
Figure 2.
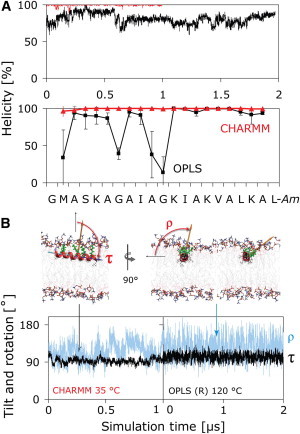
Structure (A) and orientation (B) of PGLa in the S-state. (A) The CHARMM simulation (red) reveals a full α-helix, whereas PGLa partially unfolds in the OPLS simulation (black), with loss of helicity prominent at G7 and G11. (B) The orientation is described by two angles: the tilt angle (τ) is the angle between the membrane normal and the helix long axis, which is defined to point from the N- to the C-terminus. The azimuthal rotation angle (ρ) is defined as a right-handed rotation around the helix long axis, with 0° defined to place the vector from the helix axis to the Cα atom of K12 parallel to the membrane plane. Both angles show similar behavior in the CHARMM and helically restrained (R) OPLS simulations. The rotation angle slowly fluctuates over a wide range at 35°C (orange vector, blue curves), resulting in large uncertainties in ρ. Convergence can be accelerated by use of high temperature (120°C) using a helically restrained peptide (R), which yields rapid convergence of τ and ρ without noticeably changing the averages obtained at 35°C.