope glycoproteins from South African HIV type 1 subtype C primary isolates that utilize CCR5 and/or CXCR4. AIDS Res Hum Retroviruses. 2008;24:743–751. doi: 10.1089/aid.2007.0304. [DOI] [PubMed] [Google Scholar]
- Mishra M, Vetrivel S, Siddappa NB, Ranga U, Seth P. Clade-specific differences in neurotoxicity of human immunodeficiency virus-1 B and C Tat of human neurons: significance of dicysteine C30C31 motif. Ann Neurol. 2008;63:366–376. doi: 10.1002/ana.21292. [DOI] [PubMed] [Google Scholar]
- Navia BA, Cho ES, Petito CK, Price RW. The AIDS dementia complex: II. Neuropathology. Ann Neurol. 1986;19:525–535. doi: 10.1002/ana.410190603. [DOI] [PubMed] [Google Scholar]
- Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol. 1998;72:2855–2864. doi: 10.1128/jvi.72.4.2855-2864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollakis G, Abebe A, Kliphuis A, Chalaby MI, Bakker M, Mengistu Y, Brouwer M, Goudsmit J, Schuitemaker H, Paxton WA. Phenotypic and genotypic comparisons of CCR5- and CXCR4-tropic human immunodeficiency virus type 1 biological clones isolated from subtype C-infected individuals. J Virol. 2004;78:2841–2852. doi: 10.1128/JVI.78.6.2841-2852.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power C, McArthur JC, Nath A, Wehrly K, Mayne M, Nishio J, Langelier T, Johnson RT, Chesebro B. Neuronal death induced by brain-derived human immunodeficiency virus type 1 envelope genes differs between demented and nondemented AIDS patients. J Virol. 1998;72:9045–9053. doi: 10.1128/jvi.72.11.9045-9053.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao VR, Sas AR, Eugenin EA, Siddappa NB, Bimonte-Nelson H, Berman JW, Ranga U, Tyor WR, Prasad VR. HIV-1 clade-specific differences in the induction of neuropathogenesis. J Neurosci. 2008;28:10010–10016. doi: 10.1523/JNEUROSCI.2955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel D, Ghate M, Nene M, Paranjape R, Mehendale S, Bollinger R, Sacktor N, McArthur J, Nath A. Screening for human immunodeficiency virus (HIV) dementia in an HIV clade C-infected population in India. J Neurovirol. 2006;12:34–38. doi: 10.1080/13550280500516500. [DOI] [PubMed] [Google Scholar]
- Simon V, Padte N, Murray D, Vanderhoeven J, Wrin T, Parkin N, Di Mascio M, Markowitz M. Infectivity and replication capacity of drug-resistant human immunodeficiency virus type 1 variants isolated during primary infection. J Virol. 2003;77:7736–7745. doi: 10.1128/JVI.77.14.7736-7745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DA, Cormier EG, Dragic T. CCR5 and CXCR4 usage by non-clade B human immunodeficiency virus type 1 primary isolates. J Virol. 2002;76:3059–3064. doi: 10.1128/JVI.76.6.3059-3064.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother. 2002;46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepthomi T, Paul R, Vallabhaneni S, Kumarasamy N, Tate DF, Solomon S, Flanigan T. Neurocognitive consequences of HIV in southern India: a preliminary study of clade C virus. J Int Neuropsychol Soc. 2006;12:424–430. doi: 10.1017/s1355617706060516. [DOI] [PubMed] [Google Scholar]
- Zhang H, Hoffmann F, He J, He X, Kankasa C, Ruprecht R, West JT, Orti G, Wood C. Evolution of subtype C HIV-1 Env in a slowly progressing Zambian infant. Retrovirology. 2005;2:67. doi: 10.1186/1742-4690-2-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Hoffmann F, He J, He X, Kankasa C, West JT, Mitchell CD, Ruprecht RM, Orti G, Wood C. Characterization of HIV-1 subtype C envelope glycoproteins from perinatally infected children with different courses of disease. Retrovirology. 2006;3:73. doi: 10.1186/1742-4690-3-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Tully DC, Hoffmann FG, He J, Kankasa C, Wood C. Restricted genetic diversity of HIV-1 subtype C envelope glycoprotein from perinatally infected Zambian infants. PLoSONE. 2010;5(2):e9294. doi: 10.1371/journal.pone.0009294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Lopez AL, Erichsen D, Herek S, Cotter RL, Curthoys NP, Zheng J. Mitochondrial glutaminase enhances extracellular glutamate production in HIV-1-infected macrophages: link-age to HIV-1 associated dementia. J Neurochem. 2004;88:169–180. doi: 10.1046/j.1471-4159.2003.02146.x. [DOI] [PubMed] [Google Scholar]
- Zheng J, Gendelman HE. The HIV-1 associated dementia complex: a metabolic encephalopathy fueled by viral replication in mononuclear phagocytes. Curr Opin Neurol. 1997;10:319–325. [PubMed] [Google Scholar]
- Zheng J, Thylin M, Ghorpade A, Xiong H, Persidsky Y, Cotter R, Niemann D, Che M, Zeng Y, Gelbard H, et al. Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-1-associated dementia. J Neuroimmunol. 1999;98:185–200. doi: 10.1016/s0165-5728(99)00049-1. [DOI] [PubMed] [Google Scholar]
- Zheng J, Thylin MR, Cotter RL, Lopez AL, Ghorpade A, Persidsky Y, Xiong H, Leisman GB, Che MH, Gendelman HE. HIV-1 infected and immune competent mononuclear phagocytes induce quantitative alterations in neuronal dendritic arbor: relevance for HIV-1-associated dementia. Neurotoxi Res. 2001;3:443–459. doi: 10.1007/BF03033203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.