

Abstract
Cumulative evidence indicates that activation of cyclin D-dependent kinase 4/6 (cdk4/6) represents a major trigger of cell cycle reentry and apoptosis in vertebrate neurons. We show here the existence of another mechanism triggering cell cycle reentry in differentiating chick retinal neurons (DCRNs), based on phosphorylation of E2F4 by p38MAPK. We demonstrate that the activation of p75NTR by nerve growth factor (NGF) induces nuclear p38MAPK kinase activity, which leads to Thr phosphorylation and subsequent recruitment of E2F4 to the E2F-responsive cdc2 promoter. Inhibition of p38MAPK, but not of cdk4/6, specifically prevents NGF-dependent cell cycle reentry and apoptosis in DCRNs. Moreover, a constitutively active form of chick E2F4 (Thr261Glu/Thr263Glu) stimulates G1/S transition and apoptosis, even after inhibition of p38MAPK activity. In contrast, a dominant-negative E2F4 form (Thr261Ala/Thr263Ala) prevents NGF-induced cell cycle reactivation and cell death in DCRNs. These results indicate that NGF-induced cell cycle reentry in neurons depends on the activation of a novel, cdk4/6-independent pathway that may participate in neurodegeneration.
INTRODUCTION
Neurons have classically been considered as permanently postmitotic cells, but cumulative evidence has challenged this dogma. Several studies have demonstrated that under experimental or pathological conditions, neurons can reactivate the cell cycle, a phenomenon often linked with apoptosis (30). Nevertheless, the molecular mechanism triggering cell cycle reentry in neurons is far from been completely understood. Previous studies have pointed out to the upregulation of cyclin D and the activation of cyclin-dependent kinase 4/6 (cdk4/6) as a trigger for cell cycle reentry and apoptosis in neurons (24, 45, 60). In this regard, inhibition of cdk4/6 has been shown to protect neurons against apoptosis (61), likely by preventing E2F1 activity in these cells (40). Furthermore, activation of cdk4 in cortical neurons has been shown to induce hyperphosphorylation of the retinoblastoma protein (Rb) family member p130, followed by the release of E2F4 (45), derepression of B- and C-Myb (44), and induction of proapoptotic factor Bim (6). Nevertheless, cell cycle-dependent neuronal death could also be triggered by cyclin D-independent mechanisms. Thus, cell cycle-associated events linked with neurodegeneration in Alzheimer's disease (AD) can be dissociated from classical cell cycle initiation by cyclin D-cdk4/6. While 8% of AD hippocampal neurons have been shown to express nuclear cyclin B in vivo (9), only 0.6% of these neurons express detectable levels of cyclin D (9), mainly present in the cytoplasmic compartment (53). Therefore, cyclin D seems not to participate in cell cycle reactivation in AD.
We have demonstrated that cell cycle reentry takes place in differentiating retinal ganglion cells (RGCs) during normal chick retina development (17, 51), a model system that may resemble AD-associated neurodegeneration (19). Cell cycle reentry in differentiating chick retinal neurons (DCRNs) results from the activation of the neurotrophin receptor p75 (p75NTR) by nerve growth factor (NGF) (17, 51). Interestingly, DCRNs treated with NGF undergo apoptosis several hours after p75NTR activation (17, 51), suggesting that cell death in this system is not directly triggered by a classical p75NTR-dependent pathway (58). Indeed, cell death in response to NGF is preceded by cyclin B2, but not cyclin D1, upregulation and the presence of mitotic figures in cells harboring neuronal markers (17), and it can be blocked with cdk1/cdk2 inhibitors (17). Cell cycle reentry in DCRNs can be considered as part of a physiological process taking place in the developing nervous system aimed at inducing tetraploidy in specific neuron types (48, 51). DCRNs that reactivate the cell cycle are maintained in a G2-like state in the presence of brain-derived neurotrophic factor (BDNF), which is known to inhibit the presence of cyclin B2 and mitotic figures, and the subsequent death of these cells (17, 21).
The mechanism used by p75NTR to induce cell cycle reentry remains unknown. One putative target of the p75NTR signaling pathway is E2F1, which is expressed in the absence of Rb by differentiating RGCs that reenter into the cell cycle (51). Another member of the E2F family, E2F4, is expressed in the developing mouse retina (13). Although E2F4 is often thought to act as a repressor, it can also induce gene transcription in a number of cellular systems (15, 73), being a trigger of cell cycle progression in intestinal crypt cells and colorectal cancer cells (27). Therefore, E2F4 could participate in the regulation of cell cycle reentry in DCRNs, either by inducing or by preventing cell cycle progression in these cells.
p75NTR is the founder member of the Fas/tumor necrosis factor alpha superfamily of death receptors. It can bind to all mature forms of the neurotrophins, including NGF (46). p75NTR is able to transduce proliferative and proapoptotic signals in response to ligand binding in the absence of their cognate Trk receptors (46) or in response to the precursor forms of the neurotrophins (72). Thus far, several signaling pathways have been shown to be induced by p75NTR (58), including those regulated by mitogen-activated protein kinases (MAPKs) (10, 12, 33, 70). Activation of p75NTR can also result in γ-secretase-dependent release of its intracellular domain (p75ICD) (18, 38, 65), thus inducing apoptosis (38, 47, 65). In overexpression experiments performed in differentiating neuroblastoma cells, p75ICD has been shown to bind to both Necdin and chicken MAGE (CMAGE), thus favoring E2F1 activity and apoptosis (47). Furthermore, p75NTR itself is able to sequester the MAGE proteins Necdin and MAGE-G1 when overexpressed in differentiating retinoblastoma cells, thus leading to E2F1-dependent apoptosis (43).
In the present study, we characterized the participation of E2F4 as a trigger of cell cycle reentry, followed by cell death in DCRNs. We have found that the binding of NGF to p75NTR does not lead to the translocation of p75ICD to the nucleus (47) or of CMAGE to the cell membrane (43). Instead, NGF binding to p75NTR results in nuclear activation of p38MAPK, followed by Thr phosphorylation of E2F4. This is translated into the interaction of the latter with the cdc2 promoter, used here as a paradigm of E2F-activated promoters. We present evidence that phosphorylation of the Thr261/Thr263 motif of E2F4 can mimic the signal that initiates cell cycle reentry and subsequent apoptosis in DCRNs. Finally, a dominant-negative form of E2F4 (Thr261Ala/Thr263Ala) prevents the proliferative/apoptotic effect of NGF in DCRNs.
MATERIALS AND METHODS
Chick embryos.
Fertilized eggs from White Leghorn hens were obtained from a local supplier (Granja Santa Isabel, Spain) and incubated at 38.5°C in an atmosphere of 70% humidity. The embryos were staged as described previously (29). All experiments were performed in accordance with the European Union guidelines.
Primary antibodies.
The rabbit polyclonal antiserum—9992—against the intracellular domain of human p75NTR was kindly provided by Moses V. Chao (New York University, New York, NY). This antiserum was used at 1/20,000 for Western blotting. The NC243 antiserum, raised against the 243 C-terminal amino acids of mouse Necdin fused to glutathione S-transferase (GST) (57), was a generous gift from Kazuaki Yoshikawa (Osaka University, Osaka, Japan). This antibody, which specifically recognizes CMAGE protein (47), was used at 1/20,000 dilution for Western blotting. The affinity-purified rabbit polyclonal anti-E2F1 antibody C-20 (Santa Cruz Biotechnology) was used at 0.4 μg/ml for Western blotting. The rabbit polyclonal anti-E2F4 antibody C-108 (Santa Cruz Biotechnology), raised against amino acids 108 to 300 of human E2F4, was used at 4 μg/ml for immunohistochemistry and immunocytochemistry and at 0.1 μg/ml for Western blotting. For chromatin immunoprecipitation (ChIP; EpiQuik ChIP kit; Epigentek), 3 μg of this antibody was loaded per well. The rabbit polyclonal anti-E2F4 antibody (Bethyl Laboratories), recognizing an epitope mapping to a region between residues 363 and 413 of human E2F4, was used for ChIP (EpiQuik ChIP kit) by loading 3 μg per well. The monoclonal antibody (MAb) anti-RA4, kindly provided by Stephen C. McLoon (University of Minnesota, Minneapolis, MN), recognizes an epitope expressed by RGCs soon after their final mitosis (75) and was used at 1/400 for immunohistochemistry and at 1/200 for immunocytochemistry. The mouse anti-βIII tubulin MAb (neuron specific; Chemicon) was used at 1/20,000 for Western blotting. 5-Bromo-2′-deoxyuridine (BrdU) was visualized by immunocytochemistry with a 1/4,000 dilution of the mouse MAb G3G4 (Developmental Studies Hybridoma Bank). BrdU was also detected with the rat MAb BU1/75 at a 1/200 dilution. The mouse antiphosphothreonine MAbs 1E11 and 14B3 (phosphothreonine detection kit; Calbiochem) were used (both together) at 50 ng/ml each for Western blotting. The rabbit polyclonal antibody to p38MAPK (Abcam) was used at 1/75,000 dilution for Western blotting. The mouse MAb D-8 against phospho-p38MAPK (Santa Cruz Biotechnology), recognizing the active form of the enzyme, was used at a 1/2,000 dilution for Western blotting and at 1/100 for immunocytochemistry. The rabbit polyclonal antibody to p107 (Abcam) was used at a 1/2,000 dilution for Western blotting and at a 1/100 dilution for immunohistochemistry. This antibody was used for ChIP (EpiQuik) by loading 3 μg per well. The rabbit polyclonal antibody against p130 (Santa Cruz Biotechnology) was used at a 1/200 dilution for immunohistochemistry.
Plasmids.
The luciferase reporter vector carrying the mouse c-Myc core promoter (P1 to P3), kindly provided by Kazuaki Yoshikawa (Osaka University, Osaka, Japan), was previously described (43). The luciferase reporter vector carrying the human cdc2 promoter extending from bp −3200 to +75 (68) was kindly provided by Christopher K. Glass (University of California at San Diego). The pCMVβ vector (Clontech) was used to express β-galactosidase. The pcDNA6-E2F4-Myc expression vector was obtained after amplifying the coding sequence of chick E2F4 (accession number JQ678847) with Pfu DNA polymerase from a cDNA obtained from embryonic day 6 (E6) chick retina using the oligonucleotides corresponding to bp 46 to 64 (5′-CACAAGCTTGGCGGGCAGATGGCGGAGT-3′) and complementary to bp 1276 to 1296 (5′-CTCGAATTCTCACAGATCCTCTTCAGAGATGAGTTTCTGCTCGAGGTTAAGGACAGGCACATC-3′), the latter containing a Myc tag. The amplified sequence was cloned into the pGEM-T Easy vector (Promega) and cut with EcoRI, and the resulting fragment was subcloned into the EcoRI site of pcDNA6/V5-His-A2. The sequence was confirmed by sequencing. A constitutively active form of E2F4 present in the pcDNA6-E2F4CA-Myc expression vector was obtained by introducing point mutations in the coding sequence of E2F4 (Thr261 and Thr263 were substituted by Glu residues). A dominant-negative form of E2F4 present in the pcDNA6-E2F4DN-Myc expression vector was obtained by introducing point mutations in the coding sequence of E2F4 (Thr261 and Thr263 were substituted by Ala residues). The pRFPRNAiC, pRFPRNAi Luciferase, and pRFPRNAi Egfp vectors, described by Das et al. (14), were provided by Stuart Wilson (University of Sheffield, Sheffield, United Kingdom). Two different pRFPRNAi E2f4 vectors capable of suppressing E2f4 expression were constructed using the pRFPRNAiC plasmid according to procedures described previously (14). Both constructs gave similar results (data not shown), thus indicating they are specific for the E2f4 mRNA. The target sequences used to interfere with the E2f4 mRNA corresponded to bp 657 to 678 and bp 532 to 553 (accession number XM_001231947), and they were cloned into the MluI-PaeI sites of the pRFPRNAiC plasmid (pRFPRNAi E2f4-86 and pRFPRNAi E2f4-92, respectively). Enhanced green fluorescent protein (EGFP) was expressed in the chick retinal cells by using the pEGFP-N1 plasmid (BD Biosciences Clontech).
Reverse transcription-PCR (RT-PCR).
mRNA was extracted from retinal cultures using a QuickPrep Micro mRNA purification kit (GE Healthcare). cDNA was then prepared using the first-strand cDNA synthesis kit (GE Healthcare). PCR amplification was performed using standard procedures. The PCR primers used for chick Cdc2 correspond to bp 93 to 112 and are complementary to bp 540 to 559 (accession number NM_205314). The PCR primers for chick Gapdh correspond to bp 944 to 963 and are complementary to bp 1219 to 1238 (accession number K01458). No amplification products were obtained in reactions lacking reverse transcriptase. Under the conditions used in this study, amplifications were linear.
Cell culture.
Neuronal precursors isolated from the E6 chick retina are susceptible to differentiate in the presence of laminin-1 and insulin (22, 23), mimicking the in vivo situation (17, 51). Insulin is present in the N2 supplement, and exogenous NGF can induce cell cycle reentry and apoptosis of newborn neurons through p75NTR (1, 17). Dissociated E6 retinal cells obtained as described by Frade et al. (20) were suspended in culture medium and plated (20,000 cells/cm2) on 10-mm round glass coverslips in four-well dishes (Greiner Bio-One, Germany). The coverslips were previously coated with 0.5 mg of polyornithine (PLO; Sigma)/ml and 10 μg of laminin (Invitrogen)/ml. Alternatively, E6 retinal cells were cultured on P100 tissue culture dishes (8 × 106 to 12 × 106 cells) for Western blot experiments or in T75 tissue culture flasks (6 × 106 cells) for ChIP experiments, previously treated with PLO and laminin, as described above. E6 retinal cells were cultured in 50% Dulbecco modified Eagle medium (DMEM)–50% Ham F-12 medium (Sigma) with N2 supplement (DMEM/F12/N2) (8) and maintained for 16 h at 37°C in a water-saturated atmosphere containing 5% CO2. Where stated, cultures were supplemented with 100 ng of recombinant NGF (Sigma)/ml or other drugs (see below) for an additional 30-min period. In some instances, 100 ng of NGF/ml was added at the time of plating. At E6, DCRNs are mainly newborn RGCs (17, 22). The γ-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-l-alanyl]-(S)-phenylglycine t-butyl ester (DAPT; Calbiochem) was used at 1 μM. The p38MAPK selective inhibitor SB203580 (Calbiochem) was used at 5 μM. The JNK selective inhibitor SP600125 (Calbiochem) was used at 7 μM. The selective cyclin D kinase 4/6 inhibitor PD 0332991 tetrahydrochloride (Axon Medchem) was diluted in water and used at the indicated concentrations. Pulses (30 min) with 0.5 μg of BrdU (Roche)/ml were given at the end of the culture in some cases to label cells in S phase. This BrdU analysis can be used to indirectly estimate the kinetics of the cell cycle, since a small increase in the proportion of cells incorporating BrdU usually derives from an important reduction in the length of G1 (i.e., a significant increase in the kinetics of the cell cycle). DF-1 cells (31) were grown in DMEM supplemented with 10% fetal calf serum (FCS; Invitrogen).
Explant electroporation.
In vitro electroporation of retinal explants was performed as described previously (51). Briefly, E6 chicken retinas were dissected away from the pigment epithelium and fragmented into small pieces of ∼10 mm2. These retinal fragments were laid onto a glass coverslips and subsequently immersed in 4 μl of phosphate-buffered saline (PBS) containing different plasmid combinations (see below). Electroporation was performed using four 50-ms pulses of 25 V at a 500-ms frequency. After electroporation, the explants were grown in suspension for 5 h in DMEM/F12/N2. Explants were then collected, dissociated, and cultured as described above. c-Myc or cdc2 luciferase reporter vectors were used at a concentration of 0.4 μg/μl. These vectors were electroporated, along with different combinations of E2f4 RNAi, Luciferase RNAi, Egfp RNAi, pcDNA6 (Invitrogen), E2F4, E2F4-CA, or E2F4-DN constructs at 0.5 μg/μl each. The DNA concentration was adjusted to 1.0 μg/μl with pBluescript (Stratagene) for RNA interference (RNAi) experiments and to 1.0 to 1.2 μg/μl with pcDNA6/V5-His (Invitrogen) for overexpression experiments.
Cell lipofection.
DF-1 fibroblast cells were cultured at 90% confluence and then lipofected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Luciferase reporter assays.
Luciferase assays were performed as described previously (47). Briefly, E6 retinal explants were electroporated as described above. Explants were cultured for 5 h, dissociated into single cells, and cultured on PLO/laminin-coated coverslips at a density of 70,000 cells/cm2 as described above. After 15 h in culture, transfected cells were washed with ice-cold PBS and extracted with 15 μl of Dual-Light system lysis buffer (Applied Biosystems, Foster City, CA). The luciferase and β-galactosidase activities were then measured as indicated by the manufacturer.
Immunostaining.
Immunocytochemistry was performed in cells fixed for 15 min with 4% paraformaldehyde (PFA; Merck) at room temperature and permeabilized for 30 min with phosphate-buffered saline (PBS) containing 0.05% Triton X-100 (Sigma) plus 10% FCS. The cells were then incubated for 1 h at room temperature with PBS containing 0.05% Triton X-100 plus 1% FCS and the appropriate primary antibody. After five washes in PBS containing 0.05% Triton X-100, the cells were incubated for an additional 1 h in PBS containing 0.05% Triton X-100 plus 1% FCS and either a 1/400 dilution of the Alexa Fluor 594–goat anti-mouse IgG antibody, a 1/800 dilution of the Cy2-conjugated Affinipure goat anti-rabbit IgG (H+L), or a 1/800 dilution of the Alexa Fluor 488–goat anti-rat IgG antibody. Nuclei were labeled with 1 μg of bisbenzimide/ml in PBS. Immunostained cultures were then mounted in PBS-glycerol. For BrdU immunolabeling, cultures were previously subjected to DNA denaturation by incubation for 30 min with 2 N HCl–0.33× PBS at room temperature, followed by a neutralization step consisting of three 15-min washes with 0.1 M sodium borate (pH 8.9) and two 5-min washes with PBS–0.05% Triton X-100. For immunohistochemistry, embryos were fixed for 8 to 24 h at 4°C with 4% PFA, cryopreserved overnight at 4°C in PBS containing 30% sucrose (Merck) in PBS, and embedded in the OCT compound Tissue-Tek (Sakura). Cryosections (12 μm) were permeabilized and blocked for 30 min at room temperature in PBS containing 0.5% Triton X-100 plus 10% FCS, and they were then incubated overnight at 4°C with the primary antibodies in PBS containing 0.1% Triton X-100 plus 1% FCS. After five washes with PBS containing 0.1% Triton X-100, the sections were incubated for 1 h at room temperature with an Alexa Fluor 594-coupled anti-mouse IgG antibody (Invitrogen) and Cy2-conjugated anti-rabbit IgG (H+L) antibody (Jackson Immunoresearch), each diluted 1/1,000. The sections were finally washed five times in PBS containing 0.1% Triton X-100 and once in PBS, and then they were incubated with 1 μg of bisbenzimide (Sigma)/ml in PBS before being mounted in glycerol-PBS (1:1).
Cell extracts.
Cell lysates were performed as previously described (47). Briefly, cultures were placed on ice, washed with ice-cold PBS, and incubated for 30 min with 700 μl of lysis buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% Triton X-100, 1× protease inhibitor cocktail (Roche), and 1× phosphatase inhibitor cocktail 1 (Sigma; only for Western blotting of phosphoproteins). Cell lysates were scraped with a rubber policeman and centrifuged at 13,000 × g for 10 min at 4°C. Supernatants were 10-fold concentrated with centrifugal filter units (cutoff, 3 kDa; Millipore), mixed with 1 volume of 2× Laemmli's buffer, and boiled for 5 min. DF-1 cells, isolated by fluorescence-activated cell sorting (FACS), were directly extracted with Laemmli's buffer and boiled for 5 min.
Subcellular fractions.
Subcellular fractions were performed as previously described (35). Briefly, cultures were placed on ice, washed with ice-cold PBS, and lysed with 400 μl of cold buffer containing 10 mM HEPES (pH 7.6), 10 mM NaCl, 1 mM KH2PO4, 5 mM NaHCO3, 5 mM EDTA, and 1× protease inhibitor cocktail. Cell lysates were scraped with a rubber policeman and homogenized by passage through a 22G needle. Nuclei were spun down at 300 × g for 10 min, and the resulting supernatant was then microfuged at 16,000 × g for 30 min at 4°C. The supernatant was removed (cytosol) and concentrated with centrifugal filter units (cutoff, 3 kDa), and lysis buffer containing 1% Triton X-100 was added to the pellet (membrane). Nuclear proteins were extracted in high-salt buffer containing 10 mM HEPES, 0.5 mM MgCl2, 420 mM NaCl, 0.2 mM EDTA, 25% glycerol, and 1× protease inhibitor cocktail.
Western blotting.
Cell extracts obtained as described above (corresponding to 107 E6 retinal cells or 105 DF-1 cells) were separated by SDS-PAGE on 10 to 13% acrylamide gels and transferred to Immun-Blot polyvinylidene difluoride membranes (Bio-Rad). The membranes were incubated for 1 h with 2% ECL Advance blocking agent (ECL advanced Western blotting detection kit; Amersham Biosciences) in PBS containing 0.1% Tween 20 (PBT; Sigma) and incubated for 2 h at room temperature with the appropriate antibodies in blocking buffer. After the membranes were washed five times in PBT, they were incubated for 1 h at room temperature with a 1/1,660,000 dilution horseradish peroxidase (HRP)-conjugated Affinipure goat anti-rabbit IgG antibody (Jackson Immunoresearch), 1/500,000 HRP-conjugated Affinipure goat anti-mouse IgG antibody (Jackson Immunoresearch), or 1/500,000 HRP-conjugated Affinipure donkey anti-mouse IgM antibody (Jackson Immunoresearch) in blocking buffer. Finally, they were washed again as described above, and the protein bands were visualized using ECL Advance Western blotting detection kit.
ChIP.
ChIP assay was performed with the EpiQuik ChIP kit (Epigentek) according to the manufacturer's instructions. Briefly, 6 × 106 E6 DCRNs per treatment or 3 × 106 DF-1 fibroblasts were trypsinized and washed with PBS and then cross-linked in medium containing 1% formaldehyde for 10 min at room temperature. The cells were washed in ice-cold PBS and lysed in a lysis buffer containing protease inhibitor cocktail. Chromatin was sheared to an average size of 1,000 bp by sonication (12 pulses of 10 s at level 4; Vibra-Cell, Sonics & Materials, Inc.). Anti-E2F4, anti-p107, or control antibody was coated onto 96-well plates for 90 min at room temperature. A sample of chromatin was set aside before immunoprecipitation and used to represent the input DNA. Lysates corresponding to 106 cells were added, followed by incubation for 90 min at room temperature on a rocking platform. The cross-linked DNA fragments bound to the plates were washed and reversed by proteinase K treatment. Precipitated DNA fragments were recovered by column purification and quantified by PCR using the following primer pairs adjacent to the cdc2 chick promoter (positions −527 and −508 and complementary to positions −388 and −369 from the putative transcription start, respectively): sense 5′-CCACCAACAGCAAACAAACG-3′ and antisense (5′-ATCGGGAACATCACTGGAGA-3′), which amplify a region adjacent to the putative E2F binding sites located at positions −88 and −81 and positions −51 and −44. These sequences correspond to bp 5085380 to 5085399 and bp 5085241 to 5085260 from the Gallus gallus chromosome 6 genomic contig (accession number NW_001471714.1), respectively. Thermal cycles included 5 min at 95°C, followed by 39 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 1 min. Under these conditions, the amplification was linear. PCR products were resolved in 2% agarose gels containing ethidium bromide.
FACS.
For cell sorting, a FACSAria cytometer (BD Biosciences, San Diego, CA) equipped with a double-argon (488 nm) and helium-neon (633 nm) laser was used. The data were collected by using a linear digital signal process. The emission filter used was BP 530/30 (FL1). Debris and duplets were excluded from the analysis. Cell populations were isolated in 5-ml polystyrene tubes (12 by 75 mm) using 20 lb/in2 pressure and a 100-μm nozzle aperture. The data were analyzed using FACSDiva data analysis software (BD Biosciences) and displayed using biexponential scaling. A total of 5 × 106 EGFP-positive cells were isolated as an average.
DNA and protein sequence analysis.
DNA sequence alignment was performed using the Martinez-NW method (Lasergene MegAlign; DNASTAR). The NetPhosK 1.0 software (http://www.cbs.dtu.dk/services/NetPhosK), a bioinformatic tool able to predict kinase specific phosphorylation sites in eukaryotic proteins (7), was used to determine putative p38MAPK phosphorylation sites in the amino acid sequence of different E2F members. Analysis was performed without filtering (threshold, 0.5).
Cell counting and quantification of apoptosis.
Cells were counted on a Nikon E80i microscope using an oil immersion 60× objective lens with phase-contrast and epifluorescence illumination, and an average of 300 to 500 cells were analyzed per coverslip. Quantitative data were means ± the standard error of the mean (SEM) from at least three independent experiments. The statistical differences were analyzed using a Student t test. To characterize apoptosis by morphological criteria, the DNA of paraformaldehyde-fixed cells was labeled with 1 μg of bisbenzimide (Sigma)/ml, and the number of pyknotic nuclei was established. Apoptotic cells in retinal cultures were also detected by using TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling) with an in situ cell death detection kit (POD; Roche) according to the manufacturer's instructions. TUNEL yielded results similar to those obtained using morphological criteria (results not shown).
Image analysis.
Images were processed only minimally using Photoshop CS4 Extended (v11.0). Contrast and brightness balance were equally applied to all parts of the image. Band quantifications were performed using ImageJ software.
RESULTS
NGF does not induce nuclear translocation of p75ICD in DCRNs.
To explore whether NGF binding to p75NTR can induce the release of p75ICD in our model system, dissociated E6 chick retinal cells cultured under conditions giving rise to RGCs (17, 22, 23) (i.e., DCRNs) were exposed to NGF for increasing time periods. Then, total cell extracts were subjected to Western blotting with the p75ICD-specific antiserum 9992. This analysis demonstrated that, besides the 75-kDa band, corresponding to the full-length molecule, high-mobility bands of 35 and 23 kDa were visible with similar intensity at all time points (Fig. 1A). The 23-kDa band was of identical size as that described for soluble p75ICD (18, 34, 35), whereas the 35-kDa band is likely unspecific since it was present in brain extracts from the p75NTR exon IV knockout mice (74) (data not shown, but see reference 62). The presence of the 23-kDa band under control conditions indicated that it does not seem to derive from γ-secretase-derived cleavage. Accordingly, long-lasting exposure (16 h) of DCRNs to the γ-secretase inhibitor DAPT did not change this pattern even in the presence of NGF (Fig. 1B). Subcellular fractioning of extracts from DCRNs treated for 30 min with either vehicle or NGF demonstrated that the 23-kDa band is mainly associated with the membrane fraction, whereas it is barely detectable in both cytosolic and nuclear fractions (Fig. 1C). This subcellular expression pattern was not affected by the presence of NGF (Fig. 1C), further demonstrating that NGF treatment does not induce release and nuclear translocation of p75ICD in DCRNs.
Fig 1.

NGF signal transduction in DCRNs is independent of both nuclear translocation of p75ICD and sequestration of CMAGE at the cell membrane by p75NTR. (A) The upper panel shows a Western blot with the anti-p75ICD antibody 9992 in extracts from DCRNs treated with NGF for the indicated time points. The bottom panel shows a Western blot with anti-βIII tubulin as a loading control. The 35-kDa band (asterisk) is unspecific (see the text). Arrowhead, 23-kDa band. (B) The upper panel shows a Western blot with the anti-p75ICD antibody 9992 in extracts from DCRNs treated for 16 h with NGF and/or the γ-secretase inhibitor DAPT. The bottom panel shows a Western blot with anti-βIII tubulin as a loading control. (C) Western blot with the anti-p75ICD antibody 9992 in the indicated subcellular fractions from DCRNs cultured for 30 min either in the presence (+) or absence (−) of NGF. (D) Western blot with the NC243 antibody which in the chick specifically labels CMAGE. (E) Western blot with an anti-E2F4 antibody. (F) Western blot with an anti-E2F1 antibody.
NGF does not alter the subcellular localization of CMAGE in DCRNs.
We next tested whether NGF binding to p75NTR can recruit CMAGE to the cell membrane of DCRNs, since it occurs with Necdin and MAGE-G1 in differentiating neuroblastoma cells (43). To this end, subcellular extracts from DCRNs were subjected to Western blot analysis using the anti-Necdin NC243 antibody, previously shown to specifically recognize CMAGE (47). This analysis demonstrated the existence of a band of ∼24 kDa associated with the membrane fraction (Fig. 1D), resembling the monomeric form of the molecule as described by (47). Furthermore, another band of ∼48 kDa was detected in the nuclear extract (Fig. 1D), likely equivalent to the Necdin/NRAGE heterodimer observed to interact with the Msx2 homeoprotein in the nucleus of mammalian cells (42). Interestingly, the presence of NGF did not alter the subcellular localization of both forms of CMAGE in DCRNs. Cross-contamination of the subcellular fractions in these experiments was ruled out since both full-length p75NTR and monomeric CMAGE were observed only in the membrane fraction (Fig. 1C and D), while the transcription factor E2F1 and the dimeric form of CMAGE were specific from the nuclear fraction (Fig. 1D, E, and F). Moreover, all of these proteins were absent from the cytosolic fraction (see Fig. 1C to F), and the unspecific band of 35 kDa revealed with the anti-p75NTR antibody in the cytosolic fraction was absent from the membrane-derived extract (Fig. 1C).
Overall, these results indicate that an alternative mechanism, independent of nuclear translocation of p75ICD or recruitment of CMAGE to the cell membrane, should exist in DCRNs to enhance E2F activity and trigger cell cycle reentry and apoptosis in response to NGF.
E2F4 is expressed by differentiating RGCs and it can interact with the cdc2 promoter in response to NGF.
To decipher the mechanism used by NGF to induce E2F activity in DCRNs, we analyzed the participation of E2F4 in this process. This transcription factor is expressed in the developing mouse retina (13), and double labeling in chick retina sections with an anti-E2F4 antibody and the anti-RA4 MAb, which specifically labels differentiating RGCs (75), demonstrated that virtually all RGCs expressed the E2F4 transcription factor, which was always located in the nucleus (Fig. 2A). This observation was confirmed by Western blot analysis of subcellular extracts obtained from DCRNs (Fig. 1E). These results suggest that E2F4 could regulate E2F function in differentiating chick RGCs. A faint E2F4-specific band could also be observed in the membrane fraction (Fig. 1E), suggesting that a small amount of E2F4 can be associated to a membrane compartment in DCRNs.
Fig 2.

Interaction of E2F4 with the cdc2 promoter in DCRNs. (A) E6 chick retina cryosection (12 μm) immunolabeled for E2F4 (green) and the RGC-specific marker RA4 (red) and counterstained with bisbenzimide (blue) to reveal the nuclei. Arrows indicate the E2F4-specific immunostaining in the nuclei of RA4-positive neurons. pe, pigment epithelium; v, vitreus body. The insets show high-magnification images of the dashed squares. Bar, 30 μm. (B) ChIP analysis of the occupancy of the chick cdc2 promoter by E2F4 and E2F1 in DCRNs cultured for 30 min either in the presence (NGF) or absence (Control) of NGF. The anti-E2F4 antibody C-108 (αE2F4), the anti-E2F1 antibody C-20 (αE2F1), or irrelevant (Mock) antibodies were used for immunoprecipitation. INPUT, RT-PCR amplification from extracts before immunoprecipitation. (C) ChIP analysis performed with a rabbit polyclonal anti-E2F4 antibody from Bethyl Laboratories (αE2F4) or irrelevant (Mock) antibodies. See panel B for details. (D) RT-PCR analysis of cdc2 expression in DCRNs treated for 15 h with NGF (+) or vehicle (−). Gapdh was used as a control. (E) Luciferase activity, normalized to β-galactosidase, in extracts from DCRNs cotransfected with β-galactosidase and cdc2-luciferase, and treated for 16 h with NGF or vehicle. ***, P < 0.005 (Student t test; n = 3).
The interaction of endogenous E2F4 with the promoter of cdc2, used here as a paradigm of E2F-responsive genes, was analyzed by ChIP. ChIP experiments were performed in lysates from DCRNs, immunoprecipitated with either an anti-E2F4 specific antibody or an irrelevant control antibody (mock). Immunoprecipitates were then sonicated to obtain small DNA fragments (average, 1,000 bp) and subjected to PCR using primers specific for a sequence contained within the chick cdc2 promoter. This analysis demonstrated that NGF induces the interaction of endogenous E2F4 with the cdc2 promoter in DCRNs (Fig. 2B), suggesting that E2F4 has a proliferative potential in these neurons. As a control, we performed a similar ChIP assay using an anti-E2F1-specific antibody. This analysis demonstrated that E2F1 remains constitutively bound to the cdc2 promoter irrespective of the presence or absence of NGF in the culture medium (Fig. 2B), an observation consistent with the presence of E2F1 in the nuclear compartment (Fig. 1F) (see also reference 51). To further confirm the specificity of the interaction of E2F4 with the cdc2 promoter, a ChIP assay was performed with a different antibody that recognizes an alternative E2F4 epitope using lysates from NGF-treated DCRNs versus control DCRNs. This analysis confirmed that E2F4 interacts with the cdc2 promoter in DCRNs treated with NGF (Fig. 2C). The interaction of E2F4 with the cdc2 promoter in response to NGF correlated with an increase of cdc2 expression in DCRNs, as detected by reverse transcription-PCR (RT-PCR) (Fig. 2D). Accordingly, such interaction also correlated with enhanced luciferase activity in DCRNs transfected with a luciferase reporter gene driven by the human cdc2 promoter (68), further stressing the role of E2F4 as a transcriptional activator in these neurons (Fig. 2E).
E2F4 knockdown prevents cell cycle progression in NGF-treated DCRNs.
The hypothesis that E2F4 facilitates NGF-dependent cell cycle progression in DCRNs was tested through a loss-of-function approach using two different RNAi constructs. These E2f4 RNAi constructs were electroporated into E6 retinal cells, which were subsequently cultured for 16 h under neurogenic conditions. The presence of E2f4 RNAi in the DCRNs was able to knock down the expression of E2F4, as demonstrated by anti-E2F4 immunostaining (Fig. 3A). This conclusion was confirmed by Western blotting in cell extracts from EGFP-positive, DF-1 chick fibroblasts previously colipofected with EGFP and either E2f4 RNAi or control RNAi and then isolated by fluorescence-activated cell sorting (FACS) (Fig. 3D to G). Western blot analysis performed with antibodies specific for E2F4 demonstrated a dramatic reduction of this transcription factor in extracts obtained from E2f4 RNAi-transfected fibroblasts compared to those from fibroblasts transfected with control RNAi (Fig. 3B). As a control, the levels of E2F1 protein did not change in E2f4 RNAi-transfected fibroblasts compared to control fibroblasts (Fig. 3B).
Fig 3.

E2F4 acts as a transcriptional activator in DCRNs. (A) The E2f4 RNAi-92 construct (RFP) reduced E2F4 expression (E2F4) in DCRNs (arrow). Nuclei were labeled with bisbenzimide (Bisb.). (B) Western blot with anti-E2F4 (E2F4) or anti-E2F1 (E2F1) antibodies in extracts from DF-1 chick fibroblasts coexpressing EGFP and either E2f4 RNAi-86 (E2f4 RNAi) or a Luciferase RNAi control construct (Luc RNAi). EGFP-positive fibroblasts were isolated by FACS, as shown in panels E and G. (C to G) Flow cytometric analysis of untransfected DF-1 chick fibroblasts (C) or DF-1 chick fibroblasts coexpressing EGFP and either a Luciferase RNAi control construct (D and E) or E2f4 RNAi-86 (F and G) prior to (D and F) or after (E and G) sorting for EGFP-specific labeling. The numbers indicate the proportion of EGFP-positive fibroblasts (dark gray). (H and I) Luciferase activity, normalized to β-galactosidase, in extracts from DCRNs cotransfected with either cdc2-luciferase (H) or c-Myc-luciferase (I), plus β-galactosidase and either E2f4 RNAi-92 or a control construct (Egfp RNAi), and treated for 16 h with NGF or vehicle. (J) BrdU incorporation (30-min pulse) in DCRNs transfected with either E2f4 RNAi-86 or a control construct (Lucif. RNAi) and treated for 16 h with NGF (see arrowheads in panel L) or vehicle. (K) Apoptotic nuclei in DCRNs transfected with either E2f4 RNAi-86 or a control construct (Lucif. RNAi) and treated for 16 h with NGF (see arrow in panel L) or vehicle. Bars: 20 μm (A) and 10 μm (L). *, P < 0.05; **, P < 0.001; ***, P < 0.005 (Student t test; n = 3).
In DCRNs expressing E2f4 RNAi, NGF was unable to enhance the activity of the cdc2 promoter, as demonstrated by luciferase assay (Fig. 3H). This suggests that E2F4 is involved in p75NTR-dependent cell cycle reentry in these neurons. This conclusion was confirmed by assays performed with DCRNs expressing luciferase under the control of the c-Myc core promoter, another well-known E2F-responsive element. These neurons showed an increase of luciferase activity in response to NGF under control conditions but not in the presence of E2f4 RNAi (Fig. 3I). In accordance with these observations, E2f4 RNAi was able to prevent NGF-dependent BrdU incorporation in DCRNs (Fig. 3J), thus confirming the proliferative capacity of E2F4 in these neurons. Finally, E2f4 RNAi expression also inhibited the increase of cell death induced by NGF in DCRNs cultured in the absence of BDNF, measured as the proportion of cells showing pyknotic nuclei (Fig. 3K and L).
Interestingly, E2f4 RNAi induced an opposing effect on the basal transcriptional capacity of the cdc2 and c-Myc core promoters, suggesting that the regulation of these promoters by E2F4 is not strictly equivalent. E2f4 RNAi induced an increase of luciferase expression in DCRNs transfected with the cdc2-luciferase construct (Fig. 3H). In contrast, luciferase expression was decreased by E2f4 RNAi in DCRNs transfected with c-Myc-luciferase (Fig. 3I). Since this latter result is equivalent to what was observed in E2f4 RNAi-expressing DCRNs treated with a short BrdU pulse (Fig. 3J), we decided to use the c-Myc core promoter as a driver for the rest of luciferase analyses included in the present study.
The reduction in BrdU incorporation observed in E2f4 RNAi-transfected DCRNs (Fig. 3K) was somehow modest, suggesting that retinal cells retaining proliferative capacity in the presence of E2f4 RNAi could be precursors at early stages of neuronal differentiation. Indeed, under our culture conditions only 26.12% ± 0.44% (n = 3) of the DCRNs showed strong expression of the RGC-specific neuronal marker RA4 (Fig. 4A). To specifically investigate the effect of E2f4 RNAi on RA4-positive neurons, double immunostaining for BrdU and RA4 was performed in DCRNs electroporated with either E2f4 RNAi or control RNAi constructs and pulse-labeled with BrdU for 30 min before fixation. This analysis demonstrated that only a few RA4-positive cells were able to incorporate BrdU under control conditions and that this proportion was dramatically enhanced by NGF (Fig. 4B). The E2f4 RNAi constructs were able to largely prevent this effect (Fig. 4B). These results confirm that E2F4 participates in NGF-dependent cell cycle reentry in RGCs.
Fig 4.

E2F4 acts as a transcriptional activator in RA4-positive neurons. (A) Expression of the RGC-specific neuronal marker RA4 (RA4) in DCRNs (arrows). Nuclei were labeled with bisbenzimide (Bisb.). (B) BrdU incorporation in RA4-positive DCRNs transfected with either E2f4 RNAi-92 or a control construct (Egfp RNAi) and cultured for 16 h in the presence (vehicle) or absence (NGF) of NGF. *, P < 0.05; ***, P < 0.005 (Student t test; n = 3). Bar, 10 μm.
NGF-dependent cell cycle reentry in DCRNs is independent of Rb family member regulation.
We have previously demonstrated that DCRNs reactivating the cell cycle in response to NGF are those lacking Rb expression (51). Nevertheless, other members of the Rb protein family might be involved in this process. Indeed, evidence exists that p130 is required for cell cycle reentry in differentiated neurons (45). Therefore, we hypothesized that p130 could regulate E2F4 activity in DCRNs treated with NGF. This hypothesis was tested by analyzing the presence of p130 by immunohistochemistry in the E6 chick retina, the stage we have focused on in this study. This analysis demonstrated that p130 is not expressed by the E6 chick retinal cells (Fig. 5A). In contrast, specific immunolabeling for p130 was evident in the chick retina at E10, when neurogenesis is basically finished (66) (Fig. 5B). These observations are consistent with a previous study in the quail retina showing that p130 is not expressed in this tissue before E6 (36), a stage equivalent to E7.5 in the chicken embryo (59). Overall, these results indicate that p130 does not participate in NGF-dependent cell cycle reentry.
Fig 5.
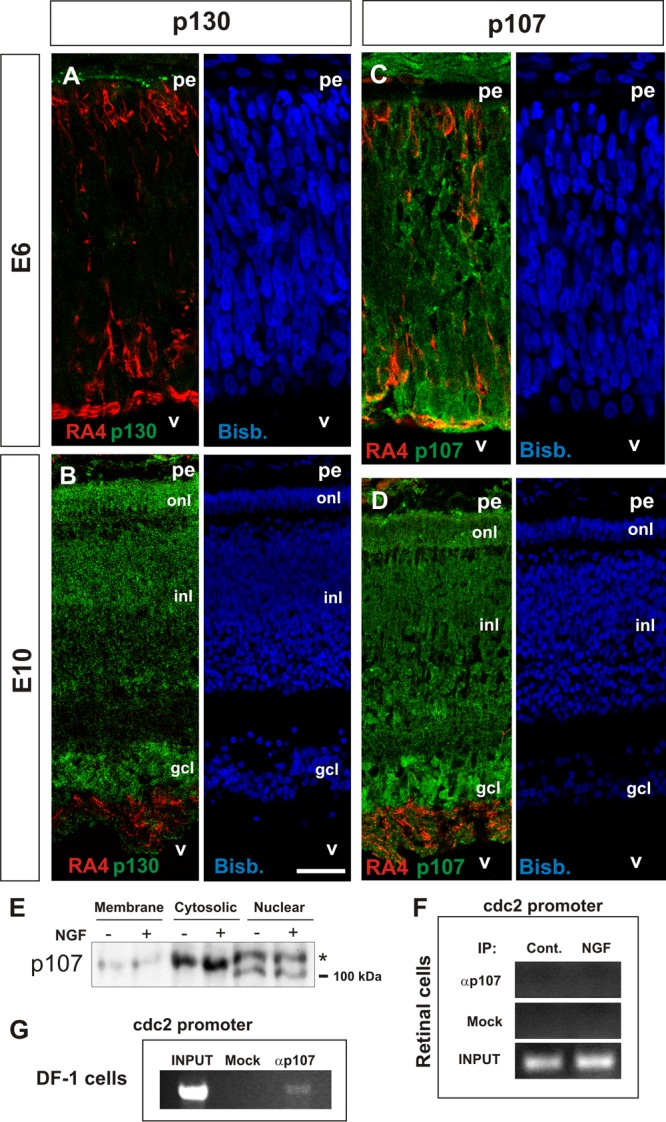
Analysis of p130 and p107 expression in DCRNs and lack of interaction of the latter with the cdc2 promoter in these cells. E6 (A and C) or E10 (B and D) chick retina cryosections (12 μm) immunolabeled for either p130 (A and B) or p107 (C and D) (green) and the RGC-specific marker RA4 (red) and counterstained with bisbenzimide (blue). (E) Western blot with the anti-p107 antibody in the indicated subcellular fractions from DCRNs cultured for 30 min either in the presence (+) or absence (−) of NGF. The asterisk indicates the hyperphosphorylated 107. (F and G) ChIP analysis of the occupancy of the chick cdc2 promoter by p107 in either DCRNs cultured for 30 min either in the presence (NGF) or absence (Control) of NGF (F) or in DF-1 chicken fibroblasts (DF-1 cells) (G). Anti-p107 (αp107) or irrelevant (Mock) antibodies were used for immunoprecipitation. INPUT, RT-PCR amplification from extracts before immunoprecipitation. pe, pigment epithelium; v, vitreus body; gcl, ganglion cell layer; inl, inner nuclear layer; onl, outer nuclear layer. Bars: 30 μm (A and C) and 60 μm (B and D).
In contrast to p130, the Rb family member p107 has been shown to be enriched in the E5/E6 quail retina, and it can be detected at lower levels in the retina of the E4 quail embryo (36), a developmental stage comparable to the E4.5 chicken embryo (59). To verify whether p107 is expressed by E6 retinal cells, retinal sections from this stage were immunostained with a specific anti-p107 antibody. This analysis indicated that p107 is readily detected in most retinal cells, including those expressing the RGC marker RA4 (Fig. 5C). The expression of p107 still remained in the chick retina at E10 (Fig. 5D). In the E6 chick retina, p107 expression was enriched in the cytoplasm (Fig. 5C). Nevertheless, it can be detected in the nuclear compartment as well, as evidenced by Western blotting performed in subcellular fractions from E6 chick retinas (Fig. 5E). The Western blot study demonstrated that nuclear p107 was in two main phosphorylation states: a low-mobility band representing the hyperphosphorylated form of p107 and a high-mobility band specific for the hypophosphorylated state of the protein (79). Importantly, addition of NGF did not alter the relative proportion between both bands, indicating that p107 is not hyperphosphorylated in response to p75NTR activation. Therefore, the phosphorylation-dependent mechanism for p107 inactivation is not active in NGF-treated DCRNs, indicating that this Rb family member does not play an active role in p75NTR-dependent cell cycle reentry in these cells.
The participation of p107 in E2F4-dependent regulation of the cdc2 promoter was further analyzed by ChIP. ChIP experiments were performed in lysates from DCRNs, immunoprecipitated with either an anti-p107 specific antibody or an irrelevant control antibody. Immunoprecipitates were sonicated and subjected to PCR using primers specific for the chick cdc2 promoter. The interaction of p107 could not be detected in both control and NGF-treated cultures even after 40 rounds of amplification (Fig. 5F). As a positive control, we performed a similar ChIP assay in lysates from DF-1 chick fibroblasts, where specific interaction of p107 with the cdc2 promoter was evident after 30 amplification rounds (Fig. 5G). Therefore, p107 does not seem to interact with E2F4 in NGF-treated DCRNs, when the latter is bound to the cdc2 promoter, as expected from the known repressive capacity of p107 on E2F transcriptional activity (5). We conclude that the interaction of E2F4 with the cdc2 promoter and the concomitant cell cycle reentry occurring in NGF-treated DCRNs is independent of p107 recruitment to the cdc2 promoter.
NGF-dependent cell cycle reentry in DCRNs is independent of cdk4/6 activity.
Cdk4/6 is known to fulfill a critical role in cell cycle regulation since phosphorylation of the Rb family members by this kinase results in the release of E2F activity (37). Therefore, the absence of a role for the Rb family proteins in NGF-dependent cell cycle reentry suggests that the mechanism used by p75NTR for signaling in DCRNs is independent of cdk4/6. This hypothesis was tested by treatment of DCRNs with the selective cdk4/6 inhibitor PD 0332991 (26), which is able to reduce BrdU incorporation by ∼90% in these cells when used at 200 nM (Fig. 6A). The presence of 200 nM PD 0332991 in the culture medium was able to prevent cell cycle progression in control DCRNs but not in NGF-treated DCRNs, as measured by either luciferase expression driven by the c-Myc promoter (Fig. 6B) or by BrdU incorporation (30 min pulse) (Fig. 6C). As expected, the presence of 200 nM PD 0332991 was not able to prevent NGF-induced apoptosis in DCRNs (Fig. 6D). These results demonstrate that the induction of cell cycle reentry by NGF is independent of cdk4/6 activity, as expected from the absence of a role for the Rb family proteins in this process.
Fig 6.

NGF-dependent cell cycle reentry and apoptosis is independent of cdk4/6 activity. (A) Dose-response curve of the effect of the selective cdk4/6 inhibitor PD 0332991 on the capacity of DCRNs to incorporate BrdU after a short pulse (30 min). The analysis was performed after 16 h of inhibitor treatment. Each experimental point is shown as mean ± the SEM (n = 3). (B) Luciferase activity, normalized to β-galactosidase, in extracts from DCRNs transfected with c-Myc-luciferase and β-galactosidase and treated for 16 h with different combinations of NGF and the cdk4/6 selective inhibitor PD 0332991. (C and D) BrdU incorporation (30-min pulse) (C) and proportion of apoptotic nuclei (D) in DCRNs treated for 16 h with different combinations of NGF and the cdk4/6 selective inhibitor PD 0332991. *, P < 0.05; ***, P < 0.005 (Student t test; n = 3).
NGF-dependent induction of p38MAPK is required for cell cycle reentry and apoptosis in DCRNs.
Neurotrophin binding to p75NTR has been shown to activate different members of the MAPK family of Ser/Thr kinases, including p38MAPK (12, 33), ERK (70), and c-Jun N-terminal kinase (JNK) (10). In accordance with these latter reports, sustained activation of p38MAPK in response to NGF was observed to rapidly occur in the nuclear compartment of DCRNs (Fig. 7A and B), affecting to both RA4- and RA4-negative cells (Fig. 7C). p38MAPK activity was observed to be crucial for cell cycle reentry and apoptosis in these cells. The addition of the p38MAPK selective inhibitor SB203580 (5 μM), but not the JNK selective inhibitor SP600125 (7 μM), was able to prevent cell cycle reentry induced by NGF in DCRNs, measured as the levels of luciferase activity driven by the c-Myc promoter (Fig. 7D), or the proportion of cells that incorporate BrdU after a short pulse (Fig. 7E). Furthermore, SB203580, but not SP600125, completely abolished the induction of apoptosis induced by NGF in these cultures (Fig. 7F). The proliferative and proapoptotic effects of JNK inhibition observed in these cultures (Fig. 7E and F) may be explained by the known capacity of JNK to prevent E2F1 activity (76).
Fig 7.

Activation of p38MAPK in response to NGF is necessary for cell cycle reentry and apoptosis in DCRNs. (A) The upper panel shows a Western blot with an anti-active p38MAPK antibody (P-p38MAPK) in extracts from DCRNs treated for the indicated time points with NGF. The bottom panel shows a Western blot with an anti-p38MAPK-specific antibody (p38MAPK). Normalized ratios between P-p38MAPK and p38MAPK are shown. (B) Immunostaining with an anti-active p38MAPK antibody (P-p38MAPK) in DCRNs cultured for 20 min in the presence of either vehicle (control) or NGF. Arrows indicate specific P-p38MAPK nuclear immunostaining. Nuclei were labeled with bisbenzimide (Bisb.). (C) DCRNs cultured for 16 h and immunolabeled with anti-phospho-p38MAPK (green) and anti-RA4 (red) antibodies. Nuclei were labeled with bisbenzimide (Bisb.). Strong nuclear labeling for phospho-p38MAPK was observed in both RA4-positive (arrow) and RA4-negative (arrowhead) cells. (D) Luciferase activity, normalized to β-galactosidase, in extracts from DCRNs transfected with c-Myc-luciferase and β-galactosidase and treated for 16 h with different combinations of NGF or the p38MAPK-selective inhibitor SB203580. (E and F) BrdU incorporation (30-min pulse) (E) and proportion of apoptotic nuclei (F) in DCRNs treated for 16 h with different combinations of NGF, SB203580, or the JNK-selective inhibitor SP600125. *, P < 0.05; ***, P < 0.005 (Student t test; n = 3). Bars: 5 μm (B) and 10 μm (C).
Activation of p38MAPK results in E2F4 phosphorylation and subsequent interaction of the latter with the cdc2 promoter.
The interaction of E2F4 with the cdc2 promoter, triggered by ligand-dependent activation of p75NTR, raises the question as to how such an interaction is induced. We reasoned that active p38MAPK could phosphorylate E2F4 in DCRNs. Therefore, immunoprecipitation with rabbit antisera specific for E2F4, followed by Western blotting with anti-phospho-Thr MAbs, was performed in cell extracts from DCRNs treated for 30 min with either NGF or vehicle. This analysis demonstrated that E2F4 became Thr phosphorylated in response to NGF, being this effect blocked by SB203580 (Fig. 8A). In contrast, a similar analysis carried out with antibodies specific for E2F1 or CMAGE demonstrated that these proteins were not Thr phosphorylated in response to NGF (Fig. 8B and c). Interestingly, the interaction of E2F4 with the cdc2 promoter triggered by NGF was abolished by SB203580, as evidenced by ChIP analysis (Fig. 8D), indicating that Thr phosphorylation is crucial for the interaction of E2F4 with the E2F-responsive cdc2 promoter. Overall, these results suggest that, in response to NGF, p38MAPK-dependent phosphorylation of E2F4 is required for the induction of cell cycle reentry in DCRNs.
Fig 8.

Thr phosphorylation of E2F4 is required for binding to the cdc2 promoter. (A) The upper panel shows a Western blot with an anti-phospho-Thr-specific antibody in E2F4-specific immunoprecipitates from DCRNs treated with different combinations of NGF and SB203580. The bottom panel shows a Western blot with an anti-E2F4-specific antibody from inputs. (B) The upper panel shows a Western blot with an anti-phospho-Thr-specific antibody in E2F1-specific immunoprecipitates from DCRNs treated with either NGF or vehicle. The bottom panel shows a Western blot with an anti-E2F1-specific antibody from inputs. (C) The upper panel shows a Western blot with an anti-phospho-Thr-specific antibody in CMAGE-specific immunoprecipitates from DCRNs treated with either NGF or vehicle. The bottom panel shows a Western blot with the CMAGE-specific NC243 antiserum from inputs. (D) ChIP analysis of the occupancy of the chick cdc2 promoter by E2F4 in DCRNs treated for 30 min with different combinations of NGF or SB203580. Anti-E2F4 (αE2F4) or irrelevant (Mock) antibodies used for immunoprecipitation. INPUT, RT-PCR amplification from extracts before immunoprecipitation.
A phosphomimetic mutant form of E2F4 induces cell cycle reentry and apoptosis in DCRNs.
NetPhosK 1.0 software was used to predict putative p38MAPK phosphorylation sites within the amino acid sequence of chick E2F4. This analysis indicated the presence of two unique Thr residues at positions 261 and 263, susceptible to being phosphorylated by p38MAPK (Fig. 9A). These residues lay within an E2F4 region in which no function has been described thus far, between the protein dimerization/marked box domain and the transactivation domain. We refer to this region as the “regulatory domain” (Fig. 9). These residues are conserved in other vertebrate E2F4s (Fig. 9B) but not in chick E2F1 (accession number NP_990550).
Fig 9.

Structure and functional domains of the E2F4 transcription factor. (A) Chick E2F4 can be divided into different functional domains: the DNA-binding domain (DB), the E2F/DP dimerization/marked box domains (DIM/MB), the regulatory domain (RD) described in the present study, and the transactivation domain (TA). Numbers indicate the amino acid positions of the transition between different domains. Arrows indicate the positions of Thr261 (T261) and Thr263 (T263), which are susceptible to be phosphorylated by p38MAPK (P). (B) Amino acid sequence comparison of the E2F4s from human E2F4 (Homo sapiens; accession number NP_001941), mouse E2F4 (Mus musculus; accession number NP_683754), chick E2F4 (Gallus gallus; accession number JQ678847), Xenopus laevis E2F4 (X. laevis; accession number NP_001086706), and zebrafish E2F4 (Danio rerio; accession number AAH56832). Alignment was performed by the CLUSTAL method to identify contiguous regions of homology. Black boxes indicate the different domains mentioned above. Boxes indicate the Thr residues susceptible to be phosphorylated by p38MAPK, as predicted by NetPhosK 1.0 software. Underlined characters indicate the consensus sequence for the p38MAPK docking site, R/KXXXXφXφ, where “φ” represent hydrophobic residues (11). Such a domain can be observed at the beginning of the DB domain. Dots represent conserved amino acids within the regulatory domain (RD).
To demonstrate that phosphorylation within the Thr261/Thr263 motif of E2F4 can trigger cell cycle reentry in DCRNs, a constitutively active E2F4 construct (E2F4-CA) was created, in which both Thr residues were substituted by Glu (Thr261Glu/Thr263Glu). This construct was able to increase the rate of proliferation in DCRNs, measured as the levels of luciferase activity driven by the c-Myc promoter (Fig. 10A), or the proportion of cells that incorporate BrdU after a short (30 min) pulse (Fig. 10B). This effect was not blocked by the p38MAPK-selective inhibitor SB203580 (Fig. 10A and B). Interestingly, the expression in DCRNs of wild-type E2F4 (E2F4-WT) could not mimic these effects (Fig. 10D and E), which were only observed in the presence of NGF. As expected, E2F4-CA was able to induce apoptosis in transfected DCRNs even in the presence of SB203580 (Fig. 10C). In contrast, E2F4-WT induced apoptosis only in the presence of NGF (Fig. 10F). These results demonstrate that the effect of p38MAPK in cell cycle reactivation and apoptosis is exclusively dependent on the phosphorylation of E2F4 within the Thr261/Thr263 motif.
Fig 10.

Phosphorylated E2F4 is able to induce cell cycle reentry and apoptosis in DCRNs. (A) Luciferase activity, normalized to β-galactosidase, in extracts from DCRNs transfected with c-Myc-luciferase, β-galactosidase, and either a constitutively active form of E2F4 (E2F4-CA) or the pcDNA6 plasmid (control) and maintained for 16 h in the presence of the p38MAPK-selective inhibitor SB203580 or vehicle. (B and C) BrdU incorporation (30-min pulse) (B) and proportion of apoptotic nuclei (C) in DCRNs transfected with either E2F4-CA or the pcDNA6 plasmid (control), and maintained for 16 h in the presence of the p38MAPK-selective inhibitor SB203580 or vehicle. (D) Luciferase activity, normalized to β-galactosidase, in extracts from DCRNs transfected with c-Myc-luciferase and β-galactosidase and either wild-type E2F4 (E2F4-WT) or the pcDNA6 plasmid (control) and maintained for 16 h in the presence of either NGF or vehicle. (E and F) BrdU incorporation (30-min pulse) (E) and proportion of apoptotic nuclei (F) in DCRNs transfected with either wild-type E2F4 (E2F4-WT) or the pcDNA6 plasmid (control) and maintained for 16 h in the presence of either NGF or vehicle. (G) Luciferase activity, normalized to β-galactosidase, in extracts from DCRNs transfected with c-Myc-luciferase and β-galactosidase and either a dominant-negative form of E2F4 (E2F4-DN) or the pcDNA6 plasmid (control) and maintained for 16 h in the presence of either NGF or vehicle. (H and I) BrdU incorporation (30-min pulse) (H) and proportion of apoptotic nuclei (I) in DCRNs transfected with either a dominant-negative form of E2F4 (E2F4-DN) or the pcDNA6 plasmid (control), and maintained for 16 h in the presence of either NGF or vehicle. *, P < 0.05; **, P < 0.001; ***, P < 0.005 (Student t test; n = 3).
A phosphorylation deficient form of E2F4 prevents NGF-dependent cell cycle reentry and apoptosis in DCRNs.
The participation of the Thr261/Thr263 motif of E2F4 in the p75NTR/p38MAPK signaling pathway was confirmed with a dominant-negative form of E2F4 (E2F4-DN). The E2F4-DN construct, in which Thr261/Thr263 were substituted by Ala (Thr261Ala/Thr263Ala), was able to prevent the effect of NGF in cell cycle reactivation, measured as either luciferase activity driven by the c-Myc promoter (Fig. 10G) or BrdU incorporation (30 min pulse) (Fig. 10H). Moreover, this construct also prevented NGF-induced apoptosis in DCRNs (Fig. 10I). This contrasts with the capacity of NGF to trigger cell cycle reentry and apoptosis in DCRNs expressing E2F4-WT (Fig. 10D to F). These results confirm the involvement of the phosphorylation within the Thr261/Thr263 motif of E2F4 as a key step to induce cell cycle reentry by NGF in DCRNs.
DISCUSSION
In this study we have provided evidence for a novel mechanism used by p75NTR to induce cell cycle reentry in differentiating RGCs, based on the activation of p38MAPK and the subsequent Thr phosphorylation of E2F4.
We have shown that in DCRNs, p75NTR does not induce nuclear translocation of p75ICD or CMAGE recruitment to the cell membrane (38, 43, 47, 65). Instead, our work confirms and extends previous findings showing that p75NTR is able to activate p38MAPK (12, 33). This effect cannot be induced by the NGF neurotrophic receptor TrkA since we have previously demonstrated that blocking antibodies against p75NTR, but not TrkA receptor bodies, prevent cell cycle reentry in DCRNs (17, 51). The activation of p38MAPK is relevant for cell cycle regulation in DCRNs since p38MAPK inhibition was observed to prevent NGF-dependent cell cycle reentry in these cells. p38MAPK is known to regulate cell cycle progression, either favoring or preventing proliferation in several cellular systems (55). In this regard, p38MAPK has been shown to phosphorylate Rb, thus facilitating Rb-E2F1 dissociation (76). p38MAPK has also been shown to be involved in the induction of cardiomyocyte hypertrophy (78), which is known to be linked to tetraploidy (28). As in the case of DCRNs (Fig. 7E and F), JNK also has an opposing effect as that of p38MAPK in cardiomyocytes (56).
We have also shown that p75NTR-dependent cell cycle reentry in DCRNs is independent of the previously described cyclin D-cdk4/6-Rb and cyclin D-cdk4/6-p130 pathways leading to neuronal death (45, 60), since the p75NTR pathway cannot be blocked by the selective cdk4/6 inhibitor PD 0332991 (26). This observation is consistent with the absence in NGF-treated DCRNs of cyclin D1 upregulation (17), enhanced p107 hyperphosphorylation (the present study), and Rb (51) and p130 (the present study) expression.
PD 0332991, used at 200 nM, was able to dramatically reduce BrdU incorporation without inducing apoptosis. This remarkable inhibition is likely due to the presence in our cultures of RA4-negative cells (see Fig. 4A), which seem to proliferate (compare Fig. 4B to Fig. 3J), likely through a cdk4/6-dependent mechanism. This may explain why PD 0332991 prevents G1/S transition in DCRNs.
We have demonstrated that E2F4 is expressed by differentiating RGCs in vivo and that it becomes Thr phosphorylated by p38MAPK in response to NGF. In this regard, a consensus sequence for the docking site of p38MAPK (11) can be detected at the N terminus of the E2F4 sequence from all of the vertebrate species analyzed here (Fig. 9B). Although the actual Thr residues phosphorylated by p38MAPK are currently unknown, we provide evidence that they should be present within the Thr261/Thr263 motif. First of all, Thr261 and Thr 263 are predicted by the NetPhosK 1.0 software to be the only residues susceptible to be phosphorylated by p38MAPK in the chick E2F4 sequence. Furthermore, unique Thr residues present in a similar region of the molecule and susceptible to be phosphorylated by p38MAPK are conserved in other vertebrate E2F4s. Finally, a constitutively active form of E2F4, double mutant for Thr261Glu/Thr263Glu, can mimic the effect of NGF on DCRNs, whereas the Thr261Ala/Thr263Ala double mutant form of E2F4 behaves as a dominant-negative factor preventing the effect of NGF on cell cycle reentry and apoptosis. The dominant-negative capacity of the latter is likely to be due to its capacity to mimic the docking site of p38MAPK while remaining unphosphorylated, thus competing with endogenous E2F4 for its interaction with p38MAPK. A similar approach has been used to prevent JNK activity by using a JIP-1 JNK-binding domain-derived peptide that interacts with JNK and functionally inhibits this kinase (4). Phosphorylation of Thr residues close to each other by p38MAPK is not an uncommon event. For instance, p38MAPK has been shown to phosphorylate two highly close residues from the transcription factor CHOP (77). Our data cannot rule out the existence of additional residues in E2F4 susceptible to be phosphorylated by p38MAPK, but if this is the case they do not seem to play a fundamental role in the induction of cell cycle reentry by NGF since the E2F4-CA construct was able to fully mimic the effect of NGF in a p38MAPK-independent manner.
E2F4 is able to bind to the promoter of E2F-responsive genes upon NGF treatment, as demonstrated by ChIP analysis using cdc2 promoter-specific primers. This interaction was disrupted by application of the p38MAPK selective inhibitor SB203580, indicating that the phosphorylation of E2F4 by p38MAPK is required for the binding of E2F4 to the E2F-responsive promoters. E2F4 is known to lack a nuclear localization signal (NLS) (49). Therefore, the question remains as to how this Rb family member accumulates in the nucleus of DCRNs. One possibility is that E2F4 binds to p107 under control conditions, whereas p107 inactivation by active p38MAPK, as previously shown in Jurkat cells (54), could lead to the release of the phosphorylated form of E2F4 in NGF-treated DCRNs. Nevertheless, this hypothesis is not consistent with the lack of changes in the phosphorylation state of p107 in response to NGF (Fig. 5E). An alternative possibility is that nuclear retention of E2F4 in DCRNs would depend on other interacting proteins. In this regard, both DP2 and an NLS-containing splice form of DP3 have both been shown to facilitate nuclear retention of E2F4 (49, 67). The activity of E2F transcription factors can be modulated by different members of the type II melanoma antigen (MAGE) protein family, known to participate in cell cycle progression, apoptosis, and neurogenetic disease (3). In this regard, both Necdin and MAGE-G1 have been shown to mimic Rb function, interacting with E2F1 and preventing its activity in postmitotic neurons (41, 43, 71). Similarly, Necdin has been shown to interact with E2F4 and modulate its function (39). In contrast to the MAGE protein family in mammals, which has dramatically expanded during evolution, only a single MAGE protein has been shown to be expressed in the chick (47). This protein, CMAGE, is expressed by RGCs, and it can prevent E2F1 activity in differentiated neuroblastoma cells (47). This suggests that, in DCRNs, CMAGE could regulate the activity of E2F1, which is observed to be constitutively bound in these cells to the cdc2 promoter.
Although E2F4 is typically classified as a repressor, in a number of cell systems it can also act as an activator of gene transcription. Indeed, the specific cellular environment is what determines whether a specific E2F family member activates or represses transcription (25). For instance, E2F4 can induce transcriptional activity in human intestinal epithelial cells (15), and hyperphosphorylated E2F4 has been suggested to participate in the transactivation of the cyclin A gene in C33A cervical cancer cells (16). In brown preadipocytes, E2F4 can induce the expression of peroxisome-proliferator-activated receptor-γ transcription (73). Interestingly, the interaction of Necdin with E2F4 in the latter cells abolishes the activity of E2F4 (73), suggesting that the interaction of E2F4 with MAGE proteins may change its function. One possibility is that a putative interaction of CMAGE with E2F4 could transform the latter transcription factor in a transcriptional activator. Future studies will test whether CMAGE actively participates in E2F1 activity regulation and facilitation of the proliferative capacity of E2F4 in DCRNs.
In sum, our results suggest that different mechanisms exist to control the postmitotic state in different neuronal populations. The mechanism described in this study could participate in cell cycle reentry, tetraploidization, and neuronal death in neurodegenerative diseases such as Alzheimer's disease (2, 19, 52). Indeed, the expression of proNGF (64), p75NTR (32), active p38MAPK (63, 69), E2F4 (50), and the type II MAGE protein Necdin (57) has been described in neurons from normal and Alzheimer's disease-affected brains.
ACKNOWLEDGMENTS
We thank K. Yoshikawa for the NC243 antibody and the c-Myc-luciferase construct, C. K. Glass for the cdc2 luciferase construct, S. Wilson for the pRFPRNAi vectors, S. C. McLoon for the RA4 MAb, M. V. Chao for antiserum 9992, and Y.-A. Barde for the p75NTR exon IV knockout mice.
This study was supported by grants from the Ministerio de Ciencia y Tecnología, Fundación La Caixa, and FUNDALUCE.
Footnotes
Published ahead of print 14 May 2012
REFERENCES
- 1. Allington C, Shamovsky IL, Ross GM, Riopelle RJ. 2001. Zinc inhibits p75NTR-mediated apoptosis in chick neural retina. Cell Death Differ. 8:451–456 [DOI] [PubMed] [Google Scholar]
- 2. Arendt T, Brückner MK, Mosch B, Lösche A. 2010. Selective cell death of hyperploids neurons in Alzheimer's disease. Am. J. Pathol. 177:15–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barker PA, Salehi A. 2002. The MAGE proteins: emerging roles in cell cycle progression, apoptosis, and neurogenetic disease. J. Neurosci. Res. 67:705–712 [DOI] [PubMed] [Google Scholar]
- 4. Barr RK, Kendrick TS, Bogoyevitch MA. 2002. Identification of the critical features of a small peptide inhibitor of JNK activity. J. Biol. Chem. 277:10987–10997 [DOI] [PubMed] [Google Scholar]
- 5. Beijersbergen RL, Carlée L, Kerkhoven RM, Bernards R. 1995. Regulation of the retinoblastoma protein-related p107 by G1 cyclin complexes. Genes Dev. 9:1340–1353 [DOI] [PubMed] [Google Scholar]
- 6. Biswas SC, Liu DX, Greene LA. 2005. Bim is a direct target of a neuronal E2F-dependent apoptotic pathway. J. Neurosci. 25:8349–8358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S. 2004. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4:1633–1649 [DOI] [PubMed] [Google Scholar]
- 8. Bottenstein JE, Sato GH. 1979. Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proc. Natl. Acad. Sci. U. S. A. 76:514–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Busser J, Geldmacher DS, Herrup K. 1998. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J. Neurosci. 18:2801–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. 1996. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature 383:716–719 [DOI] [PubMed] [Google Scholar]
- 11. Chang CI, Xu BE, Akella R, Cobb MH, Goldsmith EJ. 2002. Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol. Cell 9:1241–1249 [DOI] [PubMed] [Google Scholar]
- 12. Costantini C, et al. 2005. Characterization of the signaling pathway downstream p75 neurotrophin receptor involved in beta-amyloid peptide-dependent cell death. J. Mol. Neurosci. 25:141–156 [DOI] [PubMed] [Google Scholar]
- 13. Dagnino L, et al. 1997. Expression patterns of the E2F family of transcription factors during mouse nervous system development. Mech. Dev. 66:13–25 [DOI] [PubMed] [Google Scholar]
- 14. Das RM, et al. 2006. A robust system for RNA interference in the chicken using a modified microRNA operon. Dev. Biol. 294:554–563 [DOI] [PubMed] [Google Scholar]
- 15. Deschênes C, Alvarez L, Lizotte ME, Vézina A, Rivard N. 2004. The nucleocytoplasmic shuttling of E2F4 is involved in the regulation of human intestinal epithelial cell proliferation and differentiation. J. Cell Physiol. 199:262–273 [DOI] [PubMed] [Google Scholar]
- 16. Duro D, et al. 1999. Activation of cyclin A gene expression by the cyclin encoded by human herpesvirus-8. J. Gen. Virol. 80:549–555 [DOI] [PubMed] [Google Scholar]
- 17. Frade JM. 2000. Unscheduled cell cycle re-entry induced by NGF precedes cell death in nascent retinal neurones. J. Cell Sci. 113:1139–1148 [DOI] [PubMed] [Google Scholar]
- 18. Frade JM. 2005. Nuclear translocation of the p75 neurotrophin receptor cytoplasmic domain in response to neurotrophin binding. J. Neurosci. 25:1407–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Frade JM, López-Sánchez N. 2010. A novel hypothesis for Alzheimer's disease based on neuronal tetraploidy induced by p75NTR. Cell Cycle 9:1934–1941 [DOI] [PubMed] [Google Scholar]
- 20. Frade JM, Rodríguez-Tébar A. 2000. Neuroepithelial differentiation induced by ECM molecules. Methods Mol. Biol. 139:257–264 [DOI] [PubMed] [Google Scholar]
- 21. Frade JM, et al. 1997. Control of early cell death by BDNF in the chick retina. Development 124:3313–3320 [DOI] [PubMed] [Google Scholar]
- 22. Frade JM, et al. 1996. Insulin-like growth factor-I stimulates neurogenesis in chick retina by regulating expression of the alpha 6 integrin subunit. Development 122:2497–2506 [DOI] [PubMed] [Google Scholar]
- 23. Frade JM, Martínez-Morales JR, Rodríguez-Tébar A. 1996. Laminin-1 selectively stimulates neuron generation from cultured retinal neuroepithelial cells. Exp. Cell Res. 222:140–149 [DOI] [PubMed] [Google Scholar]
- 24. Freeman RS, Estus S, Johnson EM., Jr 1994. Analysis of cell cycle-related gene expression in postmitotic neurons: selective induction of cyclin D1 during programmed cell death. Neuron 12:343–355 [DOI] [PubMed] [Google Scholar]
- 25. Fry CJ, Farnham PJ. 1999. Context-dependent transcriptional regulation. J. Biol. Chem. 274:29583–295836 [DOI] [PubMed] [Google Scholar]
- 26. Fry DW, et al. 2004. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 3:1427–1438 [PubMed] [Google Scholar]
- 27. Garneau H, Paquin MC, Carrier JC, Rivard N. 2009. E2F4 expression is required for cell cycle progression of normal intestinal crypt cells and colorectal cancer cells. J. Cell Physiol. 221:350–358 [DOI] [PubMed] [Google Scholar]
- 28. Goodman LC, Epling S, Kelly S, Lee S, Fishbein MC. 1990. DNA flow cytometry of myocardial cell nuclei in paraffin-embedded, human autopsy, cardiac tissue. Am. J. Cardiovasc. Pathol. 3:55–59 [PubMed] [Google Scholar]
- 29. Hamburger V, Hamilton HL. 1951. A series of normal stages in the development of the chick embryo. J. Morphol. 88:49–92 [DOI] [PubMed] [Google Scholar]
- 30. Herrup K, Yang Y. 2007. Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat. Rev. Neurosci. 8:368–378 [DOI] [PubMed] [Google Scholar]
- 31. Himly M, Foster DN, Bottoli I, Iacovoni JS, Vogt PK. 1998. The DF-1 chicken fibroblast cell line: transformation induced by diverse oncogenes and cell death resulting from infection by avian leukosis viruses. Virology 248:295–304 [DOI] [PubMed] [Google Scholar]
- 32. Hu XY, et al. 2002. Increased p75NTR expression in hippocampal neurons containing hyperphosphorylated tau in Alzheimer patients. Exp. Neurol. 178:104–111 [DOI] [PubMed] [Google Scholar]
- 33. Jiang Y, et al. 2007. Nerve growth factor promotes TLR4 signaling-induced maturation of human dendritic cells in vitro through inducible p75NTR. J. Immunol. 179:6297–6304 [DOI] [PubMed] [Google Scholar]
- 34. Jung KM, et al. 2003. Regulated intramembrane proteolysis of the p75 neurotrophin receptor modulates its association with the TrkA receptor. J. Biol. Chem. 278:42161–42169 [DOI] [PubMed] [Google Scholar]
- 35. Kanning KC, et al. 2003. Proteolytic processing of the p75 neurotrophin receptor and two homologs generates C-terminal fragments with signaling capability. J. Neurosci. 23:5425–5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kastner A, Espanel X, Brun G. 1998. Transient accumulation of retinoblastoma/E2F-1 protein complexes correlates with the onset of neuronal differentiation in the developing quail neural retina. Cell Growth Differ. 9:857–867 [PubMed] [Google Scholar]
- 37. Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. 1993. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 7:331–342 [DOI] [PubMed] [Google Scholar]
- 38. Kenchappa RS, et al. 2006. Ligand-dependent cleavage of the p75 neurotrophin receptor is necessary for NRIF nuclear translocation and apoptosis in sympathetic neurons. Neuron 50:219–232 [DOI] [PubMed] [Google Scholar]
- 39. Kobayashi M, Taniura H, Yoshikawa K. 2002. Ectopic expression of Necdin induces differentiation of mouse neuroblastoma cells. J. Biol. Chem. 277:42128–42135 [DOI] [PubMed] [Google Scholar]
- 40. Konishi Y, Bonni A. 2003. The E2F-Cdc2 cell cycle pathway specifically mediates activity deprivation-induced apoptosis of postmitotic neurons. J. Neurosci. 23:1649–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kurita M, Kuwajima T, Nishimura I, Yoshikawa K. 2006. Necdin downregulates CDC2 expression to attenuate neuronal apoptosis. J. Neurosci. 26:12003–12013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kuwajima T, Taniura H, Nishimura I, Yoshikawa K. 2004. Necdin interacts with the Msx2 homeodomain protein via MAGE-D1 to promote myogenic differentiation of C2C12 cells. J. Biol. Chem. 279:40484–40493 [DOI] [PubMed] [Google Scholar]
- 43. Kuwako K, Taniura H, Yoshikawa K. 2004. Necdin-related MAGE proteins differentially interact with the E2F1 transcription factor and the p75 neurotrophin receptor. J. Biol. Chem. 279:1703–1712 [DOI] [PubMed] [Google Scholar]
- 44. Liu DX, Biswas SC, Greene LA. 2004. B-myb and C-myb play required roles in neuronal apoptosis evoked by nerve growth factor deprivation and DNA damage. J. Neurosci. 24:8720–8725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu DX, Nath N, Chellappan SP, Greene LA. 2005. Regulation of neuron survival and death by p130 and associated chromatin modifiers. Genes Dev. 19:719–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. López-Sánchez N, Frade JM. 2002. Control of the cell cycle by neurotrophins: lessons from the p75 neurotrophin receptor. Histol. Histopathol. 17:1227–1237 [DOI] [PubMed] [Google Scholar]
- 47. López-Sánchez N, González-Fernández Z, Niinobe M, Yoshikawa K, Frade JM. 2007. A single Mage gene in the chicken genome encodes CMAGE, a protein with functional similarities to mammalian type II MAGE proteins. Physiol. Genomics 30:156–171 [DOI] [PubMed] [Google Scholar]
- 48. López-Sánchez N, Ovejero-Benito MC, Borreguero L, Frade JM. 2011. Control of neuronal ploidy during vertebrate development. Results Probl. Cell Differ. 53:547–563 [DOI] [PubMed] [Google Scholar]
- 49. Magae J, Wu CL, Illenye S, Harlow E, Heintz NH. 1996. Nuclear localization of DP and E2F transcription factors by heterodimeric partners and retinoblastoma protein family members. J. Cell Sci. 109:1717–1726 [DOI] [PubMed] [Google Scholar]
- 50. Morgan KL, Chalovich EM, Strachan GD, Otis LL, Jordan-Sciutto KL. 2005. E2F4 expression patterns in SIV encephalitis. Neurosci. Lett. 382:259–264 [DOI] [PubMed] [Google Scholar]
- 51. Morillo SM, Escoll P, de la Hera A, Frade JM. 2010. Somatic tetraploidy in specific chick retinal ganglion cells induced by nerve growth factor. Proc. Natl. Acad. Sci. U. S. A. 107:109–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mosch B, et al. 2007. Aneuploidy and DNA replication in the normal human brain and Alzheimer's disease. J. Neurosci. 27:6859–6867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nagy Z, Esiri MM, Cato AM, Smith AD. 1997. Cell cycle markers in the hippocampus in Alzheimer's disease. Acta Neuropathol. 94:6–15 [DOI] [PubMed] [Google Scholar]
- 54. Nath N, Wang S, Betts V, Knudsen E, Chellappan S. 2003. Apoptotic and mitogenic stimuli inactivate Rb by differential utilization of p38 and cyclin-dependent kinases. Oncogene 22:5986–5994 [DOI] [PubMed] [Google Scholar]
- 55. Nebreda AR, Porras A. 2000. p38 MAP kinases: beyond the stress response. Trends Biochem. Sci. 25:257–260 [DOI] [PubMed] [Google Scholar]
- 56. Nemoto S, Sheng Z, Lin A. 1998. Opposing effects of Jun kinase and p38 mitogen-activated protein kinases on cardiomyocyte hypertrophy. Mol. Cell. Biol. 18:3518–3526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Niinobe M, Koyama K, Yoshikawa K. 2000. Cellular and subcellular localization of Necdin in fetal and adult mouse brain. Dev. Neurosci. 22:310–319 [DOI] [PubMed] [Google Scholar]
- 58. Nykjaer A, Willnow TE, Petersen CM. 2005. p75NTR: live or let die. Curr. Opin. Neurobiol. 15:49–57 [DOI] [PubMed] [Google Scholar]
- 59. Padgett CS, Ivey WD. 1960. The normal embryology of the Coturnix quail. Anat. Rec. 137:1–11 [DOI] [PubMed] [Google Scholar]
- 60. Padmanabhan J, Park DS, Greene LA, Shelanski ML. 1999. Role of cell cycle regulatory proteins in cerebellar granule neuron apoptosis. J. Neurosci. 19:8747–8756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Park DS, Levine B, Ferrari G, Greene LA. 1997. Cyclin-dependent kinase inhibitors and dominant-negative cyclin-dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J. Neurosci. 17:8975–8983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Paul CE, Vereker E, Dickson KM, Barker PA. 2004. A proapoptotic fragment of the p75 neurotrophin receptor is expressed in p75NTRExonIV null mice. J. Neurosci. 24:1917–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pei JJ, et al. 2001. Localization of active forms of C-jun kinase (JNK) and p38 kinase in Alzheimer's disease brains at different stages of neurofibrillary degeneration. J. Alzheimer's Dis. 3:41–48 [DOI] [PubMed] [Google Scholar]
- 64. Peng S, Wuu J, Mufson EJ, Fahnestock M. 2004. Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer's disease. J. Neuropathol. Exp. Neurol. 63:641–649 [DOI] [PubMed] [Google Scholar]
- 65. Podlesniy P, et al. 2006. Pro-NGF from Alzheimer's disease and normal human brain displays distinctive abilities to induce processing and nuclear translocation of intracellular domain of p75NTR and apoptosis. Am. J. Pathol. 169:119–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Prada C, Puga J, Pérez-Méndez L, López R, Ramírez G. 1991. Spatial and temporal patterns of neurogenesis in the chick retina. Eur. J. Neurosci. 3:559–569 [DOI] [PubMed] [Google Scholar]
- 67. Puri PL, et al. 1998. Regulation of E2F4 mitogenic activity during terminal differentiation by its heterodimerization partners for nuclear translocation. Cancer Res. 58:1325–1331 [PubMed] [Google Scholar]
- 68. Sugarman JL, Schönthal AH, Glass CK. 1995. Identification of a cell-type-specific and E2F-independent mechanism for repression of cdc2 transcription. Mol. Cell. Biol. 15:3282–3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sun A, Liu M, Nguyen XV, Bing G. 2003. p38 MAP kinase is activated at early stages in Alzheimer's disease brain. Exp. Neurol. 183:394–405 [DOI] [PubMed] [Google Scholar]
- 70. Susen K, Heumann R, Blöchl A. 1999. Nerve growth factor stimulates MAPK via the low affinity receptor p75LNTR. FEBS Lett. 463:231–234 [DOI] [PubMed] [Google Scholar]
- 71. Taniura H, Taniguchi N, Hara M, Yoshikawa K. 1998. Necdin, a postmitotic neuron-specific growth suppressor, interacts with viral transforming proteins and cellular transcription factor E2F1. J. Biol. Chem. 273:720–728 [DOI] [PubMed] [Google Scholar]
- 72. Teng KK, Felice S, Kim T, Hempstead BL. 2010. Understanding proneurotrophin actions: recent advances and challenges. Dev. Neurobiol. 70:350–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tseng YH, et al. 2005. Prediction of preadipocyte differentiation by gene expression reveals role of insulin receptor substrates and Necdin. Nat. Cell Biol. 7:601–611 [DOI] [PubMed] [Google Scholar]
- 74. Von Schack D, et al. 2001. Complete ablation of the neurotrophin receptor p75NTR causes defects both in the nervous and the vascular system. Nat. Neurosci. 4:977–978 [DOI] [PubMed] [Google Scholar]
- 75. Waid DK, McLoon SC. 1995. Immediate differentiation of ganglion cells following mitosis in the developing retina. Neuron 14:117–124 [DOI] [PubMed] [Google Scholar]
- 76. Wang S, Nath N, Minden A, Chellappan S. 1999. Regulation of Rb and E2F by signal transduction cascades: divergent effects of JNK1 and p38 kinases. EMBO J. 18:1559–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang XZ, Ron D. 1996. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science 272:1347–1349 [DOI] [PubMed] [Google Scholar]
- 78. Zechner D, Thuerauf DJ, Hanford DS, McDonough PM, Glembotski CC. 1997. A role for the p38 mitogen-activated protein kinase pathway in myocardial cell growth, sarcomeric organization, and cardiac-specific gene expression. J. Cell Biol. 139:115–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zou X, Rudchenko S, Wong K, Calame K. 1997. Induction of c-myc transcription by the v-Abl tyrosine kinase requires Ras, Raf1, and cyclin-dependent kinases. Genes Dev. 11:654–662 [DOI] [PubMed] [Google Scholar]