

Abstract
Accumulating research indicates that B cells are involved in anti-tumor immunity. Chronic alcohol consumption is associated with decreased survival of cancer patients. The effect of alcohol consumption on B cells in tumor-bearing hosts is unknown. Results in melanoma-bearing mice showed that chronic alcohol consumption does not alter the percentage and number of B cells in bone marrow, spleen and lymph nodes, but dramatically decreased B cells in the peripheral blood. Alcohol consumption did not alter the development of B cells in the bone marrow, nor did it affect follicular B cells in the spleen; however, it increased T1 B cells and decreased marginal zone B cells in the spleen. Alcohol consumption also decreased mature B cells in the blood. It did not alter the chemotactic capacity of plasma to facilitate migration of splenocytes, nor the chemotactic response of splenocytes to CXCL13 and CCL21. However, the response of splenocytes to sphingosine-1-phosphate (S1P) was impaired in alcohol-consuming, melanoma-bearing mice. The expression of sphingosine-1-phosphate receptor-1 (S1PR)1 and sphingosine-1-phosphate lyase-1 (SPL)1 in splenocytes was down regulated. Taken together, these results indicate that chronic alcohol consumption decreases peripheral blood B cells by compromising B cell egress from the spleen. The down regulation of S1PR1 and SPL1 expression in alcohol-consuming, melanoma-bearing mice could be associated with compromised egress of B cells from the spleen.
Keywords: Rodent, B cells, T cells, Chemotaxis, Tumor Immunity, Melanoma, Alcohol
Introduction
B cells are a large population of lymphocytes that originate from the bone marrow (BM) and further mature in the spleen. These cells play important roles in humoral immunity. Immature B cells in the spleen are called transitional B cells and are usually divided into two subsets, T1 B cells, which phenotypically are CD19+CD93+CD23− and T2 B cells, which phenotypically are CD19+CD93+CD23+ (1). Mature splenic B cells can also be divided into two subsets: follicular B cells and marginal zone (MZ) B cells. Folicular B cells circulate in the body and are characterized by the following phenotype: CD19+CD23hiIgDhiIgMlo. MZ B cells are non-circulating cells. These splenic B cells are the first line of defense against blood-borne antigens. MZ B cells express activated innate immune cell features and their signature cell surface markers are CD19+CD1dhiCD21hi (2, 3).
B cells not only play a pivotal role in humoral immunity by producing antibody, but also are involved in cellular immune responses by presenting antigen to T cells. B cells are the largest population of antigen-presenting cells in the immune system. They play an important role in antigen presentation, especially under low protein antigen conditions (4, 5). MZ B cells express high levels of CD1d, which is the molecule that presents lipid antigens to natural killer (NK) T cells, and they are key players in regulating the cytokine profile and activation of NKT cells (6–8). Since B cells must capture antigen to activate themselves to produce antibody and present antigen to T cells, effective recirculation among lymphatic organs and tissues is crucial for these cells to fulfill their function. B cell circulation and redistribution in lymphoid organs are directed by the CXCL13 chemokine and its receptor, CXCR5 (9, 10). The egress of B cells from lymph nodes (LN), spleen and BM is controlled by sphingosine-1-phosphate (S1P) and its sphingosine-1-phosphate receptor (S1PR) 1 (11–13).
Compared to extensive studies involving T cells in anti-tumor immunity, the knowledge of B cells in anti-tumor immune responses is limited and controversial. Some studies indicate that depletion of B cells inhibits tumor progression and enhances anti-tumor immunity, suggesting that B cells inhibit antitumor immune responses (14, 15). One of the reasons associated with the inhibitory effects of B cells on anti-tumor immunity is that some B cells produce high levels of IL-10, which inhibit T cell function (14). This suggests the existence of regulatory B cells. Recently, it was found that B cells exhibiting the CD5+CD1dhi phenotype produce IL-10 upon activation by LPS. These CD5+CD1dhi IL-10-producing cells are one subset of regulatory B cells called B10 cells (16, 17). Other studies indicate that B cells enhance T cell anti-tumor immunity. Depletion of B cells enhances B16 melanoma metastasis to the lung by inhibiting CD8+ T cell proliferation and Th1 cytokine production (18). Deficiency of mature B cells significantly compromises T cell-mediated anti-tumor immunity induced by the anti-glucocorticoid-induced TNFR-related protein monoclonal antibody, DTA-1 (19). Adaptive transfer of CpG-activated B cells inhibits tumor progression, while depletion of mature B cells increases tumor burden in the lungs of CpG-treated mice (20).
Chronic alcohol consumption in the melanoma-bearing mice leads to inhibition of memory T cell and especially tumor-specific CD8+ T cell expansion. This causes T cell dysfunction in B16BL6 melanoma-bearing mice (21). The underlying mechanism remains to be elucidated. Chronic alcohol consumption decreases B cells in the spleen of mice not inoculated with tumor (22). The relationship between chronic alcohol consumption and B cell anti-tumor immunity is unknown. Herein, using a chronic alcohol-consuming mouse model, we report that alcohol consumption 1) decreases B cells in the blood, but not in the spleen, LN or BM and 2) decreases MZ B cells and B10 cells in the spleens of mice inoculated subcutaneously with B16BL6 melanoma.
Material and Methods
Mice and alcohol administration
Female C57BL/6 mice at 6–7 weeks of age were purchased from NIH/NCI Charles River Laboratories (Wilmington, MA). After arrival, mice were single housed in filter toped transparent polycarbonate cages in the Wegner Hall Vivarium at Washington State University. Mice were allowed ad libitum access to sterilized Milli-Q water and Purina 5001 Rodent Laboratory Chow. After one week of acclimation, mice were randomly divided into two groups. One group was continuously given Milli-Q water and lab chow. The other group was given filter sterilized 20% (w/v) alcohol (Everclear, St. Louis, MO) and lab chow. The animal protocol used in this research was approved by the Institutional Animal Care and Use Committee at Washington State University.
Measurement of daily alcohol intake and blood alcohol concentration
The amount of daily alcohol intake was determined by measuring the starting weight and ending weight of alcohol in the feeding tube every other day. These values were then converted to ml by dividing the weight of alcohol consumed by the density of alcohol. The blood alcohol concentration was determined by using the NAD-ADH Reagent Multiple Test Vial kit (Sigma-Aldrich, St. Louis, MO) following the manufacturer’s instruction.
Tumor cell line and tumor cell inoculation
B16BL6 melanoma cells were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% FBS, 1% penicillin and streptomycin, and incubated at 37°C with 5% CO2. Cells were harvested when they reached 50–70% confluence, and suspended in PBS at 1×106 cells/ml. After 3–6 months of alcohol consumption mice were inoculated subcutaneously with 2×105 melanoma cells in the right hip area.
Antibodies and reagents
The following PE, FITC, and PerCP conjugated anti-mouse antibodies were purchased from BD Biosciences (San Diego, CA) and BioLegend (San Diego, CA): anti-CD1d (1B1), anti-CD3 (145-2C11), anti-CD4 (RM4-5), anti-CD5 (53-7.3), anti-CD8 (53-6.7), anti-CD19 (6D5), anti-CD21 (7E9), anti-CD23 (B3B4), anti-CD93 (AA4.1), anti-NK1.1 (PK136), anti-CXCR5 (2G8), anti-CCR7 (4B12), anti-IgD (11-26c.2a), and anti-IgM (RMM-1). Recombinant mouse CCL21 and CXCL13 were purchased from PeproTech (Rocky Hill, NJ). S1P was purchased from Avanti Polar Lipids, Inc. (Alabaster, AL).
Leukocyte isolation
Leukocytes from BM, spleen, LN, and peripheral blood were isolated following previous procedures (23). The leukocyte number was determined with the aid of the Vi-CELL cell viability analyzer (Beckman coulter, CA). Cells were collected by centrifugation and resuspended in an appropriate amount of PBS+ BSA.
Phenotype analysis
The phenotype of leukocytes from different organs was determined by flow cytometry following previously published methods (22). Briefly, 106 cells were pre-incubated with anti-CD16/CD32 monoclonal antibody (2.4G2) at 4°C for 5 min to block non-specific FC-receptor binding. Cells were stained with the appropriate monoclonal antibody at 4°C for 20 min. After washing with FACS buffer (PBS + 0.1% BSA + 0.1% NaN3), cells were analyzed using a BD FACscan flow cytometer. Data were collected and analyzed with CellQuest software.
Transwell migration assay
The transwell migration assay was conducted with the Costar 24-well plate transwell system using an insert with a diameter of 6.5 mm and pore size of 5 μm. Splenocytes were suspended in serum-free RPMI1640 medium, pre-warmed in an incubator at 37°C for 30 min before conducting the transwell migration assay, and 106 cells were added to the upper chamber of the insert. The lower chamber contained one of four different chemoattractants: 125 nM S1P, 500 ng/ml CCL21, 1 μg/ml CXCL13, or 300 μl plasma in a total volume of 600 μl of serum-free RPMI-1640 medium. A blank well containing 600 μl of serum-free RPMI-1640 medium without added chemoattractant was used as a control. The plates were incubated at 37°C in a CO2 incubator for 4–5 hr, and then placed on ice for 10 min. before cells in the lower chamber were collected and counted using a Vi-CELL cell viability analyzer (Beckman coulter, CA). The migration rate was calculated using the following formula:
RNA extraction and RT-PCR
Total RNA was extracted from the spleen using Trizol reagent (invitrogen, Carlsbad, CA) according to the manufacturer’s instruction. First-strand cDNA was prepared using M-MLV reverse transcriptase (Promega, Madison, WI) and adhering to the manufacturer’s protocol. The following primers were used to amplify S1PR1 and SPL1 cDNA fragment, respectively. S1PR1 forward primer: GGCCATTGGAGTGCACCGCT; S1PR1 reverse primer: ACAGCAGCCTCGCTCAAGCC; SPL1 forward primer: AGCCCTGGCAGCTCATTGCG; SPL1 reverse primer: GCAGCATGGGCACTCTCGGG. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as internal control of gene expression. Two primers, GAPDH1 ACCACAGTCCATGCCATCAC and GAPDH2 TACAGCAACAGGGTGGTGGA, were used to amplify GAPDH. PCR was conducted with 1 cycle of pre-denaturation at 94°C for 5 min, followed by 30 cycles of denaturation at 94°C for 30 sec, annealing at 58°C for 30 sec and elongation at 72°C for 40 sec, and 1 cycle of final elongation at 72°C for 7 min. PCR products were separated by 2% agarose gel electrophoresis with Tris-Acetate EDTA buffer. The absorbance of the PCR products was analyzed using the UVP BioImaging Systems and Labworks gel analysis software.
Statistical analysis
The data were analyzed by Microsoft Excel software or GraphPad Prism 5 and expressed as mean ± SD. The two-tailed student t-test was used to analyze differences between the two groups, and differences were considered significant at p <0.05. Except where specifically indicated, each group contained 7–10 mice.
Results
Daily alcohol intake and blood alcohol concentration
The daily intake of 20% w/v alcohol was 5.6±1.0 ml/day in the non-melanoma injected mice and 6.1±1.2 ml/day in the tumor-bearing mice. There was no significant difference in alcohol consumption between non-melanoma and melanoma-injected mice. The blood alcohol concentration determined two weeks after melanoma injection was 0.028±0.004%. This is consistent with previous determinations of blood alcohol concentration in mice not injected with melanoma (24, 25).
Chronic alcohol consumption induces lymphopenia in melanoma-bearing mice but not in non-tumor inoculated mice
We previously found that chronic alcohol consumption decreases cellularity in the spleen of non-tumor inoculated mice (22). Herein, we examined if chronic alcohol consumption affected leukocyte numbers in the spleen and blood of melanoma-bearing mice. Results indicated that although the cellularity is still lower in the spleen of melanoma-bearing, alcohol-consuming mice, compared to respective water-drinking mice, the difference is not significantly different (Fig. 1A). However, the numbers of PBL are significantly lower in alcohol-consuming, melanoma-bearing mice than in their water-drinking counterparts (Fig. 1B). We then examined if alcohol consumption decreased PBL in non-melanoma inoculated mice. The results indicated no effect of chronic alcohol consumption on total PBL number (Fig. 1C); however, alcohol consumption decreased the numbers of CD8+ T cells and NK (CD3−NK1.1+) cells in the blood of these non-melanoma inoculated mice (Fig. 1D). The percentage of CD19+ B cells increased and the percentage of CD8+ T cells decreased in the PBL (Fig. 1E). These results indicated that chronic alcohol consumption induces lymphopenia in the melanoma-bearing mice, but not in mice that are not inoculated with melanoma.
Fig. 1. Effects of chronic alcohol consumption on the splenocytes and PBL in B16BL6 melanoma-bearing mice and non-tumor injected mice.

A, Number of splenocytes in the water-drinking and alcohol-consuming mice at the indicated days after melanoma inoculation. B, Number of PBL in 700 μl of blood of the water-drinking and alcohol-consuming mice after the indicated days of tumor inoculation. C, Number of PBL in 700 μl of blood of the non-tumor inoculated water-drinking and alcohol-consuming mice. D, Number of indicated lymphocytes in700 μl of blood of the non-tumor inoculated water-drinking and alcohol-consuming mice. E, Percentage of indicated lymphocytes in PBL of the non-tumor inoculated water-drinking and alcohol-consuming mice. Each group contained 7–10 mice. The experiments were repeated once with the similar results. Water: Water-drinking mice. ETOH: Alcohol-consuming mice. Water-drinking group different from alcohol-consuming (ETOH) group on the indicated day, * p<0.05; ** p<0.01; *** p<0.001.
Chronic alcohol consumption selectively decreases B cells and T cells in blood, but not in BM, LN and spleen
We further determined if the decrease of PBL in melanoma-bearing, alcohol-consuming mice was associated with a general decline in all types of lymphocytes or due to selective reduction in specific cell types. CD4+ T (CD4+NK1.1−) cells, CD8+ T cells, B (CD19+) cells, NKT (CD3+NK1.1+) cells, and NK (CD3−NK1.1+) cells were examined. The results indicated that chronic alcohol consumption significantly decreased the percentage of B cells (Fig. 2C), increased the percentage of NKT cells (Fig. 2D), and did not significantly alter the percentage of CD4+ T cells (Fig. 2A). The presence of melanoma tumors had varying effects on CD8+ T cells (Fig. 2), and NK cells (Fig. 2E). On day 14 the percentages of CD8+ T cells were significantly lower and NK cells were significantly higher in the alcohol-consuming mice than in water-drinking mice, but not different on days 11 and 17.
Fig. 2. Effects of chronic alcohol consumption on different types of lymphocytes in the blood of B16BL6 melanoma-bearing mice.

A–E, Percentage of the indicated lymphocytes in PBL of melanoma-bearing mice at the indicated days after subcutaneous tumor inoculation. F–J, Number of specific types of lymphocytes in 700 μl of blood of the water-drinking and alcohol-consuming mice at the indicated days after subcutaneous tumor inoculation. Water: Water-drinking, melanoma-bearing mice. ETOH: Alcohol-consuming, melanoma-bearing mice. Each group contained 7–10 mice. Experiments were repeated once with the similar results. Water-drinking group different from alcohol-consuming (ETOH) group on the indicated day, * p<0.05; ** p<0.01; *** p<0.001.
Overall the major effects associated with lymphopenia in melanoma-inoculated mice were observed in B and T (CD4+ and CD8+) cells, where the numbers of these cells were decreased 2–4 fold (Figs. 2F–H). The greatest degree of decrease was in B cells (Fig. 2H) followed by CD8+ T cells (Fig. 2G) and CD4+ T cells (Fig. 2F).
As a peripheral reservoir of T cells and B cells, the spleen plays an important role in adjusting these lymphocytes in the blood. Alcohol consumption decreases T and B cells in the spleen of non-tumor inoculated mice (22). Therefore, we next examined if these cells were further altered in melanoma-inoculated mice. The results indicated that alcohol consumption had little effect on the percentage of T, B, NK and NKT cells in the spleen (Figs. 3A–E). CD4+ T cells (Fig. 3A) were increased while CD8+ T cells (Fig. 3B) were decreased in alcohol-consuming mice, but only on day 11 after melanoma inoculation.
Fig. 3. Effects of chronic alcohol consumption on different types of lymphocytes in the spleen of B16BL6 melanoma-bearing mice.

A–E, Percentage of the indicated lymphocytes in the splenocytes of melanoma-bearing mice at the indicated days after subcutaneous tumor inoculation. F–J, Number of the indicated lymphocytes in the spleen of the water-drinking and alcohol-consuming mice at the indicated days after subcutaneous tumor inoculation. Water: Water-drinking, melanoma-bearing mice. ETOH: Alcohol-consuming, melanoma-bearing mice. Each group contained 7–10 mice. Experiments were repeated once with the similar results. Water-drinking group different from alcohol-consuming (ETOH) group on the indicated day, * p<0.05.
The numbers of CD4+ T cells (Fig. 3F) and NKT cells (Fig. 3I) were not modulated by alcohol consumption and melanoma inoculation. B cell numbers did not significantly change during the early time points after melanoma inoculation; however they were significantly lower in alcohol-consuming mice on day 17 (Fig. 3H). CD8+ T cell numbers (Fig. 3G) were generally lower in alcohol-consuming, melanoma-bearing mice than in corresponding water-drinking mice throughout the evaluation period; however, differences in cell numbers were significantly different only on days 11 and 14. NK cells were also lower in alcohol-consuming compared to water-drinking mice; however, the differences were significant only on days 14 and 17.
B cells originate from the BM and further mature in the spleen. Thus, we examined if the decrease of B cells in peripheral blood was related to a defect in B cell production in the BM. We found that neither the percentage nor the number of B cells in the BM of alcohol-consuming, melanoma-bearing mice was altered compared to their water-drinking counterparts (Figs. 4A & B). We further examined LN, a secondary peripheral lymphoid organ that is abundant in B cells and T cells. Alcohol consumption also did not alter the percentage of T or B cells in LN (Fig. 4C). While the numbers of CD4+ T cells and B cells were not significantly changed, the number of CD8+ T cells was lower in the alcohol-consuming mice (Fig. 4D).
Fig. 4. Effects of chronic alcohol consumption on T cells and B cells in the BM and LN of B16BL6 melanoma-bearing mice.

A, Percentage of the indicated lymphocytes in the BM of the water-drinking and alcohol-consuming mice at 2 weeks after subcutaneous tumor inoculation. B, Number of the indicated lymphocytes in the BM from two femurs of water-drinking and alcohol-consuming mice at 2 weeks after subcutaneous tumor inoculation. C, Percentage of the indicated lymphocytes in the LN of the water-drinking and alcohol-consuming mice at 2 weeks after subcutaneous tumor inoculation. D, Number of the indicated lymphocytes in the LN from two inguinal LNs from water-drinking mice and alcohol-consuming mice at 2 week after subcutaneous tumor inoculation. Water: Water-drinking, melanoma-bearing mice. ETOH: Alcohol-consuming, melanoma-bearing mice. Each group contained 7–10 mice. Experiments were repeated once with the similar results. Water-drinking group different from alcohol-consuming (ETOH) group on the indicated day, * p<0.05.
Collectively, the data in Figs. 2–4 indicate that chronic alcohol consumption and melanoma inoculation dramatically decreases the numbers of B cells, CD8+ T cells and moderately decreases CD4+ T cells in the blood of melanoma-bearing mice. The degree of decrease in B cells is much greater than that of T cells. The numbers of B cells and CD4+ T cells in spleen, LN and BM are largely not affected by alcohol consumption and melanoma inoculation. However, alcohol consumption and melanoma inoculation decrease the numbers of CD8+ T cells in the spleen and LN.
Chronic alcohol consumption does not alter B cell differentiation in the BM and spleen, but increases the portion of immature B cells in the blood
We next determined if the decrease of B cells in blood was caused by a defect in their development in the BM. The expression of IgM and IgD on BM CD19+ B cells was used as a marker of B cell differentiation. The results indicated that B cell differentiation in the BM was not compromised by alcohol consumption and melanoma-inoculation (Figs. 5A & B). IgMhiIgDlo immature B cells significantly increased almost 2-fold in the peripheral blood of alcohol-consuming, melanoma-bearing mice (Figs. 5C & D). Since we found normal B cell development and differentiation in the BM, we hypothesized that the elevation of immature B cells in the blood resulted from a decrease in release of mature B cells from the spleen. Thus, we further examined differentiation and maturation of B cells in the spleen by determining the expression of CD93 and CD23 in these cells. We found that the percentage of CD93+CD23− T1 B cells was significantly increased in alcohol-consuming, melanoma-bearing mice (Figs. 5E & F). The portion of CD93−CD23+ follicular B cells did not decrease. To further confirm the changes in the percentages and numbers of mature B cells in BM, blood and spleen, we examined the percentages and numbers of CD23+ B cells in these tissue and organs. Alcohol consumption did not change the numbers of CD23+ B cells in the spleen and BM; however, there was a slight but significant increase in the percentages of CD23+ B cells in the spleen (Fig. 5G). The percentages and numbers of CD23+ B cells were greatly decreased in the blood of alcohol-consuming, melanoma-bearing mice (Figs. 5G & H). Collectively, these results indicate that alcohol consumption does not affect B cell development and differentiation in BM; however, it significantly decreases the percentage of mature B cells in blood. Except for a small increase in the percentages of CD23+ cells in spleen, the numbers of CD23+ follicular B cells in the spleen are normal in the alcohol-consuming, melanoma-bearing mice.
Fig. 5. Effects of chronic alcohol consumption on B cell development and distribution of B cell subsets of in BM, blood, and spleen of melanoma-bearing mice at 2 weeks after subcutaneous tumor inoculation.

A, Dot plot of IgMloIgDlo B cells (R4), IgMhiIgD+ B cells (R5), and IgMhiIgDhi B cells (R3) in gated BM CD19+ B cells. B, Percentage of indicated B cell subsets in the BM CD19+ B cells. C, Dot plot indicating IgMhiIgDlo immature B cells in the gated CD19+ peripheral blood B cells. D, Percentage of IgMhiIgDlo immature B cells in CD19+ peripheral blood B cells. E, Dot plot indicating CD23 and CD93 defined subsets of B cells in gated splenic CD19+ B cells. F, Percentage of indicated subsets of B cells in splenic CD19+ B cells. G, Percentage of CD23+CD19+ B cells in the indicated organs. H, Number of CD23+CD19+ B cells in the indicated organs. Water: Water-drinking, melanoma-bearing mice. ETOH: Alcohol-consuming, melanoma-bearing mice. Each group contained 7–10 mice. Experiments were repeated once with the similar results. ETOH group different from Water group, * p<0.05; ** p<0.01; *** p<0.001.
Chronic alcohol consumption decreases MZ B cells and CD1dhiCD5+ B cells, and down regulates CD1d expression on MZ B cells in melanoma-bearing mice
Alcohol consumption did not affect the numbers of follicular B cells in the spleen of melanoma-bearing mice. We next examined if MZ B cells and regulatory B cells were affected. We found that the percentages and numbers of MZ B cells (CD21hiCD1dhi) and CD1dhiCD5+ regulatory B cells were significantly lower in the alcohol-consuming, melanoma-bearing mice than in the respective water-drinking counterparts (Figs. 6A–D). Although the overall numbers of MZ B cells were not significantly different between the alcohol-consuming and water-drinking groups, the expression of CD1d on these cells was significantly down regulated in the alcohol-consuming, melanoma-bearing mice (Fig. 6E).
Fig. 6. Effects of chronic alcohol consumption on MZ B cells and CD5+CD1dhi regulatory B cells in the spleen of melanoma-bearing mice at 2 weeks after subcutaneous melanoma inoculation.

A, Dot plot indicating CD21hiCD1dhi MZ B cells (R1) in gated splenic CD19+ B cells. B, Contour plot indicating CD5+CD1dhi regulatory B cells in gated splenic CD19+ B cells. C, Percentage of MZ B cells (CD21hiCD1dhi) and regulatory B cells (CD5+CD1dhi) in splenic CD19+ B cells. D, Number of MZ B cells (CD21hiCD1dhi) and regulatory B cells (CD5+CD1dhi) in the spleen. E, Mean florescence intensity of CD1d in CD21hiCD1dhi MZ B cells. Water: Water-drinking, melanoma-bearing mice. ETOH: Alcohol-consuming, melanoma-bearing mice. Each group contained 7–10 mice. Experiments were repeated once with the similar results. ETOH group different from Water group, * p<0.05; ** p<0.01.
Chronic alcohol consumption does not induce T cell and B cell apoptosis in the blood
One factor that could explain the decreased number of T and B cells in the blood is loss of these cells via apoptosis facilitated by melanoma and alcohol consumption. To test this hypothesis, we determined the percentage of annexin V+ T and B cells in the blood. The results indicated that the percentage of annexin V+ cells in the CD8+ T cells and CD19+ B cells was not significantly different between the alcohol-consuming, melanoma-bearing mice and water-drinking, melanoma-bearing mice (Figs. 7A & B). These results indicate that the decrease of T and B cells in the blood of alcohol-consuming, melanoma-bearing mice is not caused by apoptosis.
Fig. 7. Effects of chronic alcohol consumption on the apoptosis of CD8+ T cells and CD19+ B cells in the blood of tumor-bearing mice.
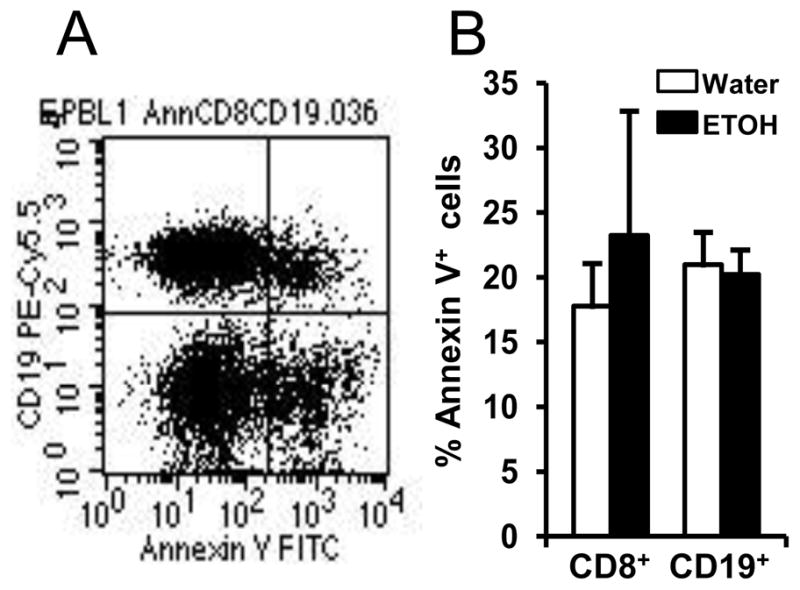
Mice were inoculated with B16BL6 melanoma subcutaneously, and euthanized 2 weeks after tumor inoculation. PBL were stained with anti-mouse CD19-PerCP, anti-mouse CD8-PE and Annexin-V-FITC. A, Dot plot indicating annexin V+ cells in CD19+ B cells. B, Percentage of annexin V+ cells in CD8+ T cells and CD19+ B cells. Water: Water-drinking mice. ETOH: Alcohol-consuming mice. Each group contained 7–10 mice.
Chronic alcohol consumption does not compromise the chemotactic ability of plasma from melanoma-bearing mice to facilitate splenocyte migration
Since the cell number and differentiation of B cells in the BM and spleen were normal in the alcohol-consuming, melanoma-bearing mice, the decrease of B cells in the blood is not caused by apoptosis. We also found that the decrease in peripheral blood B cells is mainly found in the mature B cell portion. Therefore, the decrease in the PBL is highly likely due to an inability of splenic T and B cells to navigate into the blood. One possibility is that the blood is deficient in chemotactic factors to attract T cells and B cells from spleen. To examine this possibility, we used a transwell cell migration system to test the chemotactic ability of plasma from alcohol-consuming, melanoma-bearing mice and water-drinking, melanoma-bearing mice to facilitate splenocyte migration in vitro. The results indicated that there was no significant difference in migration of splenocytes indicating that neither alcohol nor melanoma affects the chemotactic ability of plasma to attract splenocytes (Fig. 8).
Fig. 8. Effects of chronic alcohol consumption on chemotactic capacity of plasma in melanoma-bearing mice.
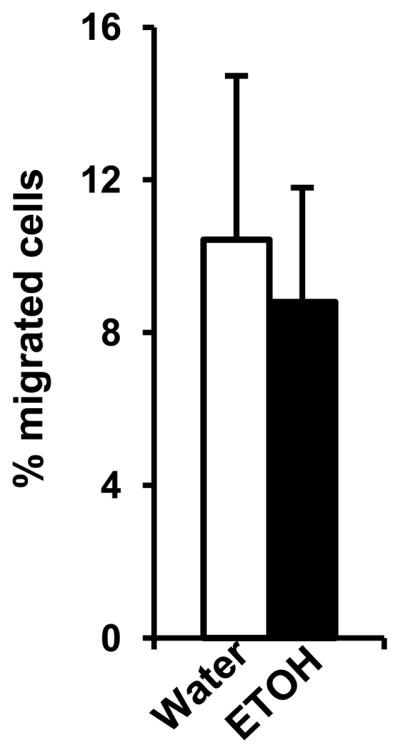
Plasma was collected two weeks after subcutaneous melanoma inoculation. The chemotactic capacity of plasma was examined in a transwell migration experiment as detailed in the Materials and Methods. The figure shows the percentage of migrated cells compared to the total number of seeded cells. The bar graphs represent the mean ± SD from six individual migration experiments conducted for six mice from each group. Water: Water-drinking, melanoma-bearing mice. ETOH: Alcohol-consuming, melanoma-bearing mice.
Chronic alcohol consumption does not impair the response of splenocytes from melanoma-bearing mice to CCL21 and CXCL13
Given that the chemotactic ability of plasma is intact in alcohol-consuming, melanoma-bearing mice, it is possible that the inability of splenic T cells and B cells to enter the blood could result from a defect in the cells themselves to respond to chemotactic factors. It is well established that the most important chemokines and receptors that control T cell migration and homing are CCL21, CCL19 and their receptor, CCR7 (26, 27). The chemokine and its receptor that are involved in B cell migration and redistribution in the follicle of the spleen is CXCL13 and CXCR5 (9). Therefore, we examined if alcohol consumption impaired the expression of the chemokine receptors, CCR7 and CXCR5, on splenic T cells and B cells, respectively. We found that chronic alcohol consumption did not alter the expression of these receptors on splenic T and B cells from melanoma-bearing mice (Figs. 9A–D). Consistent with chemokine receptor expression, the ability of splenocytes to respond to CCL21 and CXCL13 also was not altered in the alcohol-consuming, melanoma-bearing mice (Fig. 9E).
Fig. 9. Effects of chronic alcohol consumption on the expression of CXCR5 and CCR7 on B cells and T cells, and the response of splenocytes to CXCL13 and CCL21 in melanoma-bearing mice.

Mice were euthanized two weeks after tumor inoculation, and splenocytes were isolated to examine the expression of CXCR5 and CCR7 on B cells and T cells. The response of splenocytes to CXCL13- and CCL21-induced chemotaxis was examined in a transwell migration assay as outlined in the Materials and Methods. A, Histogram indicating the expression of CXCR5 in gated CD19+ splenic B cells of water-drinking, melanoma-bearing mouse. B, Percentage of CXCR5+ cells in splenic CD19+ B cells. C, Histogram indicating the expression of CCR7 in gated splenic CD8+ T cells of water-drinking, melanoma-bearing mouse. D, Percentage of CCR7+ cells in splenic CD8+ T cells. E, Percentage of migrated cells in the total seeded splenic cells to CXCR13 and CCL21. Each group contained 6–10 mice. Water: Water-drinking, melanoma-bearing mice. ETOH: Alcohol-consuming, melanoma-bearing mice.
Chronic alcohol consumption compromises migration of splenocytes responding to S1P and down regulates S1PR1 and SPL expression in the spleen of melanoma-bearing mice
T and B cell egress from the spleen and LN to the blood is controlled by S1P/S1PR1 signaling (11, 28). The fact that alcohol consumption does not compromise the chemotactic ability of the plasma from melanoma-bearing mice suggests that the S1P in the blood is not affected. Indeed, there was no difference in the concentration of S1P in the plasma between the two groups of mice (data not shown). S1P levels in the blood of mice given the alcohol-containing Leiber-DeCarli liquid diet are also not altered by chronic alcohol consumption (29). Therefore, the ability of splenocytes to respond to S1P chemotaxis could be compromised by alcohol consumption in melanoma-bearing mice. To examine this possibility, we determined migration of splenocytes from water-drinking and alcohol-consuming, melanoma-bearing mice in response to S1P. We found that chronic alcohol consumption significantly decreased S1P-induced splenocyte migration (Fig. 10A). S1PR1 controls the response of T and B cells to S1P. Cell surface S1PR1 is controlled by S1PR1 gene expression and also by S1P concentration in the microenvironment. High concentrations of S1P will induce S1PR1 internalization (30). We used RT-PCR to examine the gene expression of S1PR1 in splenocytes from water-drinking and alcohol-consuming, melanoma-bearing mice. Expression of S1PR1 was significantly down regulated in splenocytes from alcohol-consuming, melanoma-bearing mice compared to their water-drinking counterparts (Fig. 10B). Gene expression of SPL1, an enzyme catalyzing the degradation of S1P to hexadecanaldehyde and phosphoethanolamine, was also down regulated in the alcohol-consuming, melanoma-bearing mice (Fig. 10C). These data indicate that alcohol consumption compromises the splenocyte response to S1P via decreasing S1PR1 expression on splenocytes.
Fig. 10. Chronic alcohol consumption impairs the response of splenocytes to S1P and down regulates the expression of S1PR1 and SPL1 in splenocytes of melanoma-bearing mice.

A, Percentage of migrated cells relative to the total number of seeded splenocytes responding to S1P induced chemotaxis. B, Absorbance ratio of S1PR1 to GAPDH from RT-PCR analyses of splenocytes. C, Absorbance ratio of SPL1 to GAPDH from RT-PCR analyses of splenocytes. Water: Water-drinking, melanoma-bearing mice. ETOH: Alcohol-consuming, melanoma-bearing mice. Experiments were repeated once with the similar results. ETOH group different from Water group, ** p<0.01.
Discussion
The data demonstrate that alcohol consumption differentially affects lymphocyte distribution in the spleen and blood of normal and melanoma-bearing mice. Alcohol consumption in the non-tumor inoculated mice results in a general decrease in lymphocytes in the spleen (22), but does not alter PBL except for CD8+ T cells, which are decreased in both spleen and blood (Fig. 1). In the melanoma-bearing mice, however, alcohol consumption dramatically decreases PBL, but does not significantly decrease lymphocytes in the spleen except for CD8+ T cells (Figs. 2 & 3). The effects of alcohol consumption on each type of lymphocyte in the blood of melanoma-bearing mice were different. Alcohol does not significantly affect NK cells, increases NKT cells, only moderately decreases CD4+NK1.1− T cells, and dramatically decreases B cells and CD8+ T cells. Compared to B cells, CD8+ T cells are a small percentage of the total PBL population. B cells comprise approximately 45% of the PBL; thus, the decrease in B cells contributes more profoundly to the lymphopenia in the alcohol-consuming, melanoma-bearing mice.
B cells circulate in the blood, and a decrease could result from the defective production and/or release of these cells from the BM, or from impaired egress of these cells from LN and spleen. Since alcohol consumption neither alters the total numbers of B cells in the BM (Fig. 4B) nor significantly affects the development of B cells in the BM (Fig. 5B), the decreases in circulating B cells in the blood are not due to compromised B cell development, BM production, or the release from BM. Thus, the decreases of B cells in the blood highly likely result from compromised circulation of these cells among the secondary lymphoid organs such as LN and spleen.
The moderate increase in CD93+CD23− cells suggests that alcohol consumption might impair B cell maturation from the transitional stage into mature follicular B cells or MZ B cells. However, the increase in the percentage of CD23+ B cells (Fig. 5G) suggests that follicular B cells accumulate abnormally in the spleen. If the follicular B cells fail to circulate in the blood and accumulate in the spleen, then the percentage of CD23+ follicular B cells will increase and the percentage of MZ B cells, which are non-circulating cells, will decrease in the splenic B cell population as we observed (Fig. 6). Because the BM exports immature B cells into the blood, the majority of mature B cells in the blood come from secondary lymphoid organs. If the production and release of B cells from the BM is normal, and the egress of B cells from spleen is compromised, then the portion of immature B cells in blood would be expected to increase, which is also what we showed (Figs. 5D, G & H). Therefore, these data indicate an alcohol/melanoma interaction that influences the number and maturity of B cells in the blood.
If the alcohol/melanoma interaction impairs B cell egress from the spleen, then the number of B cells should be elevated in the spleen; however, we did not observe this (Fig. 3H). Alcohol consumption significantly decreases B cells in the spleen of non-tumor injected mice (22); however, the effect is diminished in melanoma-bearing mice where no significant decreases are observed until Day 17 after tumor inoculation. Thus, it is likely that the impaired egress of B cells is obscured by the more profound effect of alcohol consumption on B cell numbers before melanoma inoculation. It is worthy to note that alcohol consumption down regulates the expression of CD1d on MZ B cells. CD1d is a non-classical MHC I-like molecule that presents lipid antigen to NKT cells and plays an important role in NKT cell activation. MZ B cells are involved in NKT cell activation and the regulation of cytokine production (6, 8). Alcohol consumption increases iNKT cells in the blood of melanoma-bearing mice (Figs. 2D & I). Whether the decrease in MZ B cells and the down-regulation of CD1d on these cells affects iNKT cell function in the alcohol-consuming, melanoma-bearing mice is an interesting question that remains to be elucidated.
The current study indicates that the interaction between alcohol and melanoma does not affect the chemotactic capacity of plasma to induce migration of T and B cells nor does the interaction alter the expression of CXCR5 and CCR7 on these cells. Alcohol consumption down regulates the expression of S1PR1 and SPL1 in splenocytes from melanoma-bearing mice. Therefore, the impaired egress of T cells and B cells from the spleen is associated with a compromised S1PR1 signaling pathway. This is not only evidenced by the decreased response of splenocytes from alcohol-consuming, melanoma-bearing mice to S1P induced migration, but also by the fact that only B and T cells and not NKT and NK cells decrease in the blood. It is known that activation of the S1P/S1PR1 signaling pathway affects the cytokine production of NKT cells, but does not affect the egress and migration of these cells (31). NK cell egress from BM and LN is regulated by the S1P/S1PR5 pathway, and not the S1P/S1PR1 pathway (32, 33).
The egress of B cells from the BM is also controlled by the S1P/S1PR1 pathway (28). We found that alcohol consumption did not impair B cell egress from BM of melanoma-bearing mice. This suggests that the expression of S1PR1 in BM-derived B cells is not affected, which is consistent with our observations that alcohol consumption leads to down regulation of S1PR1 expression in spleen, but not in BM (data not shown). This further indicates an organ-specific effect associated with alcohol consumption in the melanoma-bearing mice. The cell surface expression of S1PR1is regulated by the s1pr1 gene and also regulated by the concentration of S1P in the microenvironment (34). We found that this gene is down regulated in the spleen of alcohol-consuming, melanoma-bearing mice. We also found that the expression of SPL1 is down regulated in the spleen. SPL1 catalyzes the degradation of S1P. The decrease of SPL1 could result in accumulation of S1P. The elevation of S1P will induce the internalization of S1PR1, which in turn could compromise the response to S1P-induced chemotaxis. One of the products of S1P degradation catalyzed by SPL1 is phosphoethanolamine. Chronic alcohol consumption alters the metabolism of lipids, including phosphoethanolamine (35, 36). It is likely that alcohol consumption interacting with tumor cells changes the metabolism of lipids, which lead to organ specific alternations in S1P and S1PR1. We are investigating this hypothesis and the mechanism of how alcohol consumption interacts with melanoma to regulate S1PR1 expression.
The effects of B cells in tumor immunity have attracted more attention in recent years. Some reports indicate that depletion of B cells enhances antitumor immunity, implicating B cells in the suppression of antitumor immune responses (14, 15). However, in B16 melanoma, the lack of B cells, especially mature B cells, compromises T cell function and enhances tumor progression (18). Also in B16 melanoma, depletion of CD20+ mature B cells increases lung tumor growth and burden in CpG-treated mice (20). It is also reported that the high density of infiltrated B cells in melanoma correlates with a significant survival advantage and positive prognostic outcome in melanoma patients (37). Understanding the relationship among impaired B cell circulation, dysfunction of CD8+ T cells, and decreased survival in alcohol-consuming melanoma-bearing mice will not only advance our knowledge on the effects of B cells in antitumor immunity, but also will provide new insight into how to recover immune function in alcoholics with melanoma and possibly other cancers. Research in this area is ongoing.
In summary, we conclude that chronic alcohol consumption compromises the egress of B and T cells from the spleen via impairing the S1P/S1PR1 pathway, which in turn induces severe lymphopenia in B16BL6 melanoma-bearing mice. Alcohol consumption neither compromises the development nor affects the egress of B cells from BM. The compromised egress of B cells from the spleen results in an immature B cell-dominant circulating population in the blood of melanoma-bearing mice.
Acknowledgments
This work was supported by Public Health Service Grants R01-AA07293 to GGM and HZ and K05-AA017149 to GGM
Abbreviations used in this article
- BM
bone marrow
- LN
lymph node
- MZ
marginal zone
- NK
natural killer
- S1P
sphingosine-1-phosphate
- S1PR
sphingosine-1-phosphate receptor
- SPL
sphingosine-1-phosphate lyase
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Rolink AG, Andersson J, Melchers F. Molecular mechanisms guiding late stages of B-cell development. Immunol Rev. 2004;197:41–50. doi: 10.1111/j.0105-2896.2004.0101.x. [DOI] [PubMed] [Google Scholar]
- 2.Weill JC, Weller S, Reynaud CA. Human marginal zone B cells. Annu Rev Immunol. 2009;27:267–285. doi: 10.1146/annurev.immunol.021908.132607. [DOI] [PubMed] [Google Scholar]
- 3.Pillai S, Cariappa A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat Rev Immunol. 2009;9:767–777. doi: 10.1038/nri2656. [DOI] [PubMed] [Google Scholar]
- 4.Parker DC. T cell-dependent B cell activation. Annu Rev Immunol. 1993;11:331–360. doi: 10.1146/annurev.iy.11.040193.001555. [DOI] [PubMed] [Google Scholar]
- 5.Malynn BA, Romeo DT, Wortis HH. Antigen-specific B cells efficiently present low doses of antigen for induction of T cell proliferation. J Immunol. 1985;135:980–988. [PubMed] [Google Scholar]
- 6.Bialecki E, Paget C, Fontaine J, Capron M, Trottein F, Faveeuw C. Role of marginal zone B lymphocytes in invariant NKT cell activation. J Immunol. 2009;182:6105–6113. doi: 10.4049/jimmunol.0802273. [DOI] [PubMed] [Google Scholar]
- 7.Sonoda KH, Stein-Streilein J. CD1d on antigen-transporting APC and splenic marginal zone B cells promotes NKT cell-dependent tolerance. Eur J Immunol. 2002;32:848–857. doi: 10.1002/1521-4141(200203)32:3<848::AID-IMMU848>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 8.Zietara N, Lyszkiewicz M, Krueger A, Weiss S. ICOS-dependent stimulation of NKT cells by marginal zone B cells. Eur J Immunol. 2011;41:3125–3134. doi: 10.1002/eji.201041092. [DOI] [PubMed] [Google Scholar]
- 9.Forster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87:1037–1047. doi: 10.1016/s0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- 10.Ansel KM, V, Ngo N, Hyman PL, Luther SA, Forster R, Sedgwick JD, Browning JL, Lipp M, Cyster JG. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature. 2000;406:309–314. doi: 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- 11.Pappu R, Schwab SR, Cornelissen I, Pereira JP, Regard JB, Xu Y, Camerer E, Zheng YW, Huang Y, Cyster JG, Coughlin SR. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science. 2007;316:295–298. doi: 10.1126/science.1139221. [DOI] [PubMed] [Google Scholar]
- 12.Pereira JP, Xu Y, Cyster JG. A role for S1P and S1P1 in immature-B cell egress from mouse bone marrow. PLoS One. 2010;5:e9277. doi: 10.1371/journal.pone.0009277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 14.Inoue S, Leitner WW, Golding B, Scott D. Inhibitory effects of B cells on antitumor immunity. Cancer Res. 2006;66:7741–7747. doi: 10.1158/0008-5472.CAN-05-3766. [DOI] [PubMed] [Google Scholar]
- 15.Ruddell A, Harrell MI, Furuya M, Kirschbaum SB, Iritani BM. B lymphocytes promote lymphogenous metastasis of lymphoma and melanoma. Neoplasia. 2011;13:748–757. doi: 10.1593/neo.11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity. 2008;28:639–650. doi: 10.1016/j.immuni.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 17.Horikawa M, Minard-Colin V, Matsushita T, Tedder TF. Regulatory B cell production of IL-10 inhibits lymphoma depletion during CD20 immunotherapy in mice. J Clin Invest. 2011;121:4268–4280. doi: 10.1172/JCI59266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DiLillo DJ, Yanaba K, Tedder TF. B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: therapeutic B cell depletion enhances B16 melanoma growth in mice. J Immunol. 2010;184:4006–4016. doi: 10.4049/jimmunol.0903009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou P, Qiu J, L’Italien L, Gu D, Hodges D, Chao CC, Schebye XM. Mature B cells are critical to T-cell-mediated tumor immunity induced by an agonist anti-GITR monoclonal antibody. J Immunother. 2010;33:789–797. doi: 10.1097/CJI.0b013e3181ee6ba9. [DOI] [PubMed] [Google Scholar]
- 20.Sorrentino R, Morello S, Forte G, Montinaro A, De Vita G, Luciano A, Palma G, Arra C, Maiolino P, Adcock IM, Pinto A. B cells contribute to the antitumor activity of CpG-oligodeoxynucleotide in a mouse model of metastatic lung carcinoma. Am J Respir Crit Care Med. 2011;183:1369–1379. doi: 10.1164/rccm.201010-1738OC. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H, Meadows GG. Chronic alcohol consumption enhances myeloid-derived suppressor cells in B16BL6 melanoma-bearing mice. Cancer Immunol Immunother. 2010;59:1151–1159. doi: 10.1007/s00262-010-0837-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H, Meadows GG. Chronic alcohol consumption in mice increases the proportion of peripheral memory T cells by homeostatic proliferation. J Leukoc Biol. 2005;78:1070–1080. doi: 10.1189/jlb.0605317. [DOI] [PubMed] [Google Scholar]
- 23.Zhang H, Zhu Z, Meadows GG. Chronic alcohol consumption decreases the percentage and number of NK cells in the peripheral lymph nodes and exacerbates B16BL6 melanoma metastasis into the draining lymph nodes. Cell Immunol. 2011;266:172–179. doi: 10.1016/j.cellimm.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meadows GG, Blank SE, Duncan DD. Influence of ethanol consumption on natural killer cell activity in mice. Alcohol Clin Exp Res. 1989;13:476–479. doi: 10.1111/j.1530-0277.1989.tb00359.x. [DOI] [PubMed] [Google Scholar]
- 25.Blank SE, Duncan DA, Meadows GG. Suppression of natural killer cell activity by ethanol consumption and food restriction. Alcohol Clin Exp Res. 1991;15:16–22. doi: 10.1111/j.1530-0277.1991.tb00514.x. [DOI] [PubMed] [Google Scholar]
- 26.Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 27.Gunn MD, Kyuwa S, Tam C, Kakiuchi T, Matsuzawa A, Williams LT, Nakano H. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J Exp Med. 1999;189:451–460. doi: 10.1084/jem.189.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwab SR, Cyster JG. Finding a way out: lymphocyte egress from lymphoid organs. Nat Immunol. 2007;8:1295–1301. doi: 10.1038/ni1545. [DOI] [PubMed] [Google Scholar]
- 29.Clugston RD, Jiang H, Lee MX, Piantedosi R, Yuen JJ, Ramakrishnan R, Lewis MJ, Gottesman ME, Huang LS, Goldberg IJ, Berk PD, Blaner WS. Altered hepatic lipid metabolism in C57BL/6 mice fed alcohol: a targeted lipidomic and gene expression study. J Lipid Res. 2011;52:2021–2031. doi: 10.1194/jlr.M017368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lo CG, Xu Y, Proia RL, Cyster JG. Cyclical modulation of sphingosine-1-phosphate receptor 1 surface expression during lymphocyte recirculation and relationship to lymphoid organ transit. J Exp Med. 2005;201:291–301. doi: 10.1084/jem.20041509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hwang SJ, Kim JH, Kim HY, Kim S, Chung DH. FTY720, a sphingosine 1-phosphate receptor modulator, inhibits CD1d-restricted NKT cells by suppressing cytokine production but not migration. Lab Invest. 2010;90:9–19. doi: 10.1038/labinvest.2009.109. [DOI] [PubMed] [Google Scholar]
- 32.Jenne CN, Enders A, Rivera R, Watson SR, Bankovich AJ, Pereira JP, Xu Y, Roots CM, Beilke JN, Banerjee A, Reiner SL, Miller SA, Weinmann AS, Goodnow CC, Lanier LL, Cyster JG, Chun J. T-bet-dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J Exp Med. 2009;206:2469–2481. doi: 10.1084/jem.20090525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walzer T, Chiossone L, Chaix J, Calver A, Carozzo C, Garrigue-Antar L, Jacques Y, Baratin M, Tomasello E, Vivier E. Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nat Immunol. 2007;8:1337–1344. doi: 10.1038/ni1523. [DOI] [PubMed] [Google Scholar]
- 34.Schwab SR, Pereira JP, Matloubian M, Xu Y, Huang Y, Cyster JG. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science. 2005;309:1735–1739. doi: 10.1126/science.1113640. [DOI] [PubMed] [Google Scholar]
- 35.Zhao Z, Yu M, Crabb D, Xu Y, Liangpunsakul S. Ethanol-induced alterations in fatty acid-related lipids in serum and tissues in mice. Alcohol Clin Exp Res. 2011;35:229–234. doi: 10.1111/j.1530-0277.2010.01338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carrasco MP, Jimenez-Lopez JM, Segovia JL, Marco C. Comparative study of the effects of short- and long-term ethanol treatment and alcohol withdrawal on phospholipid biosynthesis in rat hepatocytes. Comp Biochem Physiol B Biochem Mol Biol. 2002;131:491–497. doi: 10.1016/s1096-4959(02)00006-4. [DOI] [PubMed] [Google Scholar]
- 37.Ladanyi A, Kiss J, Mohos A, Somlai B, Liszkay G, Gilde K, Fejos Z, Gaudi I, Dobos J, Timar J. Prognostic impact of B-cell density in cutaneous melanoma. Cancer Immunol Immunother. 2011;60:1729–1738. doi: 10.1007/s00262-011-1071-x. [DOI] [PMC free article] [PubMed] [Google Scholar]