

Background: A cationic branched peptide was designed with antimicrobial activities against Gram-negative bacteria.
Results: B2088 penetrated the outer membrane through inducing phase transitions of LPS and caused inner membrane depolarization by lipid redistribution.
Conclusion: Our findings support the interfacial activity model and extend it to more complex interfaces.
Significance: We provide a functional structural motif for developing new antimicrobials.
Keywords: Antimicrobial Peptides, Electron Microscopy (EM), Fluorescence, Liposomes, Membrane Reconstitution, Molecular Dynamics, NMR, Phospholipid Vesicle, Pseudomonas aeruginosa, Structural Biology, HR-MAS, Lipopolysaccharide, Membrane-mimicking Liposomes and Micelles
Abstract
In recent years, interest has grown in the antimicrobial properties of certain natural and non-natural peptides. The strategy of inserting a covalent branch point in a peptide can improve its antimicrobial properties while retaining host biocompatibility. However, little is known regarding possible structural transitions as the peptide moves on the access path to the presumed target, the inner membrane. Establishing the nature of the interactions with the complex bacterial outer and inner membranes is important for effective peptide design. Structure-activity relationships of an amphiphilic, branched antimicrobial peptide (B2088) are examined using environment-sensitive fluorescent probes, electron microscopy, molecular dynamics simulations, and high resolution NMR in solution and in condensed states. The peptide is reconstituted in bacterial outer membrane lipopolysaccharide extract as well as in a variety of lipid media mimicking the inner membrane of Gram-negative pathogens. Progressive structure accretion is observed for the peptide in water, LPS, and lipid environments. Despite inducing rapid aggregation of bacteria-derived lipopolysaccharides, the peptide remains highly mobile in the aggregated lattice. At the inner membranes, the peptide undergoes further structural compaction mediated by interactions with negatively charged lipids, probably causing redistribution of membrane lipids, which in turn results in increased membrane permeability and bacterial lysis. These findings suggest that peptides possessing both enhanced mobility in the bacterial outer membrane and spatial structure facilitating its interactions with the membrane-water interface may provide excellent structural motifs to develop new antimicrobials that can overcome antibiotic-resistant Gram-negative pathogens.
Introduction
Naturally occurring antimicrobial peptides (AMPs)3 are found in virtually all living organisms and form a critical part of the innate immune system in mammals, providing protection against pathogen invasion (1, 2). More than 1000 different antimicrobial peptides have been documented with activity against viruses, bacteria, and fungi, prompting interest in using the structural principles of small cationic, amphiphilic molecules for improving and fine tuning target specificity and efficacy (3–6). Non-natural multivalent AMPs have been designed by conjugating copies of a peptide monomer to scaffold molecules via naturally occurring intermolecular disulfide bridges or unnatural scaffold linkers (7). Branched peptides have been shown to have considerable advantages over their monomeric forms, such as improved antimicrobial activity (8), maintaining high efficacy under physiological (high salt) conditions (9, 10), enhanced bacterial surface binding affinity (11), decreased susceptibility to proteolytic degradation (12, 13), and low cytotoxicity (14, 15). However, the quantitative structure-activity relationships and physiochemical properties of branched antimicrobial peptides are still poorly understood at the atomic level.
Several different models have been proposed to explain the mechanism by which antimicrobial peptides kill bacteria. These include the formation of bilayer-spanning pores by α-helical peptides (16–21), an electrostatic charge-based mechanism of β-strand peptides (22, 23), and newly proposed mechanisms like membrane phase transition (24) and specific targeting (25, 26). Recently, an interfacial activity model has been defined as the “the ability of a molecule to bind to a membrane, partition into the membrane-water interface, and alter the packing and organization of the lipids” (1, 2). The model predicts that a molecule capable of accumulating at bacterial membranes at a sufficiently high concentration might be an effective antimicrobial molecule. However, despite many years of intensive research, there is still no consensus on the mechanism of action or structure-function relationships that underlie the remarkable ability of antimicrobial peptides to kill pathogens with widely varying cell wall structures (27, 28).
Recently we designed a series of short (10 amino acids) linear peptides with strong antimicrobial activity against Gram-negative bacteria (29). Our previous structural studies showed that these peptides dimerize in the lipid phase or in aqueous solution in a cooperative manner at concentrations corresponding to their minimal inhibitory concentrations (MICs) (30). A V2 monomer peptide, which had promising activity and was very amenable to large scale production, was chosen as a functional motif to build a covalent dimer (V2 dimer) peptide. In early studies, the dimer showed significantly decreased MIC levels against reference bacteria Pseudomonas aeruginosa ATCC 9027 (29). Structure-activity relationships of the dimer peptide at an atomic level are presented in this work.
To maintain consistency in the peptide series nomenclature, we identified V2 dimer peptide as B2088. Compared with a variety of its analogous peptides and the commonly used antibiotic gentamicin, B2088 showed outstanding antimicrobial activity (see Table 1). More strikingly, no resistance developed after 15 bacterial serial passages. Neither a cytotoxic effect nor hemolysis was found at a peptide concentration of 2 mg/ml when tested against human conjunctival epithelial cells. Corneal wound healing in a rabbit model was not compromised. In an attempt to understand both the mechanism of action and the basis of cytotoxicity, structural studies were carried out using an array of biophysical methods, providing an action mechanism that extended the carpet model. Spatial structure/dynamics and mobility of the branched peptide in heterogeneous media, including live bacteria, LPS, liposomes, and mixed micelles, were studied using environment-sensitive fluorescent probes, electron microscopy, high resolution magic angle spinning (HR-MAS), solution NMR, and molecular dynamics simulations.
TABLE 1.
Minimal inhibitory concentrations of B2088 and its analogous variants against Gram-negative ATCC strains
| Peptides | Sequences | Activity (minimal inhibitory concentration)a |
|
|---|---|---|---|
| P. aeruginosa ATCC27853 | Escherichia coli ATCC 25922 | ||
| μm | |||
| V2 monomer | RGRKVVRRKK | >39 | 39 |
| B2088 (V2 dimer) | (RGRKVVRR)2KK | 2.75 | 2.75 |
| Linear V2 dimer | RGRKVVRRKKRRVVKRGR | 10.91 | 10.92 |
| Truncated V2 dimer | (KVVRR)2KK | >32.22 | 16.11 |
| V3 dimer | (RGRKVVVRR)2KK | 2.51 | 2.51 |
| V2-1A dimer | (RGRKVVRR)2KA | 5.60 | 5.60 |
| V2-1D dimer | (RGRKVVRR)2KD | 5.49 | 5.49 |
| V2-1K-amide dimer | (RGRKVVRR)2KK-NH2 | 5.46 | 5.46 |
| L2 dimer | (RGRKLLRR)2KK | 2.66 | 5.33 |
a Measurement has experiment error within 30%.
EXPERIMENTAL PROCEDURES
Labeled Peptide Synthesis
B2088, its analogous variants, and V2 monomer peptide were produced using the solid phase peptide synthesis method. HPLC and electrospray ionization-MS were subsequently performed for peptide purification and identification (29). The sequences were (RGRKVVRR)2KK for B2088 and RGRKVVRRKK for the monomer, respectively. The secondary chain of B2088 was formed through the Lys9 ϵ-NH-group on the primary chain. The underlined amino acids were 15N- and 13C-double labeled (supplemental Fig. S1). Lyophilized B2088 powder (10.3 mg) was dissolved in 219 μl of Milli-Q water, resulting in 20 mm peptide stock. Lyophilized monomer powder (12.5 mg) was dissolved in 478.3 μl of Milli-Q water, resulting in 20 mm stock.
Antimicrobial Activity Assay
MIC values were determined using the broth macrodilution method modified from that described by the Clinical and Laboratory Institute. Cells were grown in Mueller Hinton broth overnight. An aliquot of 1 ml of adjusted inoculum in Mueller Hinton broth was added to 1 ml of each dilution of peptides to yield a final count of 105 to 106 cfu/ml bacteria in each tube. The tubes were incubated at 35 °C for 16–20 h. A positive control tube containing only the broth and bacteria and a negative control tube containing only the broth were incubated. The MIC values of peptides for each clinical isolate or reference bacteria were recorded as the lowest concentration of peptide that inhibited 99% visible growth of the test organism.
Bactericidal Kinetics of B2088
Standard and gentamicin-resistant strains of P. aeruginosa (ATCC 9027 and DR 4877/07) were grown separately on tryptic soy agar plates at 35 °C for 24 h. The colonies were then picked from the plate and resuspended in United States Pharmacopia (USP) phosphate buffer (pH 7.2). The suspension was adjusted to 106/107 cfu/ml with various concentrations of B2088 (2.75, 5.5, 11, and 22 μm) in separate tubes and incubated at 35 °C. Aliquots of 100-μl samples were withdrawn at 10-, 20-, 30-, and 60-min intervals and plated on tryptic soy agar plates after serial dilution using the same buffer. Simultaneously, the bacteria in USP phosphate buffer without peptide addition was used as a control and plated in a similar fashion. After 48 h, all of the plates were checked, and the colonies were calculated to give the cfu/ml. A time kill curve was plotted with time against the logarithm of the viable count.
Resistant Assay
An experiment was designed to stimulate resistance development, using a reference strain of P. aeruginosa (ATCC 27853). Bacteria were removed from the original master stock (not more than four passages) obtained from the American Type Culture Collection (ATCC). The experiment was conducted in three different sets, one each for B2088, gentamicin, and norfloxacin. The test was initiated by growing the bacteria in the presence of various concentrations of B2088, gentamicin, or norfloxacin that encompassed their MICs. After 20 h of incubation at 35 °C, the bacteria that showed growth in the highest concentration was repassaged in a fresh dilution series of B2088 or other antibiotics. The process was repeated every 20 h for up to 18 passages, and the MICs were determined at every passage as specified earlier.
Cytotoxicity Assay
Cytotoxicity was determined by measuring the amount of ATP generated by viable cells in culture using the CellTiter-Glo luminescent cell viability assay (Promega, Madison, WI). B2088 and the monomer peptide were incubated with a human conjunctiva epithelial cell line (IOBA-NHC) as described previously (31, 32). The luminescent signal was read by a microplate reader (Tecan GeniosPro, Tecan Asia, Singapore). Blank wells containing only medium were included in each assay, and the reading was subtracted from each sample and control well. Using the luminescence of the untreated wells as 100%, the cell viability was calculated as follows.
 |
Each condition was assayed in triplicate, and the experiments were repeated three times.
Hemolysis Assay
The hemolytic activity of B2088 was determined by the amount of hemoglobin that was released from rabbit erythrocytes. Fresh rabbit red blood cells were isolated from the whole blood of New Zealand White rabbits, by centrifugation at 3000 rpm for 10 min. Red blood cells were washed with sterile PBS and diluted to 8% (v/v) stock solution in sterile PBS. B2088 was dissolved in and mixed with red blood cells to prepare the desired concentration of B2088 in red blood cells (final v/v = 4%). The mixtures were added to 96-well plates and incubated at 37 °C for 60 min. After incubation, the mixtures were transferred into 1.5-ml Eppendorf tubes and centrifuged at 3000 rpm for 3 min. The supernatant (100 μl) was transferred into a clean 96-well plate, and the amount of hemoglobin released was determined by measuring the absorbance at 576 nm with a TECAN Infinite 200 microplate reader. 2% Triton X-100 and PBS were used as positive and negative control, respectively. The percentage of hemolysis was calculated using Equation 2.
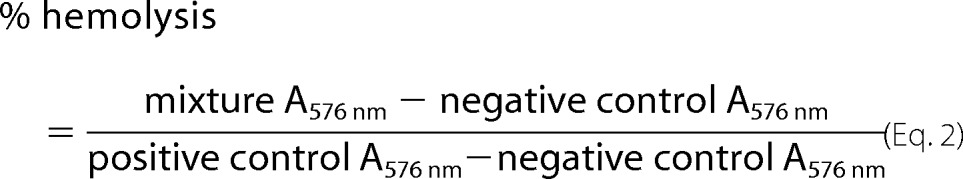 |
Fluorescence Emission Studies
Outer membrane permeabilization was studied using an N-phenyl-1-naphthylamine (NPN) assay as reported by Loh et al. (33). P. aeruginosa ATCC 9027 cultured overnight was harvested (3000 rpm, 4 °C). The pellet was washed twice using 5 mm HEPES buffer (pH 7.2) and resuspended in the same buffer to an A600 nm of 0.4. Bacteria were placed in a 10-mm stirred cuvette, and NPN was added to a final concentration of 10 μm. Appropriate concentrations of B2088 were added, and the increase in fluorescence intensity was monitored on a Quanta Master fluorescence spectrophotometer (Photon Technology International). The excitation and emission wavelengths were set at 350 and 410 nm with slit widths at 2 and 5 nm, respectively.
Live bacteria were used for the depolarization assay of transmembrane potential. Late log phase P. aeruginosa ATCC 9027 bacteria were harvested by centrifugation and washed twice with 5 mm HEPES buffer, subsequently resuspended in the same buffer (A600 = 0.4). The membrane-sensitive fluorescence probe 3,3′-dipropylthiadicarbocyanine iodide (diSC3) (5) was added to the bacterial suspension to a concentration of 0.1 μm and incubated at room temperature for 1 h. The fluorescence emission spectrum was recorded by monitoring the changes in emission intensity at 670 nm (at an excitation wavelength of 622 nm) until no further changes were seen in the intensity values. Time-dependent changes in emission intensities at 670 nm were recorded after adding various concentrations of B2088.
Transmission Electron Microscopy Studies
The effect of B2088 on the morphology of LPS was investigated by transmission electron microscopy. A suspension of LPS (extracted from P. aeruginosa 10, purchased from Sigma, product L8643) (1 mg/ml) was incubated with B2088 (70 μm) for 2.5 h. A 1-μl aliquot of this solution was placed on Formvar-coated copper grids, stained with 1% uranyl acetate, dried, and imaged by a JEOL 100 TEM machine. The same LPS suspension sample without B2088 treatment was used as a control.
NMR Sample Preparation
Lipopolysaccharide extracted from P. aeruginosa was bought from Sigma (product L8643). LPS extract powder (10 mg) was dissolved in 1 ml of Milli-Q water, resulting in 10 mg/ml stock. The average molecular mass of LPS is 10 kDa, so the stock molar concentration was about 1 mm. Different LPS/peptide ratios were tested, ranging from 1:40 to 1:5. An LPS/peptide molar ratio of 1:20 was finally used to acquire NMR spectra. The final 100-μl NMR sample (for HR-MAS probe) contained 47 μl of B2088 peptide stock and 47 μl of LPS stock, 2 μl of DSS as chemical shift reference, and 5 μl of D2O. Sample peptide concentration was 10 mm, and the LPS concentration was 5 mg/ml.
DPC (5.3 mg), together with 5.0 mg of POPG (both purchased from Avanti Polar Lipids) was directly dissolved in 0.5 ml of 30 mm phosphate buffer. This resulted in 3:1 DPC-POPG membrane mimicking micelle solutions (30 mm DPC, 13 mm POPG). The final 500-μl NMR sample contained 1 mm labeled B2088 in DPC-POPG mixed micelles.
POPG (10 mg) together with 28 mg of POPE powder were dissolved in chloroform, dried with N2, and dissolved in 500 μl of PBS buffer. After several cycles of freezing and thawing, a mixed liposome medium was prepared. Homogenous unilamellar liposomal vesicles were obtained by passing the liposome medium through a membrane (pore size, 0.4-μm diameter) 10 times. The NMR sample was prepared by mixing 7 μl of labeled B2088 peptide stock with 35 μl of liposome medium, topped up with D2O, DSS, and PBS to 70 μl. The final sample contained 2 mm peptide, 26 mm POPG, and 78 mm POPE. Following the same method, an NMR sample with a low concentration of POPE-POPG mixed liposomes was prepared (4 mm POPG and 6 mm POPE).
NMR Experiments
NMR experiments were performed using a Bruker Avance II 700-MHz spectrometer at 25 °C for aqueous and micelle solutions in cryoprobe and 30 °C for LPS, liposome, and bacterial medium in HR-MAS probe. Standard 500-μl homogenous solution samples were prepared for the cryoprobe, and 70-μl heterogeneous samples were prepared for the HR-MAS probe. Two-dimensional homonuclear TOCSY, NOESY, and ROESY experiments were conducted with mixing times of 70, 200, and 100 ms, respectively. Heteronuclear 1H,13C/15N HSQC, HNCA, 15N NOESY spectra were also acquired. T1 and T2 15N relaxation were analyzed using 10 1H,15N HSQC spectra with various delays. For T1, the delay time varied from 0.05 to 2.55 s, and for T2, the delay time varied from 0.05 to 1 s.
Residue-specific resonance assignment and assignment of NOESY cross-peaks were performed using CARA (available from the Swiss NMR Web site). Three-dimensional structures of B2088 peptide in various media were reconstructed using CYANA 3.0 (34).
pH titration experiments were performed on the sample of B2088 in DPC-POPG mixed micelles, starting from pH 8.77. For each 0.5 pH unit from pH 8 to pH 1, one-dimensional 1H and two-dimensional 15N and 13C HSQC spectra were acquired. Hydrochloride (HCl, 1 m) was used as the titrant, and 16 titration points were obtained in total.
DOSY experiments were performed with B2088 and the monomer peptide in phosphate buffer, DPC micelles (30 mm), and DPC-POPG (30 mm DPC and 3 mm POPG) mixed micelles (30). The pulsed magnetic field gradient strength was calibrated using the 1H signal in 99.9% D2O with the self-diffusion coefficient of HDO ((1.902 ± 0.002) × 10−9 m2/s). A series of one-dimensional spectra were measured with a diffusion delay of 150 ms and increasing gradient strength in 32 steps from 2 to 95% of Gmax. Diffusion coefficient DS was calculated by fitting the curve of signal intensity versus variable gradient strength. The molar fraction of peptide in lipid (FL) was calculated using Equation 3,
where DL and DF are the diffusion coefficients of lipid micelles and free peptides in solution, respectively.
Molecular Dynamics (MD) Simulations
Charge densities of B2088 and monomer peptide were calculated using methods presented previously (30). B2088 and monomer peptide structures in DPC-POPG mixed micelles were used for the calculations.
To study the impact of B2088 on membrane structure, both atomic and coarse grained simulations were performed. We first studied the interactions of B2088 with a model micelle (DPC/POPG = 3:1) using atomic MD simulations. The peptide was modeled using the CHARMM27 force field (35), the lipids were modeled using the CHARMM36 parameter set (36), and the water molecules were described by the TIP3P water model (37). The coordinates of the micelle were obtained from a 100-ns MD simulation of the self-assembly of DPC-POPG lipid mixtures. The peptide, B2088, was put close to the micelle and subjected to a 200-ns MD simulation. In addition, coarse grained simulations were also performed to study the interaction of B2088 with model membranes. For this purpose, a model bacterial membrane consisting of 384 POPE and 128 POPG lipids was constructed. The Martini force field (38) was used to model both the peptide and the lipids. Because B2088 is highly positively charged, it is expected that electrostatic interactions will play an important role in peptide-membrane interactions. Thus, we used the polarizable water model recently developed by the Martini group because this water model has been shown to correctly describe long range electrostatic interactions (39). In both atomic and coarse grained simulations, a cut-off distance of 1.2 nm was used for both the Lennard-Jones and real space electrostatic interactions, and the particle-mesh Ewald algorithm was employed to calculate the long range electrostatic interactions in reciprocal space. During the MD simulations, temperature and pressure were maintained at 300 K and 1 bar, respectively. In the case of coarse grained simulations of the peptide-membrane system, semi-isotropic coupling was used to maintain the pressure.
RESULTS
Antimicrobial Activities of B2088 and Its Analogous Peptides
Linearized B2088 (linear V2 dimer), truncated B2088 (truncated V2 dimer), B2088 with modified neutral spacers (V3 dimer and L2 dimer), and modified C termini (V2–1A dimer, V2–1D dimer, and V2–1K-amide dimer) were designed and chemically synthesized. Antimicrobial activities of those peptides against Gram-negative bacteria strains were tested and compared with B2088 (Table 1) and supplemental Table 1.
Besides V3 dimer and L2 dimer, B2088 showed lower MIC values than the rest of the peptides. Peptides with modified charge state (V2–1A dimer and V2–1D dimer) and amphiphilicity (truncated V2 dimer) showed increased MICs, indicating that amino acid composition is important for antimicrobial activity as stated in the interfacial activity model. On the other hand, peptides with similar amino acid composition but different three-dimensional structure also show lowered antimicrobial activity, like linear V2 dimer. This suggests that exploring peptide structure and peptide-lipid interactions at an atomic level was necessary for understanding the structure-function relationship of this peptide series. V3 dimer and L2 dimer showed comparable activity to B2088. The amphiphilicity and chemical composition of those peptides are similar to those of B2088, suggesting that a similar antimicrobial mechanism could be expected. We selected B2088 for further study as a representative molecule that is also comparatively easy to produce.
In experiments tracking kinetics of antibacterial activity, B2088 displayed rapid killing ability against the reference P. aeruginosa ATCC 9027, reducing an inoculum by more than 6 log cfu/ml in less than 10 min at 2 times the MIC value (Fig. 1A). Even for the gentamicin-resistant strain P. aeruginosa DR4877/07, reduction of >105 organisms could be achieved using less than 10 μm B2088 within 20 min (supplemental Fig. S2), suggesting that a membrane-targeted action is likely. Resistance of P. aeruginosa ATCC 27853 against B2088 was also tested (Fig. 1B). Over more than 17 bacterial passages, the MIC value of B2088 only rises about 4 times higher, whereas for gentamicin, bacteria developed 32 times MIC resistance, and for norfloxacin, more than 64 times higher MIC concentration was needed to inhibit the bacteria growth. Both bacterial killing kinetics and the resistance assay suggest the likelihood of a membrane-targeting action by B2088.
FIGURE 1.

A, time kill kinetics of B2088 against P. aeruginosa ATCC 9027. B, bacterial resistance assay of B2088 and conventional antibiotics against P. aeruginosa ATCC 27853. The x axis represents the bacteria serial passage number, whereas the y axis represents -fold increase in MIC (current MIC value at certain bacteria passages compared with the MIC at the first passage). Black dotted line, V2 dimer peptide; red line with square, Norfloxacin; blue line with triangle, gentamicin.
Cytotoxicity of B2088 against Mammalian Cells
Human conjunctival epithelial cells were chosen for an in vitro cytotoxicity assay (Fig. 2A). Even at a peptide concentration of 200 μg/ml (i.e. 88 μm), 16–32 times higher than B2088 MIC values for various pathogens, B2088 was found safe (86% cell viability), indicating a high therapeutic index. Hemolytic activity of B2088 was tested using rabbit red blood cells. Up to peptide concentrations of 2 mg/ml, no hemoglobin release was detected (Fig. 2B). Applying B2088 to the rabbit eye in vivo, no clinically detectable effect was seen on corneal wound healing in a rabbit model (supplemental Fig. S3).
FIGURE 2.

A, cytotoxicity assays using human conjunctival cells with the monomer or B2088 peptides showed these peptides to be safe. Reported values are means of performed experiments with error bars representing the S.D. (root mean square deviation). B, hemolysis assay of B2088 against rabbit erythrocytes.
Lipid Phase Partition of B2088 and Monomer Peptide
To characterize the affinity of B2088 and its monomeric precursors for the lipid phase, translational diffusion coefficients (DS) for free peptides and peptide micelle complexes were measured using DOSY experiments (40). Pure DPC and mixed DPC-POPG micelles were used to represent mammalian and bacterial cell membranes, respectively. Table 2 shows the molar fractions of peptides bound to micelles (FL) calculated using the measured diffusion coefficients (41). This parameter reflects the lipophilicity and affinity of the peptides for the lipid membranes. Partitioning in zwitterionic DPC micelles was only ∼50% for all studied peptides, in contrast to the full localization into the same lipid phase doped with anionic POPG lipids. This observation was consistent with their high antimicrobial activity and low cytotoxicity.
TABLE 2.
B2088 and its monomeric peptide lipid phase partitioning
| FLa | Monomer | B2088 |
|---|---|---|
| % | % | |
| DPC | 47.65 | 67.94 |
| DPC-POPG | 100 | 100 |
a FL, molar fraction of peptide that binds to lipid phase.
B2088 Interacts with Bacterial Outer and Inner Membranes
We investigated the interactions of B2088 with bacterial outer and inner membranes using membrane probes. NPN, a membrane-impermeable dye, was used to determine if the peptide acted on the outer membrane of P. aeruginosa ATCC 9027. As an impermeable hydrophobic molecule with weak fluorescence in buffer, disruption of the outer membrane allowed the dye to partition toward the inner membrane with enhanced fluorescence (42, 43). Thus, an increase in fluorescence intensity can be used to estimate permeation of the bacterial outer membrane. Fig. 3A shows that the addition of B2088 to the intact bacteria leads to an abrupt increase in the NPN fluorescence intensity. At a concentration of 5.3 ± 0.1 μm, B2088 led to a 50% increase (PC50) in NPN fluorescence emission intensity. This concentration corresponded to the previously determined MIC values, implying a correlation between outer membrane disruption and bacterial killing.
FIGURE 3.

Effect of B2088 on P. aeruginosa ATCC 9027. A, outer membrane permeabilization monitored by NPN uptake. The increase in fluorescence intensity of NPN is plotted against the concentration of B2088. The graph is fit to a sigmoidal function, and PC50 was estimated. B, depolarization of inner membrane monitored by a membrane-sensitive diSC35 assay.
We next examined the effect of B2088 on the bacterial inner membrane using the membrane potential-sensitive fluorescent dye diSC35. This dye is taken up by the bacteria and self-quenched as the dye concentrates in the cytoplasmic inner membrane. Any disturbance of the membrane potential gradient across the inner membrane will result in the release of the dye and increased fluorescence intensity (44, 45). Fig. 3B shows the peptide concentration-dependent fluorescence dequenching of diSC35 upon the addition of B2088 to intact bacterial cells. Below the MIC values, minimal change in the fluorescence intensity was observed. However, above the MIC value, upon the addition of B2088, a significant increase in fluorescence signal was observed immediately followed by a plateau phase (Fig. 3B). The maximum change in fluorescence intensity was reached in less than 10 min, under which conditions time kill assays showed a 99% reduction in bacteria viability.
B2088 Structure and Dynamic Studies with Bacterial Outer Membrane LPS Extract
Lipopolysaccharides are negatively charged molecules decorating the outer leaf of the outer membrane of Gram-negative bacteria. P. aeruginosa LPS extract was used to study peptide/bacterial outer membrane interactions. In a wide range of explored concentrations above the MIC values, the addition of B2088 immediately caused turbidity in an otherwise clear and stable solution of LPS micelles. Under TEM, we observed the LPS assembly transformed from a ribbon-like structure into large polymorphous aggregates (Fig. 4).
FIGURE 4.
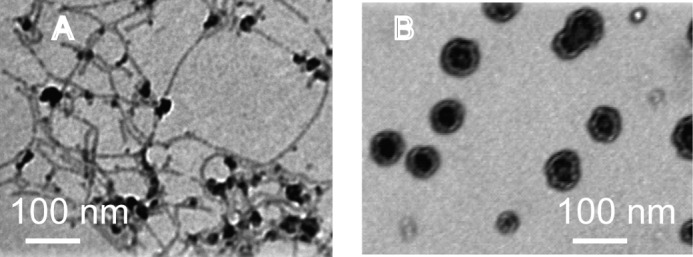
The LPS assembly transition induced by adding B2088 was observed by TEM. A, without B2088, LPS formed a ribbon-like assembly, soluble in solution; B, after adding in B2088, LPS formed large amorphous aggregates, which precipitated out from solution.
The three-dimensional structural analysis of B2088 in polymorphous LPS was carried out using NMR. Residual dipole-dipole interactions and magnetic anisotropy of the heterogeneous LPS precipitated by B2088 resulted in broadened and featureless 1H solution NMR spectra. However, the use of the HR-MAS technique in combination with residue-selective 15N and 13C labeling of B2088 enabled us to obtain the structure of B2088 in complex with LPS. Under these conditions, a 1H,15N HSQC spectrum of selectively 13C,15N-labeled B2088 showed four well resolved cross-peaks with chemical shifts displaced from those found in aqueous solution. A minor conformation of the peptide was also detectable in the spectrum (see Fig. 6BII). The observed resonances were assigned to particular residues using two-dimensional NOESY, three-dimensional HNCA, and 15N-resolved NOESY experiments. The assignment was extended to well resolved 1H resonances stemming from the unlabeled residues. The observation of multiple proton-proton NOEs under HR-MAS as well as 13C chemical shifts (Fig. 5AI) allowed us to reconstruct the three-dimensional structure of the peptide using the program CYANA 3.0 (Fig. 6AI). Intramolecular dynamics were assessed via 15N T1, T2 (supplemental Table S3), and HNOE (Fig. 5BI) relaxation measurements for four backbone amide moieties and a side-chain moiety of Arg-3. Due to the absence of a detectable compact globular fold of B2088, no attempt toward model-free modeling of the intramolecular dynamics was made.
FIGURE 6.

A, an ensemble of NMR conformers of B2088 in LPS (peptide/LPS molar ratio = 1:20) (AI) and in DPC-POPG mixed micelles (peptide/DPC/POPG = 1:30:13) (AII). Backbones of primary and secondary chains are labeled in blue and cyan, respectively. The side chains of charged amino acids are shown in orange. The Lys-9 side chain linking the primary and secondary chains is highlighted in gold. B, 1H,15N HSQC spectra of B2088 in aqueous solution (BI), LPS (BII), and DPC-POPG mixed micelles (BIII). In BI and BII, the chemical shifts of spin residing in equivalent positions in both chains are degenerate, labeled in blue. In BIII, the secondary chain that is more tightly structured is labeled in cyan with a prime. Additional peaks coming from a minor conformation are highlighted in pink. C, 1H,15N HSQC spectra of B2088 peptides in POPG-POPE liposomes. CI, peptide/POPG ratio equals 1:1. CII, peptide/POPG ratio equals 1:12. CIII, peptide/POPG ratio equals 1:13. All peaks are labeled at the same position to show the relative movement of chemical shifts.
FIGURE 5.

Secondary 13Cα chemical shifts (A) and 1H,15N HNOE (B) for the 15N backbone residues and Arg-3 side chain of B2088. I, II, and III panels correspond to the peptide in LPS, in liposomes (B2088/POPE/POPG = 1:3:1), and in DPC-POPG mixed micelles, respectively. Dark and light blue bars represent the primary and secondary backbone chains of the peptide in DPC-POPG, respectively. In LPS and liposomes, the chemical shifts of two branched chains are spectroscopically unresolved.
In polymorphous large LPS aggregates, B2088 adopted an extended conformation with spectroscopically indistinguishable primary and secondary chains. Val residues were significantly more dynamically restrained than flanking Arg and Lys residues. Arg side chains showed the highest flexibility, implying that the Arg guanidinium groups were not immobilized by electrostatic interactions with the oppositely charged LPS. Although no direct interchain NOEs were detected, it appeared likely that in water and LPS, the peptide exhibited a compact structure mediated by hydrophobic interactions between Val side chains, resulting in restricted backbone dynamics at the central hydrophobic residues. The translational diffusion rates in water indicated that the hydrodynamic radius of B2088 might be in close correspondence to that found for the structurally compact dimer of the non-covalently linked monomer peptides (supplemental Table S2) (30). In addition, the translational diffusion rates of B2088 in water showed no concentration dependence, indicating that B2088 did not further dimerize, presumably due to the absence of surface-exposed hydrophobic groups.
To test whether the aggregation of LPS was only due to the effect of electrostatic neutralization of LPS by countercharged groups of the peptide, calcium chloride was used to induce LPS aggregation. The results showed that to induce the same degree of LPS precipitation, a 300-fold higher concentration of calcium chloride was needed. These findings emphasized the importance of the underlying three-dimensional structure of the BAMP compared with linear AMPs and hydrated cations.
Although the translational diffusion rates of B2088 in aggregated LPS matrix was of considerable interest, the Z-gradient pulsed field strength available in the commercial HR-MAS probe was not sufficient for quantitative measurements of the rates. Nevertheless, data showed that while maintaining the high rotational diffusion rate, the translational diffusion of the peptide was significantly impeded in LPS but not quenched entirely. We propose that maintaining high mobility in heterogeneous condensed phase enables the peptide to penetrate through the LPS-decorated bacterial outer membrane and diffuse into the bacterial inner membrane, as was shown by environment-sensitive fluorescent assays. Serving as bacterial membrane extensions, LPS molecules effectively enlarge the reactive surface of the bacteria in relationship to the volume; interacting with LPS through strong electrostatic attractions also helps B2088 to achieve effective high concentrations at the bacterial outer membrane.
B2088 Structure and Dynamic Studies in Bacterial Inner Membrane Mimics
To explore conformational transitions and dynamics of the peptide in lipid phases, we used neutral and negatively charged micelles mimicking the lipid composition of mammalian cell membrane as well as bacterial inner membrane, respectively. The binding affinity of B2088 to mixed micelles was explored using DOSY experiments. The peptide showed only weak partitioning into the neutrally charged DPC micelles. Admixture of POPG (DPC/POPG = 3:1) increased the total negative charge of micelles, causing full partitioning of the peptide into the lipid phase. This was accompanied by accretion of a compact three-dimensional structure of the peptide shown in the HSQC spectrum (Fig. 6BIII) and HNOE data (Fig. 5BIII). Moreover, primary and secondary chains of B2088 manifested different structural associations because the degeneration of resonances stemming from those chains was abolished in the HSQC spectrum (Fig. 6BIII). We determined the detailed three-dimensional structure of B2088 in DPC/POPG micelles (Fig. 6AII) using NOEs and 13Cα chemical shifts (Fig. 5AIII). In this structure, the primary backbone chain was folded back around the secondary chain with four Val residues occupying the core positions with NOE constraints from Arg-7′ (secondary chain) to Val-5 and Arg-8′ to Arg-3 proving the spatial proximity between the two chains. Differential structural association between the two chains implied that the covalent branched linkage might provide a new structural quality surpassing just a means to facilitate chemical association.
Several studies have described a strong link between the cationic charge on a peptide and its antimicrobial activity (46). In addition, the local charge distribution and density within the a peptide have been hypothesized to be important factors (47). We had earlier developed a metric of the peptide charge density as the ratio of the total charge on the peptide and its volume (30). Our findings suggested a strong correlation between charge density and antimicrobial activity. Thus, we compared the charge density of the monomer and B2088 based on their structures in DPC-POPG micelles (supplemental Fig. S5). Remarkably, B2088 in DPC-POPG micelles had a 30% greater charge density than the monomer, implying a direct connection between three-dimensional structure and antimicrobial activity.
The nature of lipid/peptide interactions was explored by pH scanning experiments using NMR and MD simulations utilizing both atomic and coarse grained membrane models (Fig. 7). 1H chemical shifts of the peptides and lipids were recorded as a function of pH in the range from 1 to 9 in a series of one-dimensional 1H and two-dimensional 1H,15N HSQC spectra. The pKa values of peptide backbone amide moieties of individual amino acids and Arg side chains, as well as 1Hα,β of DPC/POPG were estimated via numerical data interpolation and differentiation (Fig. 7C). We found that the intrinsic basic pKa of Arg guanidinium groups and acidic pKa of lipid protons near phosphate groups (1Hα,β) were shifted toward each other in the peptide-micelle complex, suggesting electrostatic interactions or hydrogen bonding between these groups. Atomic simulations using peptides in DPC/POPG mixed micelles showed transient hydrogen bonds between peptide Arg/Lys side chains and lipid phosphate groups. Arg manifested greater hydrogen bonding ability than Lys (Fig. 7BI). This is probably due to the fact that the guanidinium group of Arg can form double hydrogen bonds with PO4 groups, which is energetically more favorable than the hydrogen bond between Lys side chain and lipids (48, 49). Coarse grained simulations using POPG-doped bacterial membrane models showed that B2088 initially solvated in water, rapidly adsorbed onto the negatively charged membrane surface, and remained at the membrane water interface (Fig. 7BII). Moreover, B2088 showed clear preferential interactions with anionic POPG rather than zwitterionic DPC and POPE, leading to redistribution of membrane lipids and the emergence of POPG-rich domains (supplemental Fig. S6). These may decrease the membrane stability, eventually resulting in membrane depolarization that is observed in the fluorescent assays.
FIGURE 7.

A, chemical groups of B2088 and DPC found in close proximity with the peptide solubilized in DPC/POPG micelles. Dashed red lines represent hydrogen bonds between the DPC PO4 group and Arg/Lys side chains. An arrow represents NOEs observed in the NOESY spectrum between Val 1Hα and DPC 1Hβ (supplemental Fig. S4). PO4 groups are shown in yellow. Arg guanidinium groups are shown in green, and the NH3+ groups of Lys are shown in cyan. BI and BII, MD simulations of B2088 in lipid bilayer and DPC/POPG micelles, respectively. CI and CII, chemical shifts as a function of pH for 1HN of Arg-13 and 1Hα of DPC, respectively. The fitted titration curves and the numerical first derivatives are shown.
Retaining high rotational mobility of B2088 in lipids enabled the extension of structural studies of the peptide to POPE/POPG mixed liposomes. In accordance with other studies of AMPs in lipid vesicles (50), at concentrations above 0.1 mm, B2088 caused immediate turbidity in an otherwise clear solution of mixed liposomes with POPE/POPG = 3:1 used to mimic bacterial anionic membranes. Due to the large size and complex morphology of the resulting magnetically anisotropic and inhomogeneous peptide/liposome solutions, HR-MAS NMR was employed in order to detect resolved NMR spectra. We found that at the stoichiometric ratio of B2088/POPE/POPG = 1:39:13, the peptide exhibits reduced rotational diffusion rates and intramolecular dynamics comparable with that found in DPC/POPG micelles (Fig. 6CIII). With an increase in the relative peptide concentration of B2088/POPE/POPG = 1:3:1, accelerated rotational diffusion of the peptide similar to that found in LPS accompanied by chemical shift changes in the 1H,15N HSQC spectra was detected (Fig. 6CI). Analysis of the spectra showed that at lower peptide concentrations, two sets of cross-peaks stemming from predominantly membrane-bound and membrane-dissociated states of the peptide, respectively, could be detected. Only one set of coalescent resonances was observed at intermediate peptide concentrations (Fig. 6CII). This indicated the slow to intermediate (in chemical shift time scale) exchange of peptide bound to and dissociated from the lipid surface. At a ratio of B2088/POPE/POPG = 1:39:13 about 0.056 peptide molecules were bound to 1 nm2 of the liposome surface (1 peptide to 32 lipids). This can be compared with ∼105 peptide molecules/1 nm2 of the bacterial surface (82 peptides to 1 lipid) estimated from our standard bacterial sterilization experiment (see supplemental material). These numbers clearly indicate a significantly lower affinity of B2088 to model liposomes in comparison with live bacteria, corroborating observations from other studies (2).
DISCUSSION
In this study, we used peptide B2088 to observe antimicrobial peptide permeation through bacterial outer membrane LPS while inducing LPS phase transition. This was followed by structural transition of the AMP upon interaction with the inner membrane, which resulted in membrane disruption leading to bacterial death. Interestingly, we noticed that this peptide exhibited different binding affinities to bacterial inner and outer membranes. To explore its significance, binding affinities of AMPs toward bacteria cells and bacterial outer/inner membranes were assessed based on B2088 experimental data and compared with previous literature. In order to establish a comprehensive comparison, both the dissociation constant, Kd, and the molecular stoichiometric ratio in the complex were analyzed, providing binding affinity data emphasizing both the “quality” (nature and strength) of binding at equilibrium and their relative quantities.
Estimates of the activities (effective concentrations) of antimicrobial peptides in typical bacterial susceptibility tests showed that AMPs have to accumulate with a tremendous excess of peptides per membrane lipid molecule in order to induce the bactericidal effect (2). The majority of known potent antimicrobial peptides exhibit bactericidal activity at or above the micromolar range of concentrations in common bacterial sterilization assays, reaching a peptide/lipid ratio of 100:1 (2). In our assays of B2088, we estimate that an excess of more than 100 peptide molecules/1 nm2 of the bacterial surface (82 peptides to 1 lipid molecule) is needed for the efficient inhibition of bacterial growth followed by cell lysis. The maximum saturation concentrations of AMP on the bacterial membranes are determined by the molecular affinities of AMPs to the interaction surfaces. Those affinities can vary wildly from the absence of interactions in the pH neutral membranes of mammals to highly sticky surfaces presented by bacterial membranes enriched with negatively charged lipids, like phosphatidylglycerol and cardiolipin. In typical membrane binding experiments, there are about 1000 AMPs bound to each large unilamellar vesicle, which in turn may contain around 100,000 lipids (52, 53). When using giant unilamellar vesicles, a small number of giant unilamellar vesicles are exposed to 1–10 μm peptides (54, 55). Even in the stoichiometric excess of AMPs in solution, the saturation (equilibrium) concentration of AMP on the surface does not exceed 1 AMP per 20 lipids (2). This is supported by our measurements of the equilibrium binding constants of B2088 to liposomes, indicating an average of 0.056 peptide molecules bound for each 1 nm2 of the liposomal surface (1 peptide to 32 lipids). Apparently, the surface of liposomes or vesicles cannot provide sufficient affinity to accommodate the tremendous excess of AMPs required to disrupt membrane integrity, even in the presence of negatively charged lipids.
Our experiments suggest that interactions with LPS may provide an additional increase in the affinity of AMPs to the complex bacterial surfaces. The net compounding effect of several concurrent electrostatically mediated weak interactions of AMPs with LPS and negatively charged lipid membranes is a significant increase in net binding affinities and concomitantly equilibrium concentrations of AMPs on the bacterial surfaces as is exemplified in combinatorial chemistry producing higher affinity ligands from lower affinity molecular fragments (56). Previously, it was estimated that the dissociation constant of AMPs interacting with LPS was in the low micromolar range (57, 58), similar to the affinity level found for the interactions between AMPs and bacterial membrane lipids (59). However, significantly stronger binding of AMP to live bacteria complex interfaces, typically in the submicromolar range, are estimated from experiments tracing AMP movements in the presence of bacterial surfaces (60). These smaller dissociation constants can usually be explained through higher stoichiometric binding ratios allowing more AMPs at their recipient surfaces. A combination of interactions with LPS resulting in ∼3 AMPs per molecule of LPS (61, 62) and inner membrane binding resulting in 1 AMP per 50 molecules of lipids (2) might enhance binding of AMPs to the composite surfaces consisting of LPS and lipids delivering a remarkably high equilibrium excess of AMPs per bacterial lipid molecule typically found in bacteria susceptibility tests (100:1). Our experimental observations are in good agreement with these estimates, suggesting that a combination of interactions of B2088 with LPS (peptide/LPS ratio = 5:1) and the inner membrane mimics (peptides/lipids = 1:32) might provide significant enhancement of affinity to reach the stoichiometric ratios required for the bactericidal effect (Table 3).
TABLE 3.
Binding affinities of AMP to bacteria-related media
| Estimated dissociation constant, Kd | Stoichiometric ratio |
||
|---|---|---|---|
| Other work | This work | ||
| AMP/LPS | 2 μm | 3:1 | 5:1 |
| AMP/membrane lipids | 2 μm | 1:50 | 1:32 |
| AMP/live bacteria | 10 nma | 100:1 | 82:1 |
a Estimated from AMP diffusion tracking (60) by assuming that the apparent concentration of AMP at which a qualitative change in AMP dynamics at the bacterial surface is observed.
Remarkably, upon interaction of AMPs with LPS, liquid expanded domains in the bacterial outer membrane were replaced by liquid condensed domain structures (45, 64–66). In atomic force microscopy, transformation of LPS assembly from ribbon-like structures into large and densely packed multilamellar aggregates was seen upon the addition of AMPs (67). In our experiments, the full absorption of B2088 into the LPS was detected. Interactions with B2088 caused an LPS phase transition from isotropic micelle solution to large insoluble aggregates. AMPs strongly attracted by LPS but locked into the polymorphous outer membrane showed no antimicrobial activity (45, 68). B2088 retains its rotational molecular mobility, slowed down translational diffusion, and three-dimensional structure resembling that found in aqueous solution. The high molecular mobility of B2088 enabled it to permeate through the bacterial outer membrane and accumulate at the surface of the inner membrane at sufficient concentrations. Although LPS can increase total affinity of AMPs to bacterial surfaces, it is not clear how AMPs shall penetrate though oppositely charged medium without being locked. Although “self-promoted” uptake via displacement of LPS-binding divalent cations has been proposed (69, 70), LPS phase transition seems critical for providing space to facilitate permeation of AMPs.
These observations support the interfacial activity model, which states that the antimicrobial activity of an antimicrobial peptide depends on “the ability of a molecule to bind to a membrane, partition into the membrane-water interface, and alter the packing and organization of the lipids.” Here we propose to extend this interfacial model to include more complex interfaces, such as LPS/mixed lipids. We showed that B2088 retained translational and rotational mobility at a complex interface. This appears to directly correlate with the peptide's efficacy, helping to achieve significantly higher surface concentrations, which are required for its antimicrobial activity.
Sufficiently high concentrations of AMPs at the bacterial membrane promotes structural compaction in B2088, as observed by NMR, and may facilitate membrane remodeling. Upon association with the bacterial inner membrane, B2088 adopts a significantly more compact structure, facilitating full absorption into the membrane through extensive transient hydrogen bonds. Rather than a highly flexible structure in aqueous solution and LPS, the primary and secondary chains of B2088 show different chemical shifts and tight packing in the inner membrane-mimicking micelles. In our MD simulation studies, such structural accretion helped to increase the strength and number of peptide-membrane lipid interactions, therefore resulting in redistribution of membrane lipids. Other studies reported similar concentration-dependent structural transitions associated with AMP activity (71). Changes of peptide orientations relative to the membrane were observed in both α-helix-forming (72–74) and β-sheet-forming (75) peptides. A two-state model of the effect of AMPs on bacterial lipid membranes was proposed (76).
In fact all previously proposed models call for sufficiently high concentrations of AMPs on the surface in order to form supramolecular structures (as in the “carpet model” or “barrel stave model”) (77, 78). In our own efforts of designing AMPs, we found that covalent linkage of monomeric AMPs contributed to a significant increase of antimicrobial activities (7, 29). Higher covalent oligomerization generally tends to increase affinities to lipid surfaces (11). However, large peptide oligomers are prone to be blocked in the LPS condensed phase, whereas a non-aggregated conformation is preferred for membrane-permeating activity (45). In our hands, higher order AMPs also tend to be more cytotoxic, reflecting their increased interactions with mammalian membranes. B2088 retains its monomeric state in various media, which is in agreement with biophysical observations of other AMPs (51, 63, 79). The trend in designing AMPs toward higher covalent order might be limited by increased cytotoxicity and can be counteracted by designing lower order AMPs, maintaining high mobility in LPS and providing higher concentrations at the lipid surfaces of bacteria.
This work was supported by NIG/NMRC/R753, NMRC/TCR/002-SER1/2088R618, BMRC-SCAMP-2/08/1/35/19/586R652, and Flagship (Exploit) funding X031. The authors also thank A* STAR Computational Resource Center for providing the computational facilities.

This article contains supplemental Tables S1–S3 and Figs. S1–S6.
- AMP
- antimicrobial peptide
- DPC
- dodecylphosphocholine
- POPG
- 1-palmitoyl-2-oleoyl-sn-glycero-3-(phospho-rac-(1-glycerol))
- POPE
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- MIC
- minimal inhibitory concentration
- HR-MAS
- high resolution magic angle spinning
- USP
- United States Pharmacopia
- NPN
- N-phenyl-1-naphthylamine
- diSC3
- 3,3′-dipropylthiadicarbocyanine iodide
- MD
- molecular dynamics
- HSQC
- heteronuclear single quantum coherence
- TOCSY
- total correlation spectroscopy
- ROESY
- rotating frame Overhauser effect spectroscopy
- DOSY
- diffusion ordered spectroscopy.
REFERENCES
- 1. Rathinakumar R., Walkenhorst W. F., Wimley W. C. (2009) Broad-spectrum antimicrobial peptides by rational combinatorial design and high-throughput screening. The importance of interfacial activity. J. Am. Chem. Soc. 131, 7609–7617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wimley W. C. (2010) Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol. 5, 905–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hamill P., Brown K., Jenssen H., Hancock R. E. (2008) Novel anti-infectives. Is host defense the answer? Curr. Opin. Biotechnol. 19, 628–636 [DOI] [PubMed] [Google Scholar]
- 4. Easton D. M., Nijnik A., Mayer M. L., Hancock R. E. (2009) Potential of immunomodulatory host defense peptides as novel anti-infectives. Trends Biotechnol. 27, 582–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hancock R. E., Sahl H. G. (2006) Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24, 1551–1557 [DOI] [PubMed] [Google Scholar]
- 6. Giuliani A., Rinaldi A. C. (2011) Beyond natural antimicrobial peptides. Multimeric peptides and other peptidomimetic approaches. Cell Mol. Life Sci. 68, 2255–2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu S. P., Zhou L., Lakshminarayanan R., Beuerman R. W. (2010) Multivalent antimicrobial peptides as therapeutics. Design principles and structural diversities. Int. J. Pept. Res. Ther. 16, 199–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu Z., Deshazer H., Rice A. J., Chen K., Zhou C., Kallenbach N. R. (2006) Multivalent antimicrobial peptides from a reactive polymer scaffold. J. Med. Chem. 49, 3436–3439 [DOI] [PubMed] [Google Scholar]
- 9. Tam J. P., Lu Y. A., Yang J. L. (2002) Antimicrobial dendrimeric peptides. Eur. J. Biochem. 269, 923–932 [DOI] [PubMed] [Google Scholar]
- 10. Campopiano D. J., Clarke D. J., Polfer N. C., Barran P. E., Langley R. J., Govan J. R., Maxwell A., Dorin J. R. (2004) Structure-activity relationships in defensin dimers. A novel link between β-defensin tertiary structure and antimicrobial activity. J. Biol. Chem. 279, 48671–48679 [DOI] [PubMed] [Google Scholar]
- 11. Pieters R. J. (2007) Intervention with bacterial adhesion by multivalent carbohydrates. Med. Res. Rev. 27, 796–816 [DOI] [PubMed] [Google Scholar]
- 12. Dewan P. C., Anantharaman A., Chauhan V. S., Sahal D. (2009) Antimicrobial action of prototypic amphipathic cationic decapeptides and their branched dimers. Biochemistry 48, 5642–5657 [DOI] [PubMed] [Google Scholar]
- 13. Falciani C., Lozzi L., Pini A., Corti F., Fabbrini M., Bernini A., Lelli B., Niccolai N., Bracci L. (2007) Molecular basis of branched peptide resistance to enzyme proteolysis. Chem. Biol. Drug Des. 69, 216–221 [DOI] [PubMed] [Google Scholar]
- 14. Pini A., Falciani C., Mantengoli E., Bindi S., Brunetti J., Iozzi S., Rossolini G. M., Bracci L. (2010) A novel tetrabranched antimicrobial peptide that neutralizes bacterial lipopolysaccharide and prevents septic shock in vivo. FASEB J. 24, 1015–1022 [DOI] [PubMed] [Google Scholar]
- 15. Pini A., Giuliani A., Falciani C., Fabbrini M., Pileri S., Lelli B., Bracci L. (2007) Characterization of the branched antimicrobial peptide M6 by analyzing its mechanism of action and in vivo toxicity. J. Pept. Sci. 13, 393–399 [DOI] [PubMed] [Google Scholar]
- 16. Dathe M., Wieprecht T. (1999) Structural features of helical antimicrobial peptides. Their potential to modulate activity on model membranes and biological cells. Biochim. Biophys. Acta 1462, 71–87 [DOI] [PubMed] [Google Scholar]
- 17. Rapaport D., Peled R., Nir S., Shai Y. (1996) Reversible surface aggregation in pore formation by pardaxin. Biophys. J. 70, 2502–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kolusheva S., Lecht S., Derazon Y., Jelinek R., Lazarovici P. (2008) Pardaxin, a fish toxin peptide interaction with a biomimetic phospholipid/polydiacetylene membrane assay. Peptides 29, 1620–1625 [DOI] [PubMed] [Google Scholar]
- 19. Hallock K. J., Lee D. K., Ramamoorthy A. (2003) MSI-78, an analogue of the magainin antimicrobial peptides, disrupts lipid bilayer structure via positive curvature strain. Biophys. J. 84, 3052–3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bommineni Y. R., Dai H., Gong Y. X., Soulages J. L., Fernando S. C., Desilva U., Prakash O., Zhang G. (2007) Fowlicidin-3 is an α-helical cationic host defense peptide with potent antibacterial and lipopolysaccharide-neutralizing activities. FEBS J. 274, 418–428 [DOI] [PubMed] [Google Scholar]
- 21. Tossi A., Sandri L., Giangaspero A. (2000) Amphipathic, α-helical antimicrobial peptides. Biopolymers 55, 4–30 [DOI] [PubMed] [Google Scholar]
- 22. Sass V., Pag U., Tossi A., Bierbaum G., Sahl H. G. (2008) Mode of action of human β-defensin 3 against Staphylococcus aureus and transcriptional analysis of responses to defensin challenge. Int. J. Med. Microbiol. 298, 619–633 [DOI] [PubMed] [Google Scholar]
- 23. Schibli D. J., Hunter H. N., Aseyev V., Starner T. D., Wiencek J. M., McCray P. B., Jr., Tack B. F., Vogel H. J. (2002) The solution structures of the human β-defensins lead to a better understanding of the potent bactericidal activity of HBD3 against Staphylococcus aureus. J. Biol. Chem. 277, 8279–8289 [DOI] [PubMed] [Google Scholar]
- 24. Epand R. M., Epand R. F. (2009) Lipid domains in bacterial membranes and the action of antimicrobial agents. Biochim. Biophys. Acta 1788, 289–294 [DOI] [PubMed] [Google Scholar]
- 25. Srinivas N., Jetter P., Ueberbacher B. J., Werneburg M., Zerbe K., Steinmann J., Van der Meijden B., Bernardini F., Lederer A., Dias R. L., Misson P. E., Henze H., Zumbrunn J., Gombert F. O., Obrecht D., Hunziker P., Schauer S., Ziegler U., Käch A., Eberl L., Riedel K., DeMarco S. J., Robinson J. A. (2010) Peptidomimetic antibiotics target outer membrane biogenesis in Pseudomonas aeruginosa. Science 327, 1010–1013 [DOI] [PubMed] [Google Scholar]
- 26. Schneider T., Kruse T., Wimmer R., Wiedemann I., Sass V., Pag U., Jansen A., Nielsen A. K., Mygind P. H., Raventós D. S., Neve S., Ravn B., Bonvin A. M., De Maria L., Andersen A. S., Gammelgaard L. K., Sahl H. G., Kristensen H. H. (2010) Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science 328, 1168–1172 [DOI] [PubMed] [Google Scholar]
- 27. Tencza S. B., Creighton D. J., Yuan T., Vogel H. J., Montelaro R. C., Mietzner T. A. (1999) Lentivirus-derived antimicrobial peptides. Increased potency by sequence engineering and dimerization. J. Antimicrob. Chemother. 44, 33–41 [DOI] [PubMed] [Google Scholar]
- 28. Eckert R. (2011) Road to clinical efficacy. Challenges and novel strategies for antimicrobial peptide development. Future Microbiol. 6, 635–651 [DOI] [PubMed] [Google Scholar]
- 29. Zhou L., Liu S. P., Chen L. Y., Li J., Ong L. B., Guo L., Wohland T., Tang C. C., Lakshminarayanan R., Mavinahalli J., Verma C., Beuerman R. W. (2011) The structural parameters for antimicrobial activity, human epithelial cell cytotoxicity, and killing mechanism of synthetic monomer and dimer analogues derived from hBD3 C-terminal region. Amino Acids 40, 123–133 [DOI] [PubMed] [Google Scholar]
- 30. Bai Y., Liu S., Jiang P., Zhou L., Li J., Tang C., Verma C., Mu Y., Beuerman R. W., Pervushin K. (2009) Structure-dependent charge density as a determinant of antimicrobial activity of peptide analogues of defensin. Biochemistry 48, 7229–7239 [DOI] [PubMed] [Google Scholar]
- 31. Liu S., Zhou L., Li J., Suresh A., Verma C., Foo Y. H., Yap E. P., Tan D. T., Beuerman R. W. (2008) Linear analogues of human β-defensin 3. Concepts for design of antimicrobial peptides with reduced cytotoxicity to mammalian cells. Chembiochem 9, 964–973 [DOI] [PubMed] [Google Scholar]
- 32. Diebold Y., Calonge M., Enriquez de Salamanca A. E., Callejo S., Corrales R. M., Sáez V., Siemasko K. F., Stern M. E. (2003) Characterization of a spontaneously immortalized cell line (IOBA-NHC) from normal human conjunctiva. Invest. Ophthalmol. Vis. Sci. 44, 4263–4274 [DOI] [PubMed] [Google Scholar]
- 33. Loh B., Grant C., Hancock R. E. (1984) Use of the fluorescent probe 1-N-phenylnaphthylamine to study the interactions of aminoglycoside antibiotics with the outer membrane of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 26, 546–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Güntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278, 353–378 [DOI] [PubMed] [Google Scholar]
- 35. MacKerell A. D., Jr., Banavali N., Foloppe N. (2000) Development and current status of the CHARMM force field for nucleic acids. Biopolymers 56, 257–265 [DOI] [PubMed] [Google Scholar]
- 36. Klauda J. B., Venable R. M., Freites J. A., O'Connor J. W., Tobias D. J., Mondragon-Ramirez C., Vorobyov I., MacKerell A. D., Jr., Pastor R. W. (2010) Update of the CHARMM all-atom additive force field for lipids. Validation on six lipid types. J. Phys. Chem. B. 114, 7830–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mackerell A. D., Jr. (2004) Empirical force fields for biological macromolecules. Overview and issues. J. Comput. Chem. 25, 1584–1604 [DOI] [PubMed] [Google Scholar]
- 38. Marrink S. J., Risselada H. J., Yefimov S., Tieleman D. P., de Vries A. H. (2007) The MARTINI force field. Coarse grained model for biomolecular simulations. J. Phys. Chem. B. 111, 7812–7824 [DOI] [PubMed] [Google Scholar]
- 39. Yesylevskyy S. O., Schäfer L. V., Sengupta D., Marrink S. J. (2010) Polarizable water model for the coarse-grained MARTINI force field. PLoS Comput. Biol. 6, e1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Morris K. F., Johnson C. S. (1992) Diffusion-ordered two-dimensional nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 114, 3139–3141 [Google Scholar]
- 41. Wang J., Schnell J. R., Chou J. J. (2004) Amantadine partition and localization in phospholipid membrane: a solution NMR study. Biochem. Biophys. Res. Commun. 324, 212–217 [DOI] [PubMed] [Google Scholar]
- 42. Falla T. J., Karunaratne D. N., Hancock R. E. (1996) Mode of action of the antimicrobial peptide indolicidin. J. Biol. Chem. 271, 19298–19303 [DOI] [PubMed] [Google Scholar]
- 43. Wu M., Hancock R. E. (1999) Interaction of the cyclic antimicrobial cationic peptide bactenecin with the outer and cytoplasmic membrane. J. Biol. Chem. 274, 29–35 [DOI] [PubMed] [Google Scholar]
- 44. Zhang L., Rozek A., Hancock R. E. (2001) Interaction of cationic antimicrobial peptides with model membranes. J. Biol. Chem. 276, 35714–35722 [DOI] [PubMed] [Google Scholar]
- 45. Papo N., Shai Y. (2005) A molecular mechanism for lipopolysaccharide protection of Gram-negative bacteria from antimicrobial peptides. J. Biol. Chem. 280, 10378–10387 [DOI] [PubMed] [Google Scholar]
- 46. Hancock R. E., Falla T., Brown M. (1995) Cationic bactericidal peptides. Adv. Microb. Physiol. 37, 135–175 [DOI] [PubMed] [Google Scholar]
- 47. Raj P. A., Dentino A. R. (2002) Current status of defensins and their role in innate and adaptive immunity. FEMS Microbiol. Lett. 206, 9–18 [DOI] [PubMed] [Google Scholar]
- 48. Li J., Garg M., Shah D., Rajagopalan R. (2010) Solubilization of aromatic and hydrophobic moieties by arginine in aqueous solutions. J. Chem. Phys. 133, 054902 [DOI] [PubMed] [Google Scholar]
- 49. Li S., Su Y., Luo W., Hong M. (2010) Water-protein interactions of an arginine-rich membrane peptide in lipid bilayers investigated by solid-state nuclear magnetic resonance spectroscopy. J. Phys. Chem. B 114, 4063–4069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rausch J. M., Marks J. R., Rathinakumar R., Wimley W. C. (2007) β-Sheet pore-forming peptides selected from a rational combinatorial library. Mechanism of pore formation in lipid vesicles and activity in biological membranes. Biochemistry 46, 12124–12139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Porcelli F., Buck-Koehntop B. A., Thennarasu S., Ramamoorthy A., Veglia G. (2006) Structures of the dimeric and monomeric variants of magainin antimicrobial peptides (MSI-78 and MSI-594) in micelles and bilayers, determined by NMR spectroscopy. Biochemistry 45, 5793–5799 [DOI] [PubMed] [Google Scholar]
- 52. Rathinakumar R., Wimley W. C. (2008) Biomolecular engineering by combinatorial design and high-throughput screening. Small, soluble peptides that permeabilize membranes. J. Am. Chem. Soc. 130, 9849–9858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mani R., Cady S. D., Tang M., Waring A. J., Lehrer R. I., Hong M. (2006) Membrane-dependent oligomeric structure and pore formation of a beta-hairpin antimicrobial peptide in lipid bilayers from solid-state NMR. Proc. Natl. Acad. Sci. U.S.A. 103, 16242–16247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tamba Y., Yamazaki M. (2005) Single giant unilamellar vesicle method reveals effect of antimicrobial peptide magainin 2 on membrane permeability. Biochemistry 44, 15823–15833 [DOI] [PubMed] [Google Scholar]
- 55. Tamba Y., Yamazaki M. (2009) Magainin 2-induced pore formation in the lipid membranes depends on its concentration in the membrane interface. J. Phys. Chem. B 113, 4846–4852 [DOI] [PubMed] [Google Scholar]
- 56. Shuker S. B., Hajduk P. J., Meadows R. P., Fesik S. W. (1996) Discovering high-affinity ligands for proteins. SAR by NMR. Science 274, 1531–1534 [DOI] [PubMed] [Google Scholar]
- 57. Thomas C. J., Surolia N., Surolia A. (1999) Surface plasmon resonance studies resolve the enigmatic endotoxin neutralizing activity of polymyxin B. J. Biol. Chem. 274, 29624–29627 [DOI] [PubMed] [Google Scholar]
- 58. Zhang L., Benz R., Hancock R. E. (1999) Influence of proline residues on the antibacterial and synergistic activities of α-helical peptides. Biochemistry 38, 8102–8111 [DOI] [PubMed] [Google Scholar]
- 59. Silvestro L., Gupta K., Weiser J. N., Axelsen P. H. (1997) The concentration-dependent membrane activity of cecropin A. Biochemistry 36, 11452–11460 [DOI] [PubMed] [Google Scholar]
- 60. Leptihn S., Har J. Y., Chen J., Ho B., Wohland T., Ding J. L. (2009) Single molecule resolution of the antimicrobial action of quantum dot-labeled sushi peptide on live bacteria. BMC Biol. 7, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bland J. M., De Lucca A. J., Jacks T. J., Vigo C. B. (2001) All-d-cecropin B. Synthesis, conformation, lipopolysaccharide binding, and antibacterial activity. Mol. Cell Biochem. 218, 105–111 [DOI] [PubMed] [Google Scholar]
- 62. Schuerholz T., Brandenburg K., Marx G. (2012) Antimicrobial peptides and their potential application in inflammation and sepsis. Crit. Care 16, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mani R., Tang M., Wu X., Buffy J. J., Waring A. J., Sherman M. A., Hong M. (2006) Membrane-bound dimer structure of a β-hairpin antimicrobial peptide from rotational-echo double-resonance solid-state NMR. Biochemistry 45, 8341–8349 [DOI] [PubMed] [Google Scholar]
- 64. Andrä J., Koch M. H., Bartels R., Brandenburg K. (2004) Biophysical characterization of endotoxin inactivation by NK-2, an antimicrobial peptide derived from mammalian NK-lysin. Antimicrob. Agents Chemother. 48, 1593–1599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Brandenburg K., Kusumoto S., Seydel U. (1997) Conformational studies of synthetic lipid A analogues and partial structures by infrared spectroscopy. Biochim. Biophys. Acta 1329, 183–201 [DOI] [PubMed] [Google Scholar]
- 66. Snyder S., Kim D., McIntosh T. J. (1999) Lipopolysaccharide bilayer structure. Effect of chemotype, core mutations, divalent cations, and temperature. Biochemistry 38, 10758–10767 [DOI] [PubMed] [Google Scholar]
- 67. Kaconis Y., Kowalski I., Howe J., Brauser A., Richter W., Razquin-Olazarán I., Iñigo-Pestaña M., Garidel P., Rössle M., Martinez de Tejada G., Gutsmann T., Brandenburg K. (2011) Biophysical mechanisms of endotoxin neutralization by cationic amphiphilic peptides. Biophys. J. 100, 2652–2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bhunia A., Saravanan R., Mohanram H., Mangoni M. L., Bhattacharjya S. (2011) NMR structures and interactions of temporin-1Tl and temporin-1Tb with lipopolysaccharide micelles. Mechanistic insights into outer membrane permeabilization and synergistic activity. J. Biol. Chem. 286, 24394–24406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bhattacharjya S., Ramamoorthy A. (2009) Multifunctional host defense peptides. Functional and mechanistic insights from NMR structures of potent antimicrobial peptides. FEBS J. 276, 6465–6473 [DOI] [PubMed] [Google Scholar]
- 70. Hancock R. E. (1984) Alterations in outer membrane permeability. Annu. Rev. Microbiol. 38, 237–264 [DOI] [PubMed] [Google Scholar]
- 71. Eun S. Y., Jang H. K., Han S. K., Ryu P. D., Lee B. J., Han K. H., Kim S. J. (2006) A helix-induced oligomeric transition of gaegurin 4, an antimicrobial peptide isolated from a Korean frog. Mol. Cells 21, 229–236 [PubMed] [Google Scholar]
- 72. Ludtke S. J., He K., Wu Y., Huang H. W. (1994) Cooperative membrane insertion of magainin correlated with its cytolytic activity. Biochim. Biophys. Acta 1190, 181–184 [DOI] [PubMed] [Google Scholar]
- 73. Huang H. W., Wu Y. (1991) Lipid-alamethicin interactions influence alamethicin orientation. Biophys. J. 60, 1079–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Heller W. T., He K., Ludtke S. J., Harroun T. A., Huang H. W. (1997) Effect of changing the size of lipid headgroup on peptide insertion into membranes. Biophys. J. 73, 239–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Heller W. T., Waring A. J., Lehrer R. I., Huang H. W. (1998) Multiple states of β-sheet peptide protegrin in lipid bilayers. Biochemistry 37, 17331–17338 [DOI] [PubMed] [Google Scholar]
- 76. Huang H. W. (2000) Action of antimicrobial peptides. Two-state model. Biochemistry 39, 8347–8352 [DOI] [PubMed] [Google Scholar]
- 77. Rapaport D., Shai Y. (1991) Interaction of fluorescently labeled pardaxin and its analogues with lipid bilayers. J. Biol. Chem. 266, 23769–23775 [PubMed] [Google Scholar]
- 78. Gazit E., Miller I. R., Biggin P. C., Sansom M. S., Shai Y. (1996) Structure and orientation of the mammalian antibacterial peptide cecropin P1 within phospholipid membranes. J. Mol. Biol. 258, 860–870 [DOI] [PubMed] [Google Scholar]
- 79. Li P., Wohland T., Ho B., Ding J. L. (2004) Perturbation of lipopolysaccharide (LPS) micelles by Sushi 3 (S3) antimicrobial peptide. The importance of an intermolecular disulfide bond in S3 dimer for binding, disruption, and neutralization of LPS. J. Biol. Chem. 279, 50150–50156 [DOI] [PubMed] [Google Scholar]