

Abstract
BACKGROUND AND PURPOSE
The TNF-related apoptosis inducing ligand (TRAIL) induces apoptosis through activation of the death receptors, TRAIL-R1 and TRAIL-R2. Recombinant human (rh) TRAIL and the TRAIL-R1 directed monoclonal antibody mapatumumab are currently clinically evaluated as anticancer agents. The objective of this study was to develop radiopharmaceuticals targeting the TRAIL-R1, suitable for clinical use to help understand and predict clinical efficacy in patients.
EXPERIMENTAL APPROACH
rhTRAIL was radioiodinated with 125I, and conjugated mapatumumab was radiolabelled with 111In. The radiopharmaceuticals were characterized, their in vitro stability and death receptor targeting capacities were determined and in vivo biodistribution was studied in nude mice bearing human tumour xenografts with different expression of TRAIL-R1.
KEY RESULTS
Labelling efficiencies, radiochemical purity, stability and binding properties were optimized for the radioimmunoconjugates. In vivo biodistribution showed rapid renal clearance of [125I]rhTRAIL, with highest kidney activity at 15 min and almost no detectable activity after 4 h. Activity rapidly decreased in almost all organs, except for the xenografts. Radiolabelled mapatumumab showed blood clearance between 24 and 168 h and a reduced decrease in radioactivity in the high receptor expression xenograft.
CONCLUSIONS AND IMPLICATIONS
rhTRAIL and mapatumumab can be efficiently radiolabelled. The new radiopharmaceuticals can be used clinically to study pharmacokinetics, biodistribution and tumour targeting, which could support evaluation of the native targeted agents in phase I/II trials.
Keywords: rhTRAIL, TRAIL receptor monoclonal antibody, radiopharmaceuticals, molecular imaging, TRAIL-R1
Introduction
The protein, TNF-related apoptosis inducing ligand (TRAIL) or Apo2L is a member of the TNF family that was originally identified from its sequence homology to Fas and TNF (Wiley et al., 1995). TRAIL is expressed as a transmembrane type II glycoprotein and can form a soluble homotrimeric form upon proteolytical cleavage of the extracellular domain from the cell surface. Both full-length surface bound TRAIL and the soluble form are biologically active. Soluble recombinant human (rh) TRAIL can induce apoptosis in numerous preclinical models, without apparent toxicity to normal tissues (Duiker et al., 2006).
The engagement of surface-expressed TRAIL receptors by TRAIL or agonist antibodies leads to triggering of the intracellular apoptosis cascade. Currently, four membrane-associated TRAIL receptors have been identified, TRAIL-R1, TRAIL-R2, TRAIL-R3 and TRAIL-R4. TRAIL-R1 and TRAIL-R2 contain a cytoplasmic death domain (DD), which is totally absent from TRAIL-R3 and is truncated in TRAIL-R4 (Debatin and Krammer, 2004). The soluble receptor osteoprotegerin, which is not exclusively a TRAIL receptor, is described as a third decoy receptor that can bind TRAIL with low affinity (Truneh et al., 2000). The TRAIL receptors are expressed by most tumour types and by a wide range of normal tissues (Chen et al., 2003; Spierings et al., 2004; Horak et al., 2005; Koornstra et al., 2005; Reesink-Peters et al., 2005).
While rhTRAIL can bind all TRAIL receptors, agonist antibodies have receptor specificity and will therefore either bind to TRAIL-R1 or TRAIL-R2. Ligand or antibody binding will lead to trimerization of TRAIL-R1 or TRAIL-R2 and recruitment of the adaptor protein Fas-associated death domain (FADD). FADD can recruit and activate the initiator proteases caspase-8 and -10, which will in turn activate downstream proteins and effector caspases, ultimately leading to apoptosis. Mapatumumab (HGS-ETR1) is a fully human IgG1 TRAIL-R1 targeting antibody, designed to mimic the activity of natural TRAIL. Both rhTRAIL and mapatumumab have shown in vitro and in vivo efficacy in a wide variety of human tumour cell lines and xenograft models (LeBlanc and Ashkenazi, 2003; Pukac et al., 2005; Marini et al., 2006).
A phase I study in patients with solid cancers and non-Hodgkin's lymphomas (NHL) showed that rhTRAIL administration is safe and well tolerated (Herbst et al., 2006). Safety and good tolerability was shown in a phase I–II mapatumumab study (Tolcher et al., 2007). Mapatumumab has also been evaluated in studies, in combination with chemotherapy and bortezomib (Leong et al., 2009; Mom et al., 2009; http://www.clinicaltrials.gov, accessed February 2010).
In patients, radiolabelled rhTRAIL or mapatumumab might be used to study receptor specificity and density, pharmacokinetics and biodistribution. This may help to determine whether rhTRAIL or mapatumumab reach the target and thereby predict clinical efficacy of the drug.
In this paper, we describe the development of iodine-125 (125I) radioiodinated rhTRAIL and indium-111 (111In) conjugated mapatumumab, suitable for clinical TRAIL-R1/R2 or TRAIL-R1 scintigraphy, their in vitro and in vivo characterization and biodistribution in human tumour-bearing mouse models.
Methods
rhTRAIL and mapatumumab
rhTRAIL was produced non-commercially in cooperation with IQ-corporation, based on a protocol described earlier (Ashkenazi et al., 1999). Briefly, a rhTRAIL expression vector (pET15b-TRAIL114-281), that harbours the gene encoding for soluble human TRAIL (amino acids 114–281) was transformed in competent Escherichia coli BL21-salt-inducible (SI) cells. Soluble rhTRAIL was recovered from host BL21-SI cell lysates by sonication and purified by SP-Sepharose Fast Flow cation exchange chromatography, nickel-nitrilotriacetic acid affinity chromatography and dialysis. The recombinant protein was formulated in a TRIS/glycerol/zinc buffer pH 7.4 (100 µM zinc sulphate and 10% glycerol).
Mapatumumab was provided by Human Genome Sciences (Rockville, MD).
The conjugation and labelling procedures described for rhTRAIL and mapatumumab were performed under Good Manufacturing Practice (GMP) conditions at our manufacturing licensed radiopharmacy unit, within the Department of Nuclear Medicine and Molecular Imaging.
Radiolabelling of [125I] rhTRAIL
A rhTRAIL solution (45 µg rhTRAIL) of 1 mg·mL−1 in a TRIS/glycerol/zinc buffer was allowed to react with 70 MBq [125I]NaI (GE Healthcare, Eindhoven, the Netherlands) and 50 µg chloramine T (Merck, Amsterdam, the Netherlands) at pH 8 for 3 min. The labelling reaction was stopped with sodium metabisulphite (Acros Organics, Geel, Belgium), and unbound 125I was removed by gel filtration chromatography, to acquire a radiochemical purity of at least 95%. The PD-10 gel filtration column (Sephadex™ G-25M, Amersham Biosciences AB, Uppsala, Sweden) was eluted with the TRIS/glycerol/zinc buffer, containing 0.5% human serum albumin (HSA, Cealb 20%, Sanquin, Amsterdam, the Netherlands).
Conjugation and radiolabelling of [111In]mapatumumab
Mapatumumab was conjugated, according to the method described earlier (Lub-de Hooge et al., 2004). Glassware, materials and solutions used for the conjugation and labelling procedures were sterile, and endotoxin- and metal-free.
Briefly, to 1 mg of antibody, an equimolar amount of cDTPA (Sigma-Aldrich, Zwijndrecht, the Netherlands) was added. After 1 h incubation at room temperature, the reaction mixture was purified by ultracentrifugation (vivaspin-2, Sartorius Mechatronics, Nieuwegein, the Netherlands) with ammonium acetate (50 mM, UMC Groningen, the Netherlands) to eliminate the excess unconjugated cDTPA. Subsequently, 1 mg purified cDTPA-mapatumumab was allowed to react with 50 MBq 111InCl3 (370 MBq·mL−1, >1.85 GBq·mg−1, 99.9% radionuclidic pure; Covidien, Zaltbommel, the Netherlands) for 1 h at room temperature. The product was purified by ultracentrifugation to obtain a radiochemical purity of at least 95%, diluted in saline and sterilized by filtration (0.2 µm Millex GV filter, Millipore, Amsterdam, the Netherlands).
Quality control
Quality control was essentially performed as described earlier (Dijkers et al., 2009). In short, radiochemical purity of the radiolabelled protein was determined by size exclusion HPLC (SEC-HPLC). Instant thin-layer chromatography (ITLC) or trichloroacetic acid (TCA) precipitation were used in addition to the SEC-HPLC to determine radiochemical purity. A Waters 1500 series SEC-HPLC equipped with an in-line radioactivity detector (Ortec, Nieuwegein, the Netherlands) and fraction collector (Bio-Rad Laboratories BV, Veenendaal, the Netherlands) was used to differentiate between labelled product, aggregates and unlabelled 111In or 125I. The TSK SW3000 XL 300 × 7.8 mm size exclusion column (Tosoh Bioscience GmbH, Stuttgart, Germany) and the Bio Silect SEC 250–5, 300 × 7.8 mm column (Bio-Rad Laboratories BV) were used for rhTRAIL and mapatumumab SEC-HPLC respectively. The mobile phase consisted of PBS (NaCl 500 mM, Na2HPO4 9.0 mM and NaH2PO4 1.3 mM; pH 7.4) with 1% methanol. Column performance was tested using a reference Bio-Rad Gel Filtration standard. At a flow of 1.0 mL·min−1 radioiodinated rhTRAIL eluted at 9.8 min, unbound radioiodine at 12 min, mapatumumab at 8 min and [111In]DTPA at 11 min. If (radiolabelled) aggregates were present they eluted early at 5–6 min. Collected fractions were used to determine 125I and establish HPLC column recovery. Radioactivity was measured by a calibrated well-type γ-counter (LKB 1282 Compugamma, LKB Wallac, Turku, Finland).
In vitro stability of the radiolabelled compounds
After optimization of the labelling procedure and assessment of (radio)chemical purity, the stability of the radiopharmaceuticals was determined in buffer (TRIS/glycerol/zinc with 0.5% HSA for [125I]rhTRAIL or ammonium acetate for [111In]mapatumumab) and in human serum. Solutions were stored at 4°C and 37°C in a humidified incubator maintained at 5% CO2 and 95% air. Frequent HPLC and TCA analyses were performed. [125I]rhTRAIL stability was assessed during the first 4 h and after 24 h. The [111In]mapatumumab stability was determined during 7 days.
Cell lines
Human colon (Colo320 and SW948) and breast (SKBR3) cancer cell lines from the ATCC (via LGC Standards GmbH, Wesel, Germany) and the small cell lung cancer (GLC4) cell line, derived from a pleural effusion in our laboratory, were used during the study. Colo320, SW948 and GLC4 cells expressTRAIL-R1 and TRAIL-R2, with the highest TRAIL-R1 and TRAIL-R2 expression in SW948 cells. SKBR3 cells displayed low TRAIL-R1 and TRAIL-R2 expression and was used as a negative control to determine non-specific binding.
All cell lines were cultured at 37°C in a humidified atmosphere with 5% CO2. GLC4 and Colo320 cells were cultured in RPMI 1640 medium (Life Technologies, Breda, the Netherlands) supplemented with 10% fetal calf serum (FCS; Bodinco BV, Alkmaar, the Netherlands). SKBR3 cells were cultured in Dulbecco's modified Eagle's medium High (Life Technologies, Rotterdam, the Netherlands) with 10% FCS. Cells were harvested by short treatment with trypsin. SW948 was cultured in Leibovitz L15-RPMI 1640 (1:1) enriched with 10% FCS, 0.05 M pyruvate, 0.1 M glutamine and 0.025% β-mercaptoethanol. SW948 cells were harvested following treatment with protease.
TRAIL-receptor membrane expression (TRAIL-R1, TRAIL-R2, TRAIL-R3 and TRAIL-R4) was analysed using flow cytometry (Epics Elite, Coulter-Electronics, Hialeah, FL, USA) as described before (van Geelen et al., 2003), to confirm TRAIL-receptor expression. In short, cells were seeded in six-well plates, left to adhere, harvested the following day with cell dissociation buffer for 5 min at 37°C and washed twice with cold PBS supplemented with 2% FCS and 0.1% sodium azide. The following antibodies were used to determine TRAIL receptor membrane expression: TRAIL-R1 (HS101), TRAIL-R3 (HS301), TRAIL-R4 (HS402) from Alexis (Alexis Corporation, Lausen, Switzerland) and secondary PE conjugated goat anti-mouse. Mouse IgG (DAKO, Glostrup, Denmark) was used as isotype control. For TRAIL-R2, phycoerythrin-conjugated mouse monoclonal TRAIL-R2 antibody was used, with mouse IgG2B as isotype control (R&D Systems, Oxon, UK). TRAIL receptor membrane expression was analysed using a flow cytometer (Epics Elite, Coulter-Electronics) and is shown as mean fluorescent intensity (MFI) of all analysed cells from at least three separate experiments.
Receptor cell binding assays
Cell binding assays were performed to confirm conservation of immunoreactivity. The receptor binding assays were essentially similar to the quality control performed during the manufacturing of the naive rhTRAIL and mapatumumab.
For [125I]rhTRAIL, the immunoreactive fraction was determined by cell-binding assays at infinite antigen excess, as described by Lindmo et al. (1984). For this assay, SW948, Colo320 and GLC4 cells were compared with the negative control (SKBR3). In short, serial dilutions in culture medium ranging from 1 to 32 were made of the cell suspension. A fixed amount of radiolabelled product was added to the cells and incubated at 21°C for 1 h. Cell suspensions were subsequently centrifuged (10 min, 167 g) and washed with PBS 5% FCS. Specific binding was calculated as the ratio of cell-bound (pellet obtained after the last centrifugation step) to total radioactivity applied minus non-specific binding, determined by the same procedure after adding a 200-fold excess of the unlabelled compound.
Antibody immunoreactivity was assessed with a surface plasmon resonance assay with the BIAcore 3000 analyser (BIAcore AB, Uppsala, Sweden) by Human Genome Sciences. The receptor molecules were directly immobilized to the surface of separate flow cells of a CM5 chip by amine coupling to a signal of approximately 900 response units (RU), with one lane treated by the amine coupling reagents as a control flow cell. A loop method was created with multiple injections cycles of 120 µL of a concentration between 1 and 480 nM of antibody, followed by a dissociation phase and a regeneration of the chip surface with a 1:1 mixture of 10 mM glycine, pH 2.0, 1.5 M NaCl, and ethylene glycol for 90 s. The experiment was performed at a flow rate of 70 µL·min−1 standard HBS-EP buffer (GE Healthcare Life Sciences, Diegem, Belgium) at 25°C.
In vivo biodistribution in tumour-bearing mice
All animal care and experimental procedures were in accordance with the Law on Animal Experimentation and local guidelines and were approved by the local ethical committee. The in vivo behaviour of radiolabelled rhTRAIL and mapatumumab was assessed using nude mice bearing SKBR3 or SW948 xenografts. The SW948 cell line was used as a TRAIL-R1 and TRAIL-R2 high expression cell line. Because there was no suitable control protein available for rhTRAIL, and we intended to use the same animal model for rhTRAIL and mapatumumab, the low TRAIL-R1 and TRAIL-R2 expression cell line SKBR3 was used as control to determine non-specific binding.
Male nude mice (Hsd:Athymic Nude-nu) were obtained from Harlan Nederland (Horst, the Netherlands) at 4–6 weeks of age (30 g). They were housed in groups of five in individually ventilated, solid bottom, polycarbonate cages with wood chips floor bedding (changed weekly), with a 12 h light–dark cycle. Mice were provided with deionized autoclaved water and standard rodent chow. After 10 days of acclimatizing, the mice were injected s.c. with 1 × 106 SKBR3 or SW948 cells, mixed equally with 0.1 mL Matrigel™ (Becton Dickinson, Bedford, MA, USA). The study was conducted approximately 1–2 weeks after inoculation.
During anaesthesia (with 4% isoflurane; Pharmachemie B.V. Haarlem, the Netherlands), 150 kBq (0.15 mL) [125I]rhTRAIL or [111In]mapatumumab (corresponding to 0.5 µg rhTRAIL or 5 µg of mapatumumab) was injected through the penile vein. At 15, 30, 60 or 240 min after [125I]rhTRAIL or 24, 72 or 168 h after [111In]mapatumumab injection, groups of mice (n= 4) bearing a SKBR3 or SW948 tumour were killed. The organs and tissues were excised, rinsed for residual blood and weighed. Tissue radioactivity was expressed as the percentage of the injected dose per gram tissue (%ID·g−1).
Statistical analysis
Data are presented as mean ± SEM. Statistical analyses were performed using a non-parametric Mann–Whitney U-test (SPSS version 18 for Windows, SPSS Inc., Chicago, IL, USA) and indicated that variances were significantly different. A P-value of ≤0.05 was considered significant.
Results
Conjugation, radiolabelling and quality control
Radioiodination of rhTRAIL resulted in a mean labelling efficiency of 70.3 ± 1.1% (n= 24). Radiochemical purity was always more than 98%, as determined by TCA precipitation.
Maximal activity was 1.3 MBq·µg−1, and mean specific activity was 0.75 ± 0.05 MBq·µg−1 (n= 24).
Mapatumumab labelling with 111In resulted in labelling efficiency of 92.0 ± 1.5% (n= 9). After ultrafiltration, the radiochemical purity was always more than 96%. The maximal specific activity was 0.27, and the mean specific activity was 0.15 ± 0.02 MBq·µg−1. No impurities were detected.
In vitro stability of the radiolabelled compounds
The stability of [125I]rhTRAIL was determined in human serum and in a TRIS/glycerol/zinc buffer, containing 0.5% HSA. At 4°C, [125I]rhTRAIL stability was maintained for 24 h. After 24 h, aggregation and dehalogenation were shown over time, indicating denaturation of the radiolabelled product. The percentage of radiolabelled aggregates (8% after 24 h), increased to 13% and 16% after 48 h and 72 h, respectively, while radiochemical purity decreased to 84% and 81% respectively. Previous analysis has indicated that these aggregates are most likely rhTRAIL dimers. These rhTRAIL dimers will not precipitate from the solution and remain soluble. Because of their molecular weight, they will not be removed by filtration.
Upon storage in human serum at 37°C, high molecular weight impurities were seen after 4 h (8%), but no free iodine could be detected. Given the short serum elimination half-life of less than 30 min in vivo (Xiang et al., 2004; Ling et al., 2006), this was considered acceptable for (pre)clinical evaluation of 125I-rhTRAIL.
Radiolabelled mapatumumab proved to be very stable. Only a small decrease (<6%) in protein-bound radioactivity was observed for mapatumumab stored during 1 week at 4°C in buffer and at 37°C in serum, allowing 111In-mapatumumab measurement and imaging during 1 week.
Receptor cell binding assays
Four different cell lines with variable TRAIL-R1 and TRAIL-R2 expression were used in this study.
There was a 13-fold and twofold difference in receptor expression between the high (SW948) and low (SKBR3) receptor-expressing cell lines for TRAIL-R1 and TRAIL-R2 respectively. Representative TRAIL-R1, TRAIL-R2, TRAIL-R3 and TRAIL-R4 flow cytometry histograms are shown in Figure 1.
Figure 1.
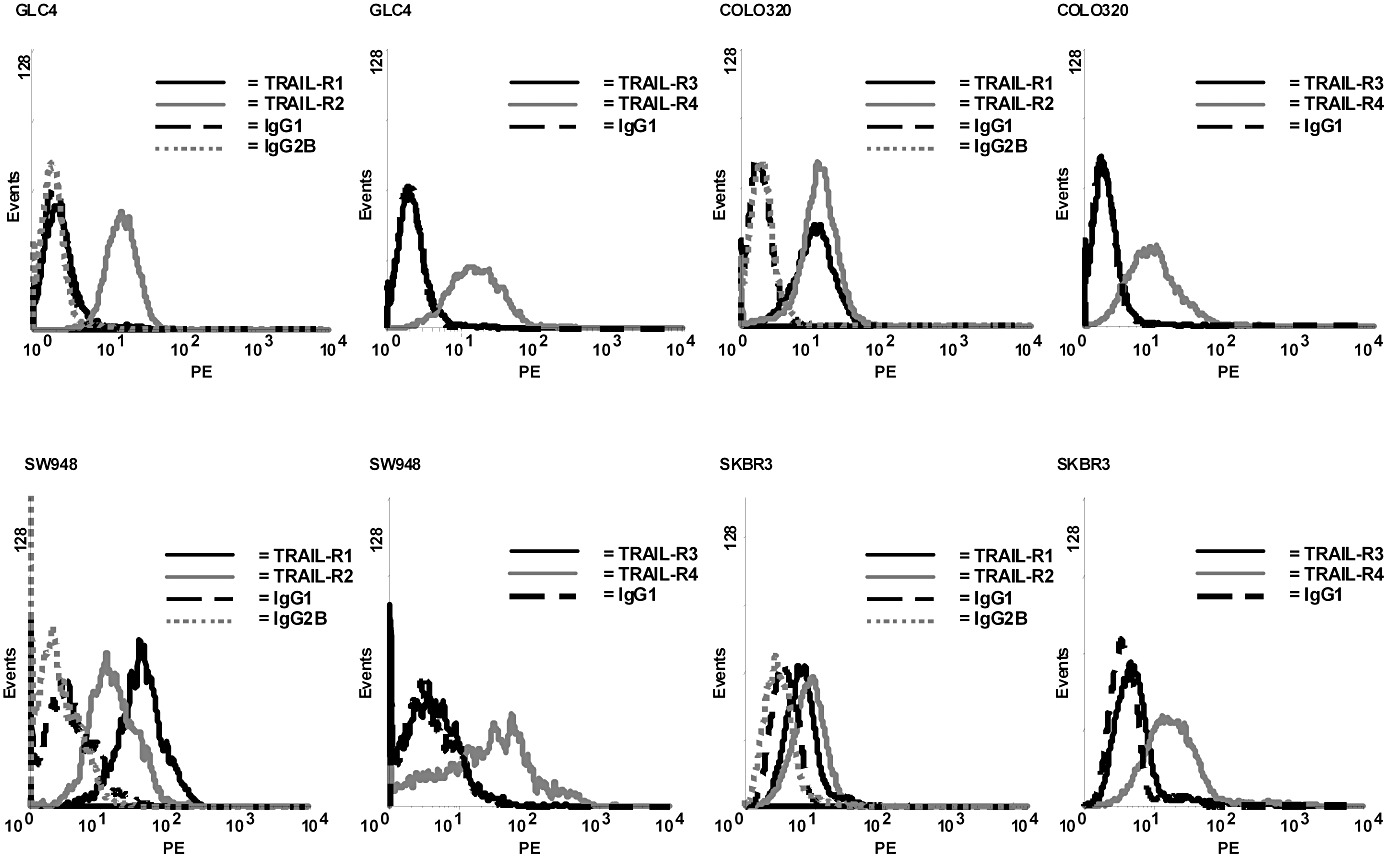
Representative TRAIL-R1, TRAIL-R2, TRAIL-R3 and TRAIL-R4 flow cytometry histograms of the cell lines used in the study.
The [125I]rhTRAIL immunoreactive fraction was calculated from the inverse plot of the cell-binding assays. The immunoreactive fraction was always >0.6 for the GLC4, Colo320 and SW948 cell lines which expressed TRAIL-R1 and TRAIL-R2 (n= 2). The immunoreactive fraction of the SKBR3 control cell line was <0.1. The [111In]mapatumumab immunoreactive fraction was determined by BIAcore analysis. The equilibrium dissociation constant (Kd) of the antibody was >0.6.
In vivo biodistribution of [125I]rhTRAIL in tumour-bearing mice
The in vivo behaviour of [125I]rhTRAIL was assessed using athymic mice bearing SW948 or SKBR3 xenografts. [125I]rhTRAIL biodistribution data of mice bearing these tumours are presented in Table 1. Similar activity in tumours and normal tissues was measured in mice with SKBR3 or SW948 xenografts. The [125I]rhTRAIL biodistribution is characterized by fast renal elimination. Already 15 min after i.v. injection considerable amounts of radioactivity were detected in the urine. SEC-HPLC revealed that no intact rhTRAIL trimer, but smaller fragments were present in the urine. Kidney uptake and total blood activity rapidly and similarly decreased over 4 h after injection in mice with SKBR3 or SW948 xenografts.(Table 1). Tumour uptake reached a similar maximum at 60 min for either tumour, although by 4h the uptake by SW948 was slightly higher (P= 0.06) than that in SKBR3 tumours (Table 1). Uptake in other all well-vascularized organs (lungs, liver and spleen) decreased simultaneously with the blood pool activity. Stomach activity slightly increased over time, and no activity could be detected in the brain, as was expected because of the presence of the blood–brain barrier. Histological analysis showed some core necrosis in the SW948 tumours but not in the SKBR3 tumours, which were smaller in size.
Table 1.
Biodistribution of 125I-rhTRAIL in mice bearing human SKBR3 (A) or SW948 (B) xenografts (n= 4). Data are expressed as the %ID·g−1 tissue ± SEM
| (A) | ||||||||
|---|---|---|---|---|---|---|---|---|
| t = 15 min | t = 30 min | t = 60 min | t = 240 min | |||||
| 125I-rhTRAIL | (±SEM) | 125I-rhTRAIL | (±SEM) | 125I-rhTRAIL | (±SEM) | 125I-rhTRAIL | (±SEM) | |
| Heart | 2.4 | 0.2 | 0.8 | 0.1 | 1.1 | 0.2 | 0.2 | 0.0 |
| Blood | 6.3 | 0.3 | 2.1 | 0.4 | 2.9 | 0.2 | 0.4 | 0.1 |
| Lung | 3.8 | 0.2 | 1.7 | 0.3 | 2.3 | 0.2 | 0.4 | 0.1 |
| Liver | 4.9 | 0.3 | 1.5 | 0.3 | 1.4 | 0.1 | 0.2 | 0.0 |
| Kidney | 43.3 | 2.5 | 6.5 | 1.4 | 6.2 | 0.6 | 0.5 | 0.1 |
| Bladder | 3.7 | 1.3 | 7.5 | 5.0 | 10.2 | 4.0 | 1.1 | 0.3 |
| Stomach | 2.8 | 0.5 | 4.1 | 1.3 | 6.5 | 0.9 | 1.6 | 0.5 |
| Pancreas | 2.0 | 0.3 | 1.1 | 0.3 | 1.7 | 0.4 | 0.2 | 0.0 |
| Spleen | 3.1 | 0.1 | 1.6 | 0.3 | 2.0 | 0.3 | 0.3 | 0.1 |
| Small intestine | 2.7 | 0.3 | 1.9 | 0.6 | 3.2 | 1.3 | 0.3 | 0.1 |
| Colon | 2.4 | 0.6 | 1.0 | 0.1 | 2.1 | 0.6 | 0.3 | 0.2 |
| Muscle | 0.5 | 0.0 | 0.4 | 0.1 | 0.7 | 0.1 | 0.1 | 0.0 |
| Bone | 1.3 | 0.1 | 0.9 | 0.2 | 1.2 | 0.3 | 0.3 | 0.1 |
| Brain | 0.3 | 0.0 | 0.1 | 0.0 | 0.2 | 0.0 | 0.0 | 0.0 |
| Tumour | 1.5 | 0.2 | 2.1 | 0.4 | 2.8 | 0.4 | 0.4 | 0.1 |
| (B) | ||||||||
|---|---|---|---|---|---|---|---|---|
| t = 15 min | t = 30 min | t = 60 min | t = 240 min | |||||
| 125I-rhTRAIL | (±SEM) | 125I-rhTRAIL | (±SEM) | 125I-rhTRAIL | (±SEM) | 125I-rhTRAIL | (±SEM) | |
| Heart | 2.5 | 0.2 | 2.3 | 0.1 | 1.7 | 0.1 | 0.3 | 0.1 |
| Blood | 6.6 | 0.7 | 4.6 | 0.6 | 3.6 | 0.1 | 0.8 | 0.2 |
| Lung | 4.2 | 0.5 | 3.7 | 0.3 | 3.1 | 0.2 | 0.7 | 0.1 |
| Liver | 6.7 | 0.6 | 5.7 | 0.2 | 1.8 | 0.2 | 0.4 | 0.1 |
| Kidney | 41.0 | 4.8 | 19.5 | 3.2 | 8.8 | 2.5 | 0.8 | 0.2 |
| Bladder | 4.7 | 1.5 | 8.3 | 1.0 | 4.1 | 0.9 | 2.3 | 0.6 |
| Stomach | 3.7 | 1.1 | 6.6 | 0.7 | 11.3 | 1.4 | 2.5 | 1.0 |
| Pancreas | 1.8 | 0.4 | 2.1 | 0.1 | 1.5 | 0.1 | 0.3 | 0.1 |
| Spleen | 4.0 | 0.6 | 4.2 | 0.5 | 2.5 | 0.3 | 0.6 | 0.1 |
| Small intestine | 3.9 | 0.4 | 3.9 | 0.6 | 2.4 | 0.3 | 0.8 | 0.1 |
| Colon | 1.6 | 0.2 | 1.9 | 0.2 | 1.5 | 0.1 | 0.4 | 0.1 |
| Muscle | 0.7 | 0.1 | 0.9 | 0.1 | 0.8 | 0.1 | 0.2 | 0.0 |
| Bone | 1.9 | 0.3 | 2.1 | 0.3 | 1.6 | 0.1 | 0.4 | 0.1 |
| Brain | 0.3 | 0.0 | 0.2 | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 |
| Tumour | 2.0 | 0.5 | 2.9 | 0.3 | 3.6 | 0.4 | 1.0 | 0.1 |
In vivo biodistribution of [111In]mapatumumab in tumour-bearing mice
Table 2 shows [111In]mapatumumab biodistribution in mice bearing SKBR3 or SW948 xenografts. As seen for rhTRAIL in Table 1, there was similar normal, non-tumour, tissue biodistribution of mapatumumab for mice with a SW948 or SKBR3 xenograft.
Table 2.
Biodistribution of 111In-mapatumumab in mice bearing human SKBR3 (A) or SW948 (B) xenografts (n= 4). Data are expressed as the %ID·g−1 tissue ± SEM
| (A) | ||||||
|---|---|---|---|---|---|---|
| t = 24 h | t = 72 h | t = 168 h | ||||
| 111In-mapatumumab | (±SEM) | 111In-mapatumumab | (±SEM) | 111In-mapatumumab | (±SEM) | |
| Heart | 3.8 | 0.4 | 2.3 | 0.6 | 1.3 | 0.3 |
| Blood | 8.7 | 2.5 | 4.7 | 2.2 | 1.2 | 1.0 |
| Lung | 3.5 | 0.8 | 2.0 | 0.7 | 1.3 | 0.5 |
| Liver | 9.9 | 2.2 | 7.1 | 1.3 | 3.8 | 1.6 |
| Kidney | 12.9 | 0.2 | 15.5 | 2.3 | 10.2 | 1.0 |
| Bladder | 4.2 | 1.9 | 3.6 | 0.8 | 1.5 | 0.6 |
| Stomach | 2.0 | 0.4 | 1.6 | 0.6 | 0.9 | 0.3 |
| Pancreas | 1.0 | 0.4 | 2.3 | 0.8 | 1.5 | 0.6 |
| Spleen | 14.2 | 3.5 | 10.3 | 4.5 | 5.5 | 3.3 |
| Small intestine | 7.5 | 3.6 | 6.6 | 3.1 | 3.2 | 1.2 |
| Colon | 1.8 | 0.6 | 1.5 | 0.6 | 0.6 | 0.2 |
| Muscle | 0.4 | 0.1 | 0.4 | 0.1 | 0.6 | 0.2 |
| Bone | 3.4 | 1.1 | 6.7 | 2.3 | 2.9 | 1.0 |
| Brain | 0.1 | 0.0 | 0.1 | 0.0 | 0.1 | 0.0 |
| Tumour | 6.2 | 1.1 | 7.5 | 3.5 | 3.7 | 1.8 |
| (B) | ||||||
|---|---|---|---|---|---|---|
| t = 24 h | t = 72 h | t = 168 h | ||||
| 111In-mapatumumab | (±SEM) | 111In-mapatumumab | (±SEM) | 111In-mapatumumab | (±SEM) | |
| Heart | 3.0 | 0.8 | 3.5 | 0.5 | 1.1 | 0.4 |
| Blood | 11.9 | 3.1 | 9.1 | 1.9 | 0.9 | 0.7 |
| Lung | 2.2 | 0.9 | 3.0 | 0.8 | 1.0 | 0.4 |
| Liver | 5.1 | 1.4 | 9.1 | 0.7 | 6.2 | 0.7 |
| Kidney | 10.2 | 3.4 | 17.1 | 2.9 | 8.7 | 1.8 |
| Bladder | 1.3 | 0.2 | 3.2 | 1.1 | 2.0 | 0.4 |
| Stomach | 0.7 | 0.3 | 1.1 | 0.5 | 1.2 | 0.1 |
| Pancreas | 1.0 | 0.3 | 3.0 | 0.7 | 0.8 | 0.4 |
| Spleen | 1.7 | 0.2 | 1.2 | 0.3 | 3.7 | 2.9 |
| Small intestine | 3.5 | 1.4 | 3.5 | 1.6 | 2.5 | 1.4 |
| Colon | 0.6 | 0.1 | 1.1 | 0.4 | 0.9 | 0.3 |
| Muscle | 0.4 | 0.2 | 0.5 | 0.3 | 0.6 | 0.3 |
| Bone | 2.6 | 1.2 | 4.9 | 1.6 | 5.6 | 2.7 |
| Brain | 0.1 | 0.1 | 0.1 | 0.0 | 0.1 | 0.0 |
| Tumour | 3.4 | 0.9 | 6.2 | 2.0 | 5.1 | 2.1 |
Uptake in the well-vascularized organs as heart, liver and lung was approximately similar for SKBR3 and SW948, except for the spleen, which showed an unexplained higher uptake in the low compared with the high TRAIL-R1 and TRAIL-R2 receptor expressing cell line (as shown in Table 2). At 24 h post injection, the SW948 tumour uptake was lower than that of SKBR3, but at end of the experiment at 168 h, there was higher uptake in SW948 then in SKBR3. This difference in tumour uptake was however not significantly different (P= 0.63), due to the variance. Maximal tumour accumulation was seen at 72 h post injection. This indicated that the [111In]mapatumumab accumulation in time was greater for the high TRAIL-R1 and TRAIL-R2 expressing cell line than for the low TRAIL-R1 and TRAIL-R2 expressing cell line.
There was considerable liver and kidney uptake, with highest activity in the kidneys.
Discussion
The present study shows the promising development of radioiodinated cytokine rhTRAIL and 111In radiolabelled monoclonal antibody mapatumumab for clinical use. The new radiopharmaceuticals have appropriate radiochemical purity, stability and receptor binding capacity. The biodistribution studies in nude mice bearing human xenografts showed specific tumour uptake for both radiolabelled rhTRAIL and mapatumumab in time, although the differences in tumour uptake between the low and high TRAIL-R1 and TRAIL-R2 expressing xenografts are generally small for both compounds. Borderline significant differences were reached for 1[125I]TRAIL. No significance was reached for the [111In]mapatumumab, although there was a small difference in tumour uptake at 168h after injection in favour of SW948. Based on the experiments described in this paper, it is concluded that the developed radiopharmaceuticals are stable and provide an interesting starting point for further evaluation in humans.
Radioiodination of rhTRAIL was conducted with an electrophilic substitution reaction, in contrary to a previous study that made use of the mild lactoperoxidase method for the radiolabelling of rhTRAIL (Xiang et al., 2004). We considered enzymatic radioiodination undesirable for clinical applications, as the enzymes themselves can be radiolabelled and have to be separated from other radioiodinated proteins before administration. Only chloramine T and iodogen have therefore been tested for the radiolabelling of rhTRAIL. Chloramine T showed the highest labelling efficiency and least impurity, possibly related to its short reaction time. Radioiodinated rhTRAIL was stable, without signs of aggregation. Zinc was used to protect against disulphide bond formation, which may occur in oxidative environments when free cysteines on rhTRAIL monomers, giving rise to free-standing dimers as well as disulphide-linked rhTRAIL dimer species within trimeric forms of rhTRAIL. This may lead to aggregation, precipitation and/or deactivation of rhTRAIL (patent WO01/00832-PCT/US00/17579, Ashkenazi).
Considering the rapid half-life in humans and animals (Ling et al., 2006), the small amount of aggregates present after storage of radiolabelled rhTRAIL at 37°C was not a matter for concern, as was confirmed in the in vivo biodistribution study. The liver uptake rapidly decreased in time, indicating non-specific and blood-pool-related uptake.
The immunoreactive fraction showed that radiolabelling of rhTRAIL or mapatumumab scarcely affected the receptor binding potential of the proteins.
Some stomach accumulation was observed after injection with [125I]rhTRAIL. This could be explained by endocytosis and lysosomal catabolism of rhTRAIL and subsequent release of the non-residualizing iodine isotope into the circulation (Duncan and Welch, 1993; Press et al., 1996; Kohlhaas et al., 2007), together with some dehalogenation. This is not considered a major issue, because in humans, rhTRAIL is very rapidly cleared. In addition, in clinical settings, potassium perchlorate can be used to block free iodine and prevent thyroid toxicity, as is already done for other radiopharmaceuticals.
In contrast to rhTRAIL, the use of a residualizing isotope is mandatory for the long-circulating internalizing antibody mapatumumab. This was the main reason for using 111In, although iodination of mapatumumab resulted in loss of immunoreactivity.
The biodistribution of [125I]rhTRAIL in nude human xenograft bearing mice showed that rhTRAIL biodistribution was characterized by fast renal clearance, with highest activity in the kidneys at 15 min. This is in accordance with earlier reports (Kelley et al., 2001; Xiang et al., 2004). In most other organs, activity decreased from 15 min onwards, following blood pool activity. The steady decrease of radioactivity in non-tumour tissue suggests absence of specific accumulation in these tissues. The [125I]rhTRAIL tumour uptake in both xenografts reached a maximum at 60 min post injection, with the highest values for the xenograft expressing high levels of TRAIL-R1 and TRAIL-R2.
Regarding the biodistribution of [111In]mapatumumab, there was, in the non-tumour tissues, clear uptake in liver, kidneys, spleen and bone, with the highest activity in liver and kidneys at 168 h post injection. The high liver and spleen uptake was in agreement with known antibody metabolism and retention of 111In in the reticuloendothelial system (Clarke et al., 2000), although the difference in spleen uptake between the low and high TRAIL-R1 and TRAIL-R2 expressing xenografts could not be explained.
[111In]mapatumumab kidney uptake was not expected as the molecular size of an antibody excludes renal filtration. Although dissociation of [111In]cDTPA might also have caused kidney activity, this would have resulted in fast renal 111In clearance and high bone uptake, which was not the case in our animals. In addition, because the mapatumumab kidney accumulation increased in time, this suggested specific antigen binding. A possible explanation for the kidney uptake could be the binding to the mouse killer (MK) death domain containing receptor, which has been identified at high expression levels in the heart, lung and kidney of mice (Wu et al., 1999; Schneider et al., 2003). The MK death domain possesses 76% and 79% amino acid homology with TRAIL-R1 and TRAIL-R2, respectively, and can bind human TRAIL.
The accumulation of [111In]mapatumumab in xenografts at the earliest time point was higher in SKBR3 than in the SW948 xenograft, possible due to a difference in tumour pressure, vasculature and size. After immunohistochemical analysis, core necrosis was seen in the SW948 tumours, which could be an explanation for the difference in tumour penetration velocity for large molecules (mapatumumab). At later time points, there was a steady increase in SW948 tumour accumulation, and a decrease in the SKBR3 tumour.
Our results are comparable with another animal study (Gong et al., 2006), in which mice bearing a colorectal carcinoma were treated with 60 mg·kg−1 paclitaxel i.v., which gave rise to a 20-fold increase in TRAIL-R1 and TRAIL-R2 xenograft levels. For comparison, in our study, there was a 13-fold difference in TRAIL-R1 receptor expression between the SKBR3 and the SW948 cell lines, based on flow cytometry. Imaging and biodistribution with 111In radiolabelled mapatumumab before and after paclitaxel administration showed results comparable to our data, with high uptake in liver and spleen. A difference is that there was less [111In]mapatumumab uptake in the kidneys than we observed. After paclitaxel administration, there was a 1.4-fold difference in xenograft uptake of radiolabelled mapatumumab. In our study, a similar 1.4-fold difference between the SKBR3 and SW948 xenografts was found in vivo.
The flow cytometry data indicate that the cell lines we used also express TRAIL-R3 and TRAIL-R4. Although, in theory, decoy receptors could cause rhTRAIL binding and thus increase rhTRAIL accumulation in the tumour xenografts, such a mechanism has not been described so far. Moreover, in general a clear correlation between decoy receptor expression and resistance to rhTRAIL has not been shown (Held and Schulze-Osthoff, 2001; Holoch and Griffith, 2009).
For both TRAIL-R1 and TRAIL-R2 targeting radiopharmaceuticals, the absolute tumour uptake was low, compared to the levels seen for HER2 imaging with radiolabelled trastuzumab (Lub-de Hooge et al., 2004; Dijkers et al., 2009). It should, however, be taken into account that the tumour receptor overexpression levels that have been seen for HER2 (up to 100-fold due to gene amplification), do not occur for the TRAIL receptors (Pegram et al., 2000).
With [111In]trastuzumab, there was a good correlation between the actual HER2 expression and [111In]trastuzumab uptake, only if the uptake was corrected for non-specific and circulating radioactivity (McLarty et al., 2009). This indicates that there is always a certain amount of non-specific tumour uptake. Correcting for this amount is especially necessary in case of low antigen expression levels. Therefore, it would be advisable to include a radiolabelled control protein (such as IgG for antibodies) in future (pre)clinical studies to allow better discrimination between specific and non-specific tumour uptake. Because in this study, there was no suitable control protein available for rhTRAIL, and we wanted to use the same animal model for rhTRAIL and mapatumumab, we used the low and high TRAIL-R1 and TRAIL-R2 expressing cell lines to discriminate between target specific and non-specific radiopharmaceutical tumour uptake.
Currently, a number of clinical phase II studies have been performed with mapatumumab or are presently active. At this moment, physicians have no good biomarkers to predict mapatumumab response. Radiolabelled rhTRAIL and mapatumumab might help to determine if mapatumumab tumour uptake in patients is variable, due to, for example, tumour pressure, vasculature and size and related to therapy efficacy. Therefore, a clinical study with [111In]mapatumumab is currently ongoing in our centre.
Acknowledgments
This study was supported by grant 2003–2922 of the Dutch Cancer Society and grant 02–17 of the ‘JK de Cock Stichting’, Groningen.
Glossary
- %ID·g−1
percentage injected dose per gram tissue
- cDTPA
cyclic diethylenetriaminepentaacetic acid dianhydride
- DD
death domain
- FADD
Fas-associated death domain
- FCS
fetal calf serum
- ITLC
instant thin-layer chromatography
- MK
mouse killer
- NHL
non-Hodgkin's lymphoma
- rh
recombinant human
- RU
response unit
- SEC-HPLC
size exclusion HPLC
- SI
salt-inducible
- TCA
trichloroacetic acid
- TRAIL
TNF-related apoptosis inducing ligand
Conflicts of interest
The authors state no conflict of interest. Mapatumumab was kindly provided by Human Genome Sciences.
References
- Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–162. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XP, He SQ, Wang HP, Zhao YZ, Zhang WG. Expression of TNF-related apoptosis-inducing Ligand receptors and antitumor effects of TNF-related apoptosis-inducing Ligand in human hepatocellular carcinoma. World J Gastroenterol. 2003;9:2433–2440. doi: 10.3748/wjg.v9.i11.2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke K, Lee FT, Brechbiel MW, Smyth FE, Old LJ, Scott AM. In vivo biodistribution of a humanised anti-Lewis Y monoclonal antibody (hu3S193) in MCF-7 xenografted BALB/c nude mice. Cancer Res. 2000;60:4804–4811. [PubMed] [Google Scholar]
- Debatin KM, Krammer PH. Death receptors in chemotherapy and cancer. Oncogene. 2004;23:2950–2966. doi: 10.1038/sj.onc.1207558. [DOI] [PubMed] [Google Scholar]
- Dijkers EC, Kosterink JG, Rademaker AP, Perk LR, van Dongen GA, Bart J, et al. Development and characterization of clinical-grade 89Zr-trastuzumab for HER2/neu immunoPET imaging. J Nucl Med. 2009;50:974–981. doi: 10.2967/jnumed.108.060392. [DOI] [PubMed] [Google Scholar]
- Duiker EW, Mom CH, de Jong S, Willemse PHB, Gietema JA, van der Zee AGJ, et al. The clinical trail of TRAIL. Eur J Cancer. 2006;42:2233–2240. doi: 10.1016/j.ejca.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Duncan JR, Welch MJ. Intracellular metabolism of indium-111-DTPA-labeled receptor targeted proteins. J Nucl Med. 1993;34:1728–1738. [PubMed] [Google Scholar]
- van Geelen CM, de Vries EG, Le TK, van Weeghel RP, de Jong S. Differential modulation of the TRAIL receptors and the CD95 receptor in colon carcinoma cell lines. Br J Cancer. 2003;89:363–373. doi: 10.1038/sj.bjc.6601065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J, Yang D, Kohanim S, Humphreys R, Broemeling L, Kurzrock R. Novel in vivo imaging shows up-regulation of death receptors by paclitaxel and correlates with enhanced antitumor effects of receptor agonist antibodies. Mol Cancer Ther. 2006;12:2991–3000. doi: 10.1158/1535-7163.MCT-06-0188. [DOI] [PubMed] [Google Scholar]
- Held J, Schulze-Osthoff K. Potential and caveats of TRAIL in cancer therapy. Drug Resist Updat. 2001;4:243–252. doi: 10.1054/drup.2001.0208. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Mendelson DS, Ebbinghaus S, Gordon MS, O'Dwyer P, Lieberman G, et al. A phase I safety and pharmacokinetic (PK) study of recombinant Apo2L/TRAIL, an apoptosis-inducing protein in patients with advanced cancer. J Clin Oncol. 2006;24:3013. [Google Scholar]
- Holoch PA, Griffith TS. TNF-related apoptosis-inducing ligand (TRAIL): a new path to anti-cancer therapies. Eur J Pharmacol. 2009;625:63–72. doi: 10.1016/j.ejphar.2009.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horak P, Pils D, Kaider A, Pinter A, Elandt K, Sax C, et al. Perturbation of the tumor necrosis factor–related apoptosis-inducing ligand cascade in ovarian cancer: overexpression of FLIPL and deregulation of the functional receptors DR4 and DR5. Clin Cancer Res. 2005;11:8585–8591. doi: 10.1158/1078-0432.CCR-05-1276. [DOI] [PubMed] [Google Scholar]
- Kelley SK, Harris LA, Xie D, DeForge L, Totpal K, Bussiere J, et al. Preclinical studies to predict the disposition of Apo2L/tumor necrosis factor-related apoptosis-inducing ligand in humans: characterization of in vivo efficacy, pharmacokinetics, and safety. J Pharmacol Exp Ther. 2001;299:31–38. [PubMed] [Google Scholar]
- Kohlhaas SL, Craxton A, Sun XM, Pinkoski MJ, Cohen GM. Receptor-mediated endocytosis is not required for TRAIL-induced apoptosis. J Biol Chem. 2007;282:12831–12841. doi: 10.1074/jbc.M700438200. [DOI] [PubMed] [Google Scholar]
- Koornstra JJ, Jalving M, Rijcken FE, Westra J, Zwart N, Hollema H, et al. Expression of tumour necrosis factor-related apoptosis-inducing ligand death receptors in sporadic and hereditary colorectal tumours: potential targets for apoptosis induction. Eur J Cancer. 2005;41:1195–1202. doi: 10.1016/j.ejca.2005.02.018. [DOI] [PubMed] [Google Scholar]
- LeBlanc HN, Ashkenazi A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003;10:66–75. doi: 10.1038/sj.cdd.4401187. [DOI] [PubMed] [Google Scholar]
- Leong S, Cohen RB, Gustafson DL, Langer CJ, Camidge DR, Padavic K, et al. Mapatumumab, an antibody targeting TRAIL-R1, in combination with paclitaxel and carboplatin in patients with advanced solid malignancies: results of a phase I and pharmacokinetic study. J Clin Oncol. 2009;26:4413–4421. doi: 10.1200/JCO.2008.21.7422. [DOI] [PubMed] [Google Scholar]
- Lindmo T, Boven E, Cuttitta F, Fedorko J, Bunn PA., Jr Determination of the immunoreactive fraction of radiolabeled monoclonal antibodies by linear extrapolation to binding at infinite antigen excess. J Immunol Methods. 1984;72:77–89. doi: 10.1016/0022-1759(84)90435-6. [DOI] [PubMed] [Google Scholar]
- Ling J, Herbst RS, Mendelson DS, Eckhardt SG, O'Dwyer P, Ebbinghaus S, et al. Apo2L/TRAIL pharmacokinetics in a phase Ia trial in advanced cancer and lymphoma. J Clin Oncol Proc Am Soc Clin Oncol. 2006;24:3047. [Google Scholar]
- Lub-de Hooge MN, Kosterink JG, Perik PJ, Nijnuis H, Tran L, Bart J, et al. Preclinical characterisation of 111In-DTPA-trastuzumab. Br J Pharmacol. 2004;143:99–106. doi: 10.1038/sj.bjp.0705915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLarty K, Cornelissen B, Scollard DA, Done SJ, Chun K, Reilly RM. Associations between the uptake of 111In-DTPA-trastuzumab, HER2 density and response to trastuzumab (Herceptin) in athymic mice bearing subcutaneous human tumour xenografts. Eur J Nucl Med Mol Imaging. 2009;36:81–93. doi: 10.1007/s00259-008-0923-x. [DOI] [PubMed] [Google Scholar]
- Marini P, Denzinger S, Schiller D, Kauder S, Welz S, Humphreys R, et al. Combined treatment of colorectal tumours with agonistic TRAIL receptor antibodies HGS-ETR1 and HGS-ETR2 and radiotherapy: enhanced effects in vitro and dose-dependent growth delay in vivo. Oncogene. 2006;25:5145–5154. doi: 10.1038/sj.onc.1209516. [DOI] [PubMed] [Google Scholar]
- Mom CH, Verweij J, Oldenhuis CN, Gietema JA, Fox NL, Miceli RL, et al. Mapatumumab, a fully human agonistic monoclonal antibody that targets TRAIL-R1, in combination with gemcitabine and cisplatin: a phase I study. Clin Cancer Res. 2009;17:5584–5590. doi: 10.1158/1078-0432.CCR-09-0996. [DOI] [PubMed] [Google Scholar]
- Pegram MD, Konecny G, Slamon DJ. The molecular and cellular biology of HER2/neu gene amplification/overexpression and the clinical development of herceptin (trastuzumab) therapy for breast cancer. Cancer Treat Res. 2000;103:57–75. doi: 10.1007/978-1-4757-3147-7_4. [DOI] [PubMed] [Google Scholar]
- Press OW, Shan D, Howell-Clark J, Eary J, Appelbaum FR, Matthews D, et al. Comparative metabolism and retention of iodine-125, yttrium-90, and indium-111 radioimmunoconjugates by cancer cells. Cancer Res. 1996;56:2123–2129. [PubMed] [Google Scholar]
- Pukac L, Kanakaraj P, Humphreys R, Alderson R, Bloom M, Sung C, et al. HGS-ETR1, a fully human TRAIL-receptor 1 monoclonal antibody, induces cell death in multiple tumour types in vitro and in vivo. Br J Cancer. 2005;92:1430–1441. doi: 10.1038/sj.bjc.6602487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reesink-Peters N, Hougardy BM, van den Heuvel FA, Ten Hoor KA, Hollema H, Boezen HM, et al. Death receptors and ligands in cervical carcinogenesis: an immunohistochemical study. Gynecol Oncol. 2005;96:705–713. doi: 10.1016/j.ygyno.2004.10.046. [DOI] [PubMed] [Google Scholar]
- Schneider P, Olson D, Tardivel A, Browning B, Lugovskoy A, Gong D, et al. Identification of a new murine tumor necrosis factor receptor locus that contains two novel murine receptors for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) J Biol Chem. 2003;278:5444–5454. doi: 10.1074/jbc.M210783200. [DOI] [PubMed] [Google Scholar]
- Spierings DC, de Vries EG, Vellenga E, van den Heuvel FA, Koornstra JJ, Wesseling J, et al. Tissue distribution of the death ligand TRAIL and its receptors. J Histochem Cytochem. 2004;52:821–831. doi: 10.1369/jhc.3A6112.2004. [DOI] [PubMed] [Google Scholar]
- Tolcher AW, Mita M, Meropol NJ, von Mehren M, Patnaik A, Padavic K, et al. Phase I pharmacokinetic and biologic correlative study of mapatumumab, a fully human monoclonal antibody with agonist activity to tumor necrosis factor-related apoptosis-inducing ligand receptor-1. J Clin Oncol. 2007;25:1390–1395. doi: 10.1200/JCO.2006.08.8898. [DOI] [PubMed] [Google Scholar]
- Truneh A, Sharma S, Silverman C, Khandekar S, Reddy MP, Deen KC, et al. Temperature-sensitive differential affinity of TRAIL for its receptors. DR5 is the highest affinity receptor. J Biol Chem. 2000;275:23319–23325. doi: 10.1074/jbc.M910438199. [DOI] [PubMed] [Google Scholar]
- Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–682. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- Wu GS, Burns TF, Zhan Y, Alnemri ES, El Deiry WS. Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res. 1999;59:2770–2775. [PubMed] [Google Scholar]
- Xiang H, Nguyen CB, Kelley SK, Dybdal N, Escandon E. Tissue distribution, stability, and pharmacokinetics of Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand in human colon carcinoma COLO205 tumor-bearing nude mice. Drug Metab Dispos. 2004;32:1230–1238. doi: 10.1124/dmd.104.000323. [DOI] [PubMed] [Google Scholar]