

Abstract
Cuprizone inhibits mitochondrial function and induces demyelination in the corpus callosum, which resembles pattern III lesions in multiple sclerosis patients. However, the molecular and cellular mechanism by which cuprizone induces demyelination remains unclear. Interleukin-17 (IL-17) secreted by T helper 17 cells and γδT cells are essential in the development of experimental autoimmune encephalomyelitis. In this study, we examined the importance of IL-17 signaling in cuprizone-induced demyelination. We found that mice deficient in IL-17A, IL-17 receptor C (IL-17RC), and adaptor protein Act1 (of IL-17R) all had reduced demyelination accompanied by lessened microglial and polydendrocyte cellular reactivity compared with that in wild-type mice in response to cuprizone feeding, demonstrating the essential role of IL-17-induced Act1-mediated signaling in cuprizone-induced demyelination. Importantly, specific deletion of Act1 in astrocytes reduced the severity of tissue injury in this model, indicating the critical role of CNS resident cells in the pathogenesis of cuprizone-induced demyelination. In cuprizone-fed mice, IL-17 was produced by CNS CD3+ T cells, suggesting a source of IL-17 in CNS upon cuprizone treatment.
Introduction
Multiple sclerosis (MS) is a chronic disease of the CNS with pathological characteristics including inflammation, demyelination, gliosis, and axonal injury (Steinman, 2001; Sospedra and Martin, 2005). The pathology of the actively demyelinating lesions was analyzed and categorized into distinct patterns (Lucchinetti et al., 1999, 2000). While experimental autoimmune encephalomyelitis (EAE) can effectively model the pattern I and pattern II lesions (Storch et al., 1998), cuprizone-induced demyelination resembles pattern III lesions. Cuprizone inhibits mitochondrial function, resulting in oligodendrocyte apoptosis and demyelination, which can yield mechanistic insights into the pathogenesis of pattern III lesions (Matsushima and Morell, 2001; Kipp et al., 2009).
Interleukin-17A (IL-17A), also called IL-17, is produced by the T helper 17 (Th17) subsets of CD4+ T cells and also secreted by natural killer (NK) T cells, CD8+ T cells, and γδ T cells (Iwakura et al., 2011). IL-17 is the index member of the IL-17 cytokine family, which includes IL-17A to IL-17F (Gaffen, 2011). IL-17 is involved in the pathogenesis of human and animal autoimmune diseases as well as allergen-specific immune responses (Cua et al., 2003; Aujla et al., 2008; Ishigame et al., 2009; Lin et al., 2009; Cua and Tato, 2010). IL-17 levels are elevated in CNS diseases such as motor neuron disease (Fiala et al., 2010), neuroborreliosis (Nordberg et al., 2011), and MS (Brucklacher-Waldert et al., 2009). EAE is markedly suppressed in mice lacking IL-17 or IL-17 receptor (IL-17R) (Komiyama et al., 2006; Ho et al., 2010). Additionally, IL-17 has been implicated in other nonimmune neuroinflammatory processes including stroke, ischemia–reperfusion and oxygen–glucose deprivation, peripheral nerve injury or spinal cord contusion injury as well as both viral and bacterial cerebral infection (Bai et al., 2008; Lees et al., 2008; Reboldi et al., 2009; Shichita et al., 2009; Sutton et al., 2009; van Leeuwen et al., 2009; Wang et al., 2009; Lee et al., 2010). In summary, IL-17 is expressed in CNS innate as well as adaptive immune processes and appears to constitute an intrinsic neuroinflammatory cytokine.
Both receptor subunits of IL-17 (IL-17 RA and IL-17RC) belong to a newly defined protein family with a conserved cytoplasmic termed SEF/IL-17R (SEFIR) domain (Novatchkova et al., 2003). We previously reported that Act1 is a key component in IL-17 signaling (Leonardi et al., 2000; Li et al., 2000; Qian et al., 2002, 2007). Act1 contains a SEFIR domain at the C terminus and is therefore a member of the SEFIR protein family (Novatchkova et al., 2003). Upon IL-17 stimulation, Act1 is recruited to IL-17R through the SEFIR domain, followed by recruitment of the kinase TAK1 and E3 ubiquitin ligase TRAF6 that mediate downstream nuclear factor-κB activation. Act1 deficiency results in reduced EAE severity (Kang et al., 2010). Remarkably, mice lacking Act1 in myeloid or endothelial cells were EAE susceptible, while those deficient for Act1 in neuroepithelial (derived from Nestin-positive) cells were resistant to disease induced by Th17 cells (Kang et al., 2010). These results suggested that direct signaling by IL-17 to resident CNS cells was neurotoxic in the context of EAE. In this study, we aimed to determine the role of IL-17-induced Act1-mediated signaling for cuprizone-induced demyelination, which mimics the pattern III lesions of MS.
Materials and Methods
Mice and cuprizone treatment.
Act1-deficient C57BL/6 mice were generated as described previously (Qian et al., 2004). C57BL/6J mice (B6 mice) were purchased from The Jackson Laboratory. IL-17RC-deficient mice were provided by Dr. Wenjun Ouyang (Genentech) (Zheng et al., 2008). IL-17-deficient mice were provided by Dr. Yoichiro Iwakura (The Institute of Medical Science, The University of Tokyo, Japan) (Nakae et al., 2002). All the strains are C57BL/6 background (by backcrossing with C57BL/6 mice for at least 12 generations) and were housed under specific pathogen-free conditions. For all experimental groups, age- and sex- matched mice were used (8–10 weeks old). Unless specified, female mice were used for the experiments. Experimental protocols were approved by the Institutional Animal Care and Use Committee of the Cleveland Clinic. Eight- to 10-week-old mice were fed cuprizone (0.2%, w/w; TD.06172; Harlan) to induce demyelination for 1–4 weeks while matched mice fed regular food were included as controls (Liu et al., 2010).
Myelin staining and quantification.
Black-gold staining was performed according to a protocol adapted from the manufacturer (Millipore Bioscience Research Reagents). Briefly, three free-floating sections from each mouse were stained in Eppendorf tubes with 300 μl of 0.2% black-gold solution at 65°C for 10 min. After staining with black-gold, sections were carefully matched and imaged by a three-charge-coupled device video camera interfaced with an Image-Pro Plus Analysis System (version 4.1.0.0; Media Cybernetics) and analyzed with NIH Image J1.34 s. The threshold was set to black-gold staining within each gated corpus callosum and held constant for images obtained at equal objectives and light intensities from slides that were processed in one session. Data represent demyelinated tissue areas (void of black-gold staining) expressed as percentages of total corpus callosum areas.
Electron microscopy.
For electron microscopy, mice were anesthetized and perfused with 1× Sorenson's buffer followed by solution containing 4% paraformaldehyde (PFA) and 2.5% glutaraldehyde. Brains were sliced into 1 to 2 mm sections, the sections containing the hippocampus area were trimmed, and cross sections of the corpus callosum were obtained. Sections were postfixed in the same solution overnight at 4°C, then sections were embedded in Araldite resin. Semithin sections were stained with 1% toluidine blue staining at 65°C for a few seconds, washed several times with water, and then dehydrated and mounted with Permount (Fisher Scientific). Thin sections were cut and stained with uranyl acetate and lead citrate. G-ratios, defined as the diameter of the axon divided by the diameter of the axon and myelin, were calculated by ImageJ. Ten pictures were randomly chosen for each mouse, and 50–100 fibers per picture were calculated. At least three mice per strain per time point were analyzed. The data are shown as G-ratio and total number of myelinated fibers.
TUNEL assay.
Sections were processed for TUNEL staining using a kit from the manufacturer (Roche). Briefly, free-floating tissue sections from controls or cuprizone-treated mice were incubated with Target Retrieval Solution, pH 6.0 (DAKO) for 30 min in an 85°C water bath and then stained with the TUNEL kit following the manufacturer's instruction. Then sections were counterstained with 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI). Data are representative of at least three mice per group.
ELISA.
IL-17 levels were assayed by a mouse IL-17A ELISA kit (BioLegend). All procedures followed the manufacturer's instruction.
Intracellular staining and flow cytometry.
Fluorescence-conjugated CD4 [fluorescein isothiocyanate (FITC; RM4-5)], CD8 (PE,53.6.7), T-cell receptor-γδ [phycoerythrin (PE), GL3)], CD45 (allophycocyanin, 30-F11), Ly6G (PE, 1A8), IL-17 (PE, TC11-18H10.1), and CD3 (FITC, 145–2C11) monoclonal antibodies and isotype controls were all purchased from BD Biosciences or eBiosciences. F4/80 (FITC, A3-1) was obtained from Serotec. For cell surface staining, cells were blocked with 1% BSA plus 2 μg/ml CD16/32 FcR blocker (BD Biosciences) for 30 min on ice. Then cells were incubated with antibodies for 30 min on ice. Cells were then washed three times and fixed by 1% PFA. For intracellular staining, cells were restimulated with 30 ng/ml phorbol 12-myristate 13-acetate (PMA) (Sigma) and 1 μg/ml ionomycin (Sigma) for 5 h with the addition of 1 μm monensin (BD Biosciences) for the last 2 h. Then cells were fixed with Cytofix/Cytoperm buffer (BD Biosciences) before incubation with anti-IL-17, anti-CD4, and anti-δ chain for 30 min on ice. Cells were then washed three times and analyzed on a FACSCalibur flow cytometer (Becton Dickinson). Data were analyzed by FlowJo software (Tree Star).
Histological staining and analysis.
For histological and immunohistochemical analysis of CNS tissues, mice were transcardially perfused with ice-cold PBS, followed by 4% PFA solution, under anesthesia. Thirty-micrometer-thick coronal sections of the brain were cut on a sliding microtome (Leica Microsystems). For immunostaining, sections were pretreated as necessary in a water bath (90°C) for 20 min in sodium citrate antigen retrieval buffer (Dako), pH 6.0, and then cooled for 30 min. Next, sections were incubated overnight with primary antibodies at 4°C. Rat anti-mouse CD45 monoclonal antibody (Serotec) and rabbit anti-mouse PDGF receptor α (PDGFR-α; gift from Dr. William Stallcup, Burnham Institute for Medical Research, La Jolla, CA) were used as primary antibodies. Antigens were visualized following incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies (Molecular Probes). HRP-conjugated antibodies were visualized by the addition of DAB (3,3′-diaminobenzidine, SK-4100; Vector Laboratories) as substrate. Quantification of the percentage of area occupied by immunoreactivity, the number of cells in lesions (number per unit area), or the percentage of immunoreactive cells among total cells was performed as previously described (Liu et al., 2006).
Real-time PCR.
Total RNA was extracted from corpus callosum with TRIzol (Invitrogen) according to the manufacturer's instructions. The SuperScript First-Strand Synthesis System (Invitrogen) was used to synthesize cDNA from 2 μg of total RNA. The cDNA was resuspended in 200 μl of H2O, and 5 μl of cDNA samples were used for real-time PCR in a total volume of 25 μl of SYBR Green Reagent (Invitrogen) and specific primers. All gene expression results are expressed as arbitrary units relative to expression of the gene encoding β-actin. Fold induction of gene expression in the corpus callosum after cuprizone treatment was determined by dividing the relative gene expression of the experimental samples by the mean relative gene expression of the control samples from naive mice.
Statistical analysis.
p Values were determined by Student's t tests. Unless otherwise specified, all results are shown as mean ± SEM; p < 0.05 was considered significant.
Results
Cuprizone-induced demyelination was reduced in Act1-deficient mice
To determine the impact of Act1 deficiency on cuprizone-induced demyelination, we fed age- and sex-matched wild-type and Act1-deficient mice cuprizone-containing food and killed the mice at different time points (2–4 weeks). Histochemical staining of myelin with aurohalophosphate complexes (black-gold staining) detected extensive demyelination in corpus callosum of wild-type mice, especially following 4 weeks of cuprizone feeding (Fig. 1A). Cuprizone-induced demyelination was much reduced in Act1-deficient mice compared with that in wild-type mice. Demyelination is often accompanied by remyelination in MS and cuprizone-induced lesions. The dynamic demyelination–remyelination process is also observed during the development of cuprizone-induced lesions. In particular, cuprizone treatment for more than 3 weeks causes the appearance of large numbers of apoptotic cells. Repair of myelin damage in the adult CNS requires oligodendrocyte progenitor cell (OPC) proliferation and subsequent differentiation into remyelinating oligodendrocytes (Rivers et al., 2008). PDGFR-α+ cells in CNS represent a population of OPCs. OPC proliferation and oligodendrocyte regeneration are impaired in PDGFR-α heterozygotes after cuprizone treatment (Murtie et al., 2005). We thus analyzed PDGFR-α+ OPCs 4 weeks after cuprizone treatment in both groups. PDGFR-α+ OPCs (also termed polydendrocytes) dramatically increased in the corpus callosum of wild-type mice 3–4 weeks after beginning cuprizone feeding (Fig. 1B). It was reported that cuprizone induced cell death of mature oligodendrocytes in corpus callosum (Jha et al., 2010). By in situ TUNEL assay, we found that cuprizone-induced cell death in corpus callosum was much reduced in Act1-deficient mice compared with that in control mice (Fig. 1C). Electron microscopic analysis showed more healthy-appearing myelinated axons in the corpus callosum of Act1-deficient mice than in wild-type controls (Fig. 1D). Collectively, these results demonstrated that cuprizone-induced demyelination was much more severe in wild-type mice than in Act1-deficient mice.
Figure 1.
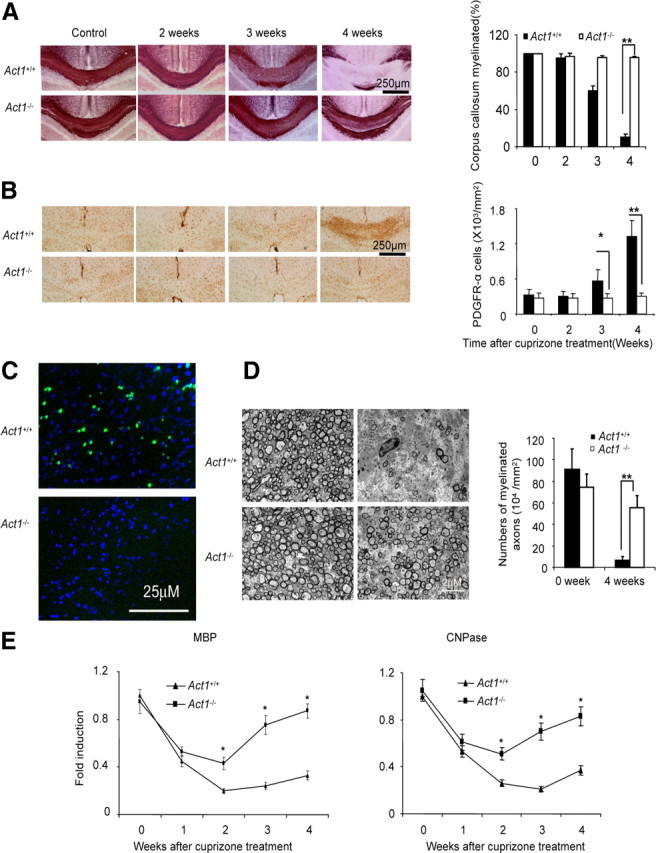
Act1-deficient (Act1−/−) mice are less susceptible to cuprizone-induced demyelination. A, Act1+/+ or Act1−/− mice were fed 0.2% cuprizone food for 2, 3, or 4 weeks. Black-gold staining shows demyelination after cuprizone treatment (left). Percentage of myelinated corpus callosum at different time points is shown (right). B, Corpus callosum from cuprizone-treated mice was stained with anti-PDGFR-α antibodies (left). Quantification of PDGFR-α-positive cells is shown (right). C, Cell death in the corpus callosum of Act1+/+ or Act1−/− mice (fed cuprizone for 4 weeks) was measured by TUNEL assay. Green indicates dead cells, and blue indicates DAPI staining. D, Electron microscopy of corpus callosum of Act1+/+ or Act1−/− mice after 4 weeks of cuprizone feeding. E, Act1+/+ or Act1−/− mice were fed 0.2% cuprizone food for 0–4 weeks. Corpus callosum was dissected under a dissecting microscope, and the expressions of MBP and CNPase were analyzed by real-time PCR. Quantification of myelinated axons is shown. The experiment was repeated three times, and the data are shown as mean ± SEM; n = 5/group. *p < 0.05, **p < 0.005.
Cuprizone-induced damage of oligodendrocytes was transient in Act1-deficient mice
We next examined the direct toxic impact of cuprizone feeding on the differentiated functions of oligodendrocytes, as monitored by levels of myelin protein messages, encoding myelin basic protein (MBP) and 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase) in the corpus callosum of the wild-type and Act1-deficient mice. As shown in Figure 1E, cuprizone treatment led to similar rapid loss of MBP and CNPase in the corpus callosum of wild-type and Act1-deficient mice during the first week of treatment, indicating equivalent direct toxicity of cuprizone for oligodendrocytes in both groups of animals. However, myelin protein mRNAs showed a substantial and dramatic rebound in the corpus callosum of Act-deficient mice but not in wild-type control mice, despite continuous cuprizone feeding of both groups. These data suggested that cuprizone-induced damage to oligodendrocytes in wild-type mice was severe and prolonged, whereas the toxicity of cuprizone was transient and reversible in Act1-deficient mice.
Cuprizone-induced microglial accumulation and leukocyte infiltration are reduced in Act1-deficient mice
Cuprizone-induced demyelination is accompanied by robust myeloid-cell accumulation mainly derived from proliferation of resident microglia (Remington et al., 2007). As shown in Figure 2A, abundant CD45+ cell accumulation was observed in the corpus callosum of wild-type mice, especially following 3–4 weeks of cuprizone treatment, whereas only a mild increase of CD45+ cells was detected in Act1-deficient mice (Fig. 2B). To determine whether these CD45+ cells included infiltrated leukocytes, we performed flow cytometric analysis of mononuclear cells from the brain 4 weeks after cuprizone feeding. As shown in Figure 2C, we detected infiltrating leukocytes (CD45hi cells) in the brain after cuprizone feeding, including macrophages, neutrophils, conventional T cells, and γδ T cells. Infiltrated leukocytes were substantially more numerous in wild-type mice than in Act1-deficient mice (Fig. 2D). More than 60% of CD45+ cells were microglia cells in both groups by flow cytometric analysis, as previously reported (Remington et al., 2007). Together with our previous report (Liu et al., 2010), these data implicate leukocyte infiltration in the pathogenesis of cuprizone-induced demyelination, and it is also plausible that Act1 signaling is critical for leukocyte infiltration after cuprizone feeding. Consistent with this hypothesis, we detected inflammatory gene expression in the corpus callosum of wild-type mice, including tumor necrosis factor-α (TNF-α), IL-1β, IL-18, NLRP3, and CCL2, the last of which may play a critical role in promoting the infiltration of inflammatory monocytes after cuprizone treatment. Importantly, cuprizone-induced inflammatory gene expression in the corpus callosum was substantially reduced in Act1-deficient mice compared with that in wild-type mice as shown in Figure 2, E and F.
Figure 2.
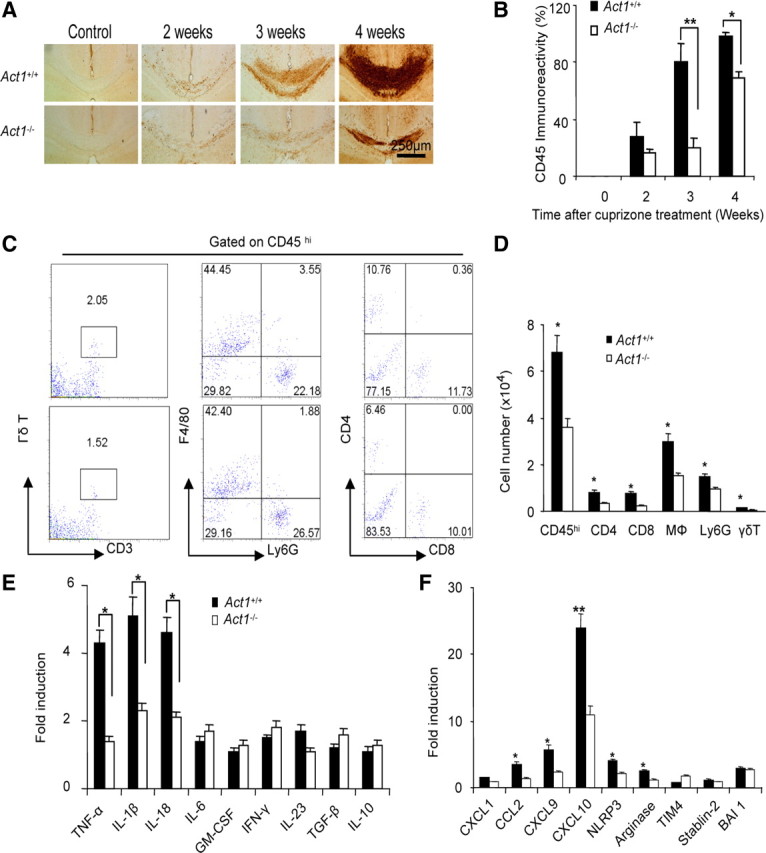
Decreased inflammatory reaction in the CNS of Act1-deficient mice. A, Act1+/+ or Act1−/− mice were fed 0.2% cuprizone food for 2, 3, or 4 weeks. Corpus callosum was stained with anti-CD45 antibody. B, Quantification of CD45 immunoreactivity is shown (right). C, MNCs were isolated from Act1+/+ or Act1−/− mice after 4 weeks of cuprizone feeding and analyzed by flow cytometry. Cells were gated on the CD45hi population, and the frequency of cell subsets is shown. Data are representative of five mice for each group. D, Absolute number of different cell subsets from one brain is shown. E, Gene expression in corpus callosum from each group was measured by real-time PCR after 4 weeks of cuprizone feeding. The experiment was repeated three times, and the data are shown as mean ± SEM, except when specified; n = 5/group. *p < 0.05, **p < 0.005.
IL-17A-Act1 axis is important for cuprizone-induced demyelination
Receptor subunits for IL-17A and IL-17F (IL-17 RA and IL-17RC) belong to the IL-17R family, including three additional members: IL-17RB, IL-17RD, and IL-17RE. While IL-17E and IL-17B bind IL-17RB (Chang and Dong, 2011; Swaidani et al., 2011), to induce Th2 cytokines, IL-17C is the ligand for IL-17RE (Chang et al., 2011; Ramirez-Carrozzi et al., 2011; Song et al., 2011). Importantly, it has now been well documented that Act1 is the adaptor protein for signaling mediated by the different members of IL-17 and IL-17R family. To determine the role of the IL-17A–Act1 axis in cuprizone-induced demyelination, we fed age- and sex-matched wild-type, IL-17A-, and IL-17RC-deficient mice cuprizone-containing food. Cuprizone-induced demyelination was much reduced in IL-17A- and IL-17RC-deficient mice compared with that in wild-type mice (Fig. 3A,D). The response of the OPCs to cuprizone (PDGFR-α+ cell accumulation) (Fig. 3B,E) and cuprizone-induced microglial activation were much reduced in IL-17A- and IL-17RC-deficient mice compared with that in wild-type mice (Fig. 3C,F). Together, these data demonstrate that IL-17A-induced Act1-mediated signaling via IL-17RC is critical for cuprizone-induced demyelination.
Figure 3.
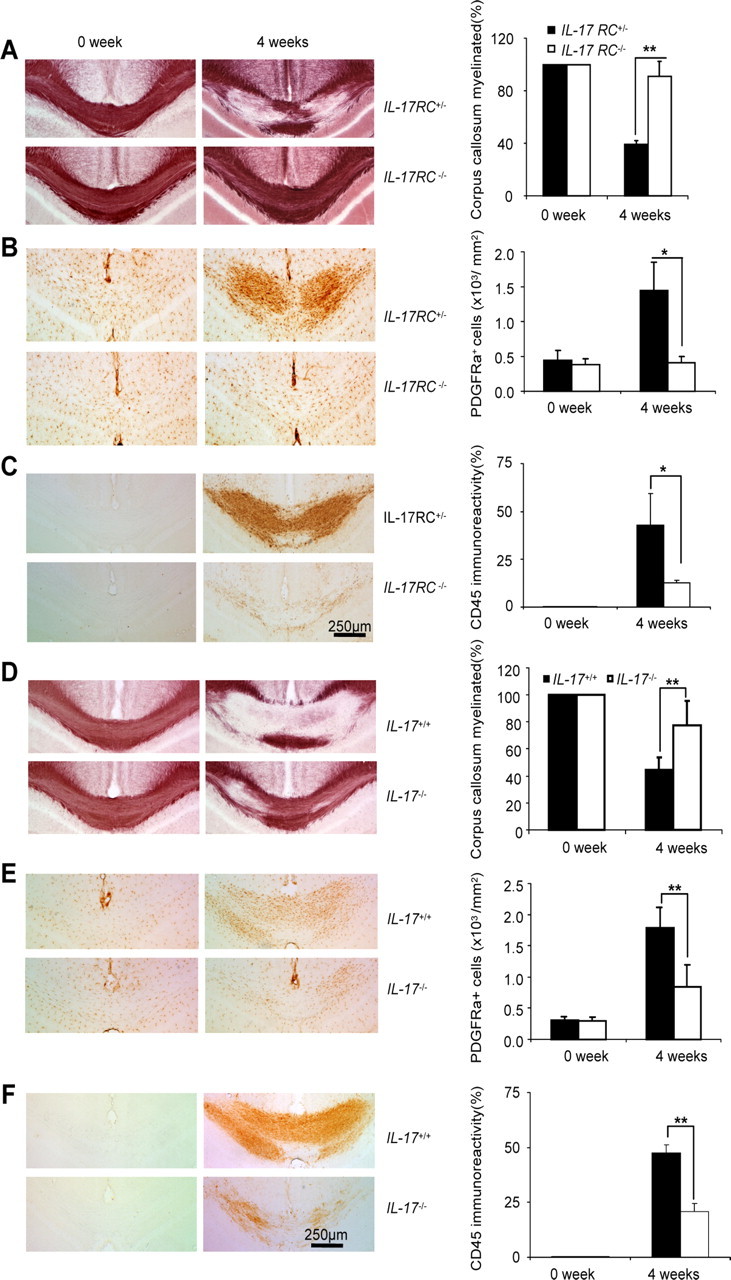
IL-17RC-mediated signaling is critical for cuprizone-induced demyelination. IL-17RC+/− or IL-17RC−/− mice were fed 0.2% cuprizone food or regular food for 4 weeks. A, Black-gold staining shows demyelination after cuprizone treatment (left). The percentage of myelinated corpus callosum at different time points is shown (right). B, Corpus callosum from cuprizone-treated mice was stained with anti-PDGFR-α antibody (left). Quantification of PDGFR-α positive cells is shown (right). C, Corpus callosum was stained with anti-CD45 antibody (left). Quantification of CD45 immunoreactivity is shown (right). D, IL-17+/+ or IL-17−/− mice were fed 0.2% cuprizone food for 4 weeks, and mice fed regular food were included as controls. Black-gold staining shows demyelination after cuprizone treatment (left). Percentage of myelinated corpus callosum at different time points is shown (right). E, Corpus callosum from cuprizone-treated mice was stained with anti-PDGFRα antibody (left). Quantification of PDGFR-α positive cells is shown (right). F, Corpus callosum was stained with anti-CD45 antibody (left). Quantification of CD45 immunoreactivity is shown (right). The data are shown as mean ± SEM; n = 5/group. *p < 0.05, **p < 0.005.
Specific deletion of Act1 in astrocytes reduced the severity of cuprizone-induced demyelination
These studies left unresolved which cells were direct targets for IL-17 action during cuprizone-induced demyelination. Our previous data indicated that IL-17-induced Act1-mediated signals to neuroepithelial cells derived from Nestin+ progenitors were critical for the pathogenesis of EAE (Kang et al., 2010). Of neuroepithelial cells, astrocytes constitute the most cytokine-reactive elements. In our previous study (Kang et al., 2010), we demonstrated that astrocytes are highly responsive to IL-17. Upon IL-17 stimulation, astrocytes produce high levels of inflammatory cytokines (IL-6, TNF, and granulocyte macrophage-colony stimulating factor) and chemokines (CCL2, CXCL1, and CXCL2), which might mediate the recruitment of leukocytes as well as neurotoxicity. To determine the role of Act1 signaling to astrocytes in cuprizone-induced demyelination, we fed wild-type and astrocyte-specific Act1-deficient mice cuprizone chow for 4 weeks and analyzed the severity of demyelination by histochemical black-gold staining. Astrocyte-specific Act1-deficient mice showed less demyelination in corpus callosum (Fig. 4A) than wild-type controls. Cuprizone-induced PDGFR-α+ cell accumulation (Fig. 4B) and microgliosis (Fig. 4C) were not observed in astrocyte-specific Act1-deficient mice. These results indicate that specific deletion of Act1 in astrocytes reduced the severity of cuprizone-induced demyelination.
Figure 4.

Specific deletion of Act1 in astrocytes reduced the severity of cuprizone-induced demyelination. Astrocyte-specific Act1-deficient mice or wild-type control mice were fed 0.2% cuprizone food for 4 weeks and mice fed regular food were included as controls. A, Black-gold staining shows demyelination after cuprizone treatment (left). The percentage of myelinated corpus callosum at different time points is shown (right). B, Corpus callosum from cuprizone-treated mice was stained with anti-PDGFRα antibody (left). Quantification of PDGFR-α positive cell is shown (right). C, Corpus callosum was stained with anti-CD45 antibody (left). Quantification of CD45 immunoreactivity is shown (right). The experiment was repeated three times, and the data are shown as mean ± SEM; *p < 0.05.
CD3+ T cells are a source of IL-17 in response to cuprizone treatment
Since IL-17-induced Act1-mediated signaling is crucial for cuprizone-induced demyelination, it was of interest to define the CNS cellular source of IL-17 in response to cuprizone. We performed flow cytometry on the mononuclear cells (MNCs) isolated from the brains of cuprizone-treated wild-type mice. As shown in Figure 5A, only 0.1% CD45intSSClow cells produced IL-17, while 4.4% CD45hiSSClow cells secreted IL-17, suggesting that IL-17 was mainly derived from infiltrating lymphocytes. Importantly, whereas 12.4% CD45hiCD3+ cells in the brain produced IL-17, only 0.9% CD45hiCD3+ cells in the spleen were IL-17 positive, suggesting that IL-17 was induced in CNS CD3+ T cells rather than in the periphery in response to cuprizone (Fig. 5B). In support of this hypothesis, we detected high levels of IL-17 (∼100 pg/ml) in the supernatant of brain MNCs restimulated with PMA and ionomycin compared with 20 pg/ml from peripheral MNCs. Together, these data suggest that IL-17 produced by CNS CD3+ T cells is one of the sources of IL-17 upon cuprizone treatment.
Figure 5.

CNS CD3+ cells are a source of IL-17 upon cuprizone treatment. C57BL/6 mice were fed 0.2% cuprizone food for 4 weeks, and mononuclear cells were isolated from the brain. Spleen cells were also collected. Cells were restimulated with PMA and ionomycin for 6 h. Golgi inhibitors were added in the last 2 hours. Cells were then stained with the indicated surface markers and anti-IL-17. A, Flow cytometric analysis shows the frequency of IL-17-producing leukocytes (CD45hi) and microglia cells (CD45int). Data are representative of 10 mice per group. B, The frequency of IL-17-producing CD45hi CD3+ cells in the CNS and in the splenocytes is shown. Data are representative of 10 mice per group.
Discussion
Previous studies have clearly demonstrated the indispensable role of IL-17 in the induction and pathogenesis of EAE, which resembles the pattern I and II MS lesions driven by T cell-mediated autoimmune inflammatory response. In this study, we show for the first time that IL-17-mediated signaling plays a critical role in cuprizone-induced demyelination, which shares similarities with pattern III MS lesions associated with apoptosis of myelin-forming oligodendrocytes and pronounced loss of oligodendrocytes. Interestingly, mice deficient in IL-17A, IL-17RC, and adaptor protein Act1 (of IL-17R) displayed diminished demyelination, microglial accumulation, and leukocyte infiltration compared with that in wild-type mice in response to cuprizone. Importantly, astrocytes are highly responsive to IL-17 in vitro, and selective deletion of Act1 in astrocytes ameliorated cuprizone-induced demyelination. Together, these results suggest that IL-17-mediated signaling in astrocytes contributes to the pathogenesis of cuprizone-induced demyelination.
IL-17 is an inflammatory cytokine that upregulates the expression of stimulus-responsive genes in various tissue cells, including astrocytes. Cuprizone-induced inflammatory cytokines (including TNF-α, IL-1β, and IL-18) in the corpus callosum were substantially reduced in vivo in Act1-deficient mice compared with wild-type mice. Previous studies showed delayed demyelination and oligodendrocyte loss in IL-18−/− (Mason et al., 2001) and TNF-α−/− (Arnett et al., 2001) mice during cuprizone-induced demyelination and lack of IL-1β signaling reduced remyelination in this disease model (Mason et al., 2001). Therefore, it is possible that IL-17-mediated signaling impacts cuprizone-induced demyelination at the level of induction of inflammatory cytokines such as TNF-α, IL-1β, and IL-18. It has indeed been shown that astrocytes can produce both TNF-α and IL-1β in response to IL-17 stimulation, which may be partially responsible for the impact of astrocyte-specific IL-17 signaling on cuprizone-induced demyelination.
IL-17 derived from CD4+ Th17 lymphocytes plays an important role in human and mouse autoimmune and inflammatory diseases, including MS and EAE. IL-17 is produced by varied cell types, including CD8+ T cells, γδT, and NK cells as well as CD4+ Th17 cells. Interestingly, we found that IL-17 was produced by CNS CD3+ T cells (including CD4, CD8, and γδ T cells) in response to cuprizone. It is worthwhile to consider how cuprizone feeding leads to accumulation of IL-17-producing T cells. Cuprizone (bis-cyclohexanone-oxaldihydrazone) is a copper chelator that inhibits mitochondrial function. Recent studies showed that the inflammasome sensor NLRP3 plays an important role in cuprizone-induced demyelination (Jha et al., 2010). Nlrp3−/−, casp1−/−, and IL-18−/− mice all showed reduced demyelination in response to cuprizone feeding compared with that in wild-type control mice. These results suggest that the inflammasome sensor NLRP3 probably promotes CNS inflammation and demyelination via caspase-1 and IL-18, implicating NLRP3 as a sensor, directly or indirectly, for cuprizone-induced cellular stress. Indeed, IL-18 (lying downstream of NLRP3) induces IL-17 production by T cells, promoting CNS inflammation and demyelination (Lalor et al., 2011). Whereas CD3+ cells were shown as a source of IL-17 upon cuprizone treatment, it does not rule out other possible sources of IL-17 in this model, as suggested by previous studies (Tzartos et al., 2008). An important question is whether the neurotoxic effects of cuprizone are sufficient to mediate demyelination without additional stressors for oligodendrocytes. It was previously reported that oligodendrocytes could be incubated in vitro with cuprizone without undergoing apoptosis, but that addition of inflammatory cytokines induced oligodendrocyte cell death (Pasquini et al., 2007), strongly suggesting that cuprizone was not sufficient to mediate demyelination and also that the apoptotic event was not entirely cell autonomous (since the inflammatory cytokines came from microglia-conditioned media). Furthermore, NOS−/− mice are resistant to cuprizone-induced demyelination (Liñares et al., 2006), showing that demyelination in vivo is not mediated entirely by the toxic effects of cuprizone. Further, these data imply that cuprizone induces demyelination by a neurotoxic process that is not cell autonomous with respect to oligodendrocytes since NOS1 is expressed by neurons. As we previously published, the direct neurotoxic effects of cuprizone can be assayed by the drastic reduction of myelin protein message expression within corpus callosum at weeks 1–2 of cuprizone feeding, consistent with literature reports of this model (Matsushima and Morell, 2001; Liu et al., 2010). However, our present finding that these neurotoxic changes are reversible in cuprizone astrocyte-specific Act1-deficient mice indicates that a “second hit” is required, in addition to cuprizone-induced mitochondrial dysfunction, to bring about oligodendrocyte cell death and demyelination. Our data in the present study indicate that this second hit must involve signaling of IL-17 to astrocytes. An intriguing observation in the cuprizone model is that oligodendrocytes do survive the initial loss of differentiated function (reflected by myelin protein gene expression). However, the mechanism by which they survive and recover full differentiated function remains under investigation. Empirically, however, it is clear that in an environment in which oligodendrocytes do not undergo apoptosis [as in Cxcr2−/− mice (Liu et al., 2010)] or in NOS1−/− mice (Liñares et al., 2006)], the oligodendrocytes do recover. Here we report that oligodendrocytes recover in Act1-deficient mice despite continued exposure to cuprizone, suggesting IL-17 signaling through its receptor and the adaptor Act1 are implicated in the induction of oligodendrocyte apoptosis in cuprizone-fed mice.
Our data suggested that specific deletion of Act1 in astrocytes reduced the severity of cuprizone-induced demyelination. However, this does not exclude the possibility that Act1 in other CNS resident cell types (such as oligodendrocyte lineages) may also be important for cuprizone-induced demyelination. Future studies are required to clarify this issue by generating oligodendrocyte-specific Act1-deficient mice.
In summary, we propose that cuprizone treatment, directly or indirectly, activates the NLRP3 inflammasome, resulting in the release IL-18 and IL-1, which elicit IL-17 production by CNS infiltrating T cells, etc. In turn, IL-17 acts on CNS resident cells such as astrocytes to amplify the inflammatory cascade, contributing to cuprizone-induced oligodendrocyte cell death and demyelination as shown in Figure 6.
Figure 6.

“Two-hit ” model for cuprizone-induced demyelination. First hit: Upon cuprizone treatment, most of the oligodendrocytes lose differentiated function and partially undergo apoptosis, leading to the release of intracellular substances such as heat shock proteins (HSPs), ATP, reactive oxygen species (ROS), and nucleic acids. While HSPs and nucleic acids activate TLR signaling in microglia to upregulate the expression of pro-IL-1β and pro-IL-18, ATP and ROS activate inflammasomes in microglia to promote the release of IL-18 and IL-1β, which then in turn act on T cells to polarize them to IL-17-producing cells. Second hits: IL-17 targets CNS resident cells such as astrocytes to produce chemokines, TNF-α, and ROS. While chemokines lead to the recruitment of neutrophils and monocytes, TNF and ROS can promote cell death in the CNS and enhance cuprizone-induced demyelination.
Footnotes
This work was supported by National Institutes of Health Grants R01NS071996 (X.L.) and R01NS32151 and the Williams Family Fund for MS Research (R.M.R.). We thank William B. Stallcup (Burnham Institute for Medical Research, La Jolla, CA) for generously providing antibodies to PDGFR-α.
References
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001;4:1116–1122. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburg MM, Mason CA, Iwakura Y, Kolls JK. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Liu R, Huang D, La Cava A, Tang YY, Iwakura Y, Campagnolo DI, Vollmer TL, Ransohoff RM, Shi FD. CCL2 recruitment of IL-6-producing CD11b+ monocytes to the draining lymph nodes during the initiation of Th17-dependent B cell-mediated autoimmunity. Eur J Immunol. 2008;38:1877–1888. doi: 10.1002/eji.200737973. [DOI] [PubMed] [Google Scholar]
- Brucklacher-Waldert V, Stuerner K, Kolster M, Wolthausen J, Tolosa E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain. 2009;132:3329–3341. doi: 10.1093/brain/awp289. [DOI] [PubMed] [Google Scholar]
- Chang SH, Dong C. Signaling of interleukin-17 family cytokines in immunity and inflammation. Cell Signal. 2011;23:1069–1075. doi: 10.1016/j.cellsig.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SH, Reynolds JM, Pappu BP, Chen G, Martinez GJ, Dong C. Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity. 2011;35:611–621. doi: 10.1016/j.immuni.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10:479–489. doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- Fiala M, Chattopadhay M, La Cava A, Tse E, Liu G, Lourenco E, Eskin A, Liu PT, Magpantay L, Tse S, Mahanian M, Weitzman R, Tong J, Nguyen C, Cho T, Koo P, Sayre J, Martinez-Maza O, Rosenthal MJ, Wiedau-Pazos M. IL-17A is increased in the serum and in spinal cord CD8 and mast cells of ALS patients. J Neuroinflammation. 2010;7:76. doi: 10.1186/1742-2094-7-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffen SL. Recent advances in the IL-17 cytokine family. Curr Opin Immunol. 2011;23:613–619. doi: 10.1016/j.coi.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AW, Shen F, Conti HR, Patel N, Childs EE, Peterson AC, Hernández-Santos N, Kolls JK, Kane LP, Ouyang W, Gaffen SL. IL-17RC is required for immune signaling via an extended SEF/IL-17R signaling domain in the cytoplasmic tail. J Immunol. 2010;185:1063–1070. doi: 10.4049/jimmunol.0903739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, Iwakura Y. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity. 2011;34:149–162. doi: 10.1016/j.immuni.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Jha S, Srivastava SY, Brickey WJ, Iocca H, Toews A, Morrison JP, Chen VS, Gris D, Matsushima GK, Ting JP. The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J Neurosci. 2010;30:15811–15820. doi: 10.1523/JNEUROSCI.4088-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Z, Altuntas CZ, Gulen MF, Liu C, Giltiay N, Qin H, Liu L, Qian W, Ransohoff RM, Bergmann C, Stohlman S, Tuohy VK, Li X. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity. 2010;32:414–425. doi: 10.1016/j.immuni.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipp M, Clarner T, Dang J, Copray S, Beyer C. The cuprizone animal model: new insights into an old story. Acta Neuropathol. 2009;118:723–736. doi: 10.1007/s00401-009-0591-3. [DOI] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- Lalor SJ, Dungan LS, Sutton CE, Basdeo SA, Fletcher JM, Mills KH. Caspase-1-processed cytokines IL-1beta and IL-18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J Immunol. 2011;186:5738–5748. doi: 10.4049/jimmunol.1003597. [DOI] [PubMed] [Google Scholar]
- Lee J, Sandor M, Heninger E, Fabry Z. Mycobacteria-induced suppression of autoimmunity in the central nervous system. J Neuroimmune Pharmacol. 2010;5:210–219. doi: 10.1007/s11481-010-9199-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees JR, Iwakura Y, Russell JH. Host T cells are the main producers of IL-17 within the central nervous system during initiation of experimental autoimmune encephalomyelitis induced by adoptive transfer of Th1 cell lines. J Immunol. 2008;180:8066–8072. doi: 10.4049/jimmunol.180.12.8066. [DOI] [PubMed] [Google Scholar]
- Leonardi A, Chariot A, Claudio E, Cunningham K, Siebenlist U. CIKS, a connection to Ikappa B kinase and stress-activated protein kinase. Proc Natl Acad Sci U S A. 2000;97:10494–10499. doi: 10.1073/pnas.190245697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Commane M, Nie H, Hua X, Chatterjee-Kishore M, Wald D, Haag M, Stark GR. Act1, an NF-kappa B-activating protein. Proc Natl Acad Sci U S A. 2000;97:10489–10493. doi: 10.1073/pnas.160265197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Ritchea S, Logar A, Slight S, Messmer M, Rangel-Moreno J, Guglani L, Alcorn JF, Strawbridge H, Park SM, Onishi R, Nyugen N, Walter MJ, Pociask D, Randall TD, Gaffen SL, Iwakura Y, Kolls JK, Khader SA. Interleukin-17 is required for T helper 1 cell immunity and host resistance to the intracellular pathogen Francisella tularensis. Immunity. 2009;31:799–810. doi: 10.1016/j.immuni.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liñares D, Taconis M, Maña P, Correcha M, Fordham S, Staykova M, Willenborg DO. Neuronal nitric oxide synthase plays a key role in CNS demyelination. J Neurosci. 2006;26:12672–12681. doi: 10.1523/JNEUROSCI.0294-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Graham GJ, Damodaran A, Hu T, Lira SA, Sasse M, Canasto-Chibuque C, Cook DN, Ransohoff RM. Cutting edge: the silent chemokine receptor D6 is required for generating T cell responses that mediate experimental autoimmune encephalomyelitis. J Immunol. 2006;177:17–21. doi: 10.4049/jimmunol.177.1.17. [DOI] [PubMed] [Google Scholar]
- Liu L, Belkadi A, Darnall L, Hu T, Drescher C, Cotleur AC, Padovani-Claudio D, He T, Choi K, Lane TE, Miller RH, Ransohoff RM. CXCR2-positive neutrophils are essential for cuprizone-induced demyelination: relevance to multiple sclerosis. Nat Neurosci. 2010;13:319–326. doi: 10.1038/nn.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. A quantitative analysis of oligodendrocytes in multiple sclerosis lesions: a study of 113 cases. Brain. 1999;122:2279–2295. doi: 10.1093/brain/122.12.2279. [DOI] [PubMed] [Google Scholar]
- Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Mason JL, Suzuki K, Chaplin DD, Matsushima GK. Interleukin-1β promotes repair of the CNS. J Neurosci. 2001;21:7046–7052. doi: 10.1523/JNEUROSCI.21-18-07046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11:107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtie JC, Zhou YX, Le TQ, Vana AC, Armstrong RC. PDGF and FGF2 pathways regulate distinct oligodendrocyte lineage responses in experimental demyelination with spontaneous remyelination. Neurobiol Dis. 2005;19:171–182. doi: 10.1016/j.nbd.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- Nordberg M, Forsberg P, Johansson A, Nyman D, Jansson C, Ernerudh J, Ekerfelt C. Cytotoxic mechanisms may play a role in the local immune response in the central nervous system in neuroborreliosis. J Neuroimmunol. 2011;232:186–193. doi: 10.1016/j.jneuroim.2010.09.028. [DOI] [PubMed] [Google Scholar]
- Novatchkova M, Leibbrandt A, Werzowa J, Neubüser A, Eisenhaber F. The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem Sci. 2003;28:226–229. doi: 10.1016/S0968-0004(03)00067-7. [DOI] [PubMed] [Google Scholar]
- Pasquini LA, Calatayud CA, Bertone Uña AL, Millet V, Pasquini JM, Soto EF. The neurotoxic effect of cuprizone on oligodendrocytes depends on the presence of pro-inflammatory cytokines secreted by microglia. Neurochem Res. 2007;32:279–292. doi: 10.1007/s11064-006-9165-0. [DOI] [PubMed] [Google Scholar]
- Qian Y, Zhao Z, Jiang Z, Li X. Role of NF kappa B activator Act1 in CD40-mediated signaling in epithelial cells. Proc Natl Acad Sci U S A. 2002;99:9386–9391. doi: 10.1073/pnas.142294499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Qin J, Cui G, Naramura M, Snow EC, Ware CF, Fairchild RL, Omori SA, Rickert RC, Scott M, Kotzin BL, Li X. Act1, a negative regulator in. Immunity. 2004;21:575–587. doi: 10.1016/j.immuni.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, Xiao J, Lu Y, Giltiay N, Liu J, Kordula T, Zhang QW, Vallance B, Swaidani S, Aronica M, Tuohy VK, Hamilton T, Li X. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, Lesch J, Hackney J, Kim J, Zhou M, Lai J, Modrusan Z, Sai T, Lee W, Xu M, Caplazi P, Diehl L, de Voss J, Balazs M, Gonzalez L, Jr, Singh H, et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol. 2011;12:1159–1166. doi: 10.1038/ni.2156. [DOI] [PubMed] [Google Scholar]
- Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B, Sallusto F. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10:514–523. doi: 10.1038/ni.1716. [DOI] [PubMed] [Google Scholar]
- Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson WD. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–1401. doi: 10.1038/nn.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, Iwaki T, Okada Y, Iida M, Cua DJ, Iwakura Y, Yoshimura A. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15:946–950. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- Song X, Zhu S, Shi P, Liu Y, Shi Y, Levin SD, Qian Y. IL-17RE is the functional receptor for IL-17C and mediates mucosal immunity to infection with intestinal pathogens. Nat Immunol. 2011;12:1151–1158. doi: 10.1038/ni.2155. [DOI] [PubMed] [Google Scholar]
- Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- Steinman L. Multiple sclerosis: a two-stage disease. Nat Immunol. 2001;2:762–764. doi: 10.1038/ni0901-762. [DOI] [PubMed] [Google Scholar]
- Storch MK, Stefferl A, Brehm U, Weissert R, Wallström E, Kerschensteiner M, Olsson T, Linington C, Lassmann H. Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol. 1998;8:681–694. doi: 10.1111/j.1750-3639.1998.tb00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Swaidani S, Bulek K, Kang Z, Gulen MF, Liu C, Yin W, Abbadi A, Aronica M, Li X. T cell-derived Act1 is necessary for IL-25-mediated Th2 responses and allergic airway inflammation. J Immunol. 2011;187:3155–3164. doi: 10.4049/jimmunol.1002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–155. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen WM, Lehto M, Karisola P, Lindholm H, Luukkonen R, Sallinen M, Härmä M, Porkka-Heiskanen T, Alenius H. Sleep restriction increases the risk of developing cardiovascular diseases by augmenting proinflammatory responses through IL-17 and CRP. PLoS One. 2009;4:e4589. doi: 10.1371/journal.pone.0004589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DD, Zhao YF, Wang GY, Sun B, Kong QF, Zhao K, Zhang Y, Wang JH, Liu YM, Mu LL, Wang DS, Li HL. IL-17 potentiates neuronal injury induced by oxygen-glucose deprivation and affects neuronal IL-17 receptor expression. J Neuroimmunol. 2009;212:17–25. doi: 10.1016/j.jneuroim.2009.04.007. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]