

Abstract
A novel lipolytic enzyme was isolated from a metagenomic library obtained from tidal flat sediments on the Korean west coast. Its putative functional domain, designated MPlaG, showed the highest similarity to phospholipase A from Grimontia hollisae CIP 101886, though it was screened from an emulsified tricaprylin plate. Phylogenetic analysis showed that MPlaG is far from family I.6 lipases, including Staphylococcus hyicus lipase, a unique lipase which can hydrolyze phospholipids, and is more evolutionarily related to the bacterial phospholipase A1 family. The specific activities of MPlaG against olive oil and phosphatidylcholine were determined to be 2,957 ± 144 and 1,735 ± 147 U mg−1, respectively, which means that MPlaG is a lipid-preferred phospholipase. Among different synthetic esters, triglycerides, and phosphatidylcholine, purified MPlaG exhibited the highest activity toward p-nitrophenyl palmitate (C16), tributyrin (C4), and 1,2-dihexanoyl-phosphatidylcholine (C8). Finally, MPlaG was identified as a phospholipase A1 with lipase activity by cleavage of the sn-1 position of OPPC, interfacial activity, and triolein hydrolysis. These findings suggest that MPlaG is the first experimentally characterized phospholipase A1 with lipase activity obtained from a metagenomic library. Our study provides an opportunity to improve our insight into the evolution of lipases and phospholipases.
INTRODUCTION
Microorganisms produce different classes of carboxylic ester hydrolases (EC 3.1.1) affiliated with the structural superfamily of α/β-hydrolases, including lipases, esterases, and phospholipases. Lipases (EC 3.1.1.3) catalyze the hydrolysis of water-insoluble medium- and long-chain triglycerides, although they also display high activity against emulsions of slightly water-soluble short-chain triglycerides that are better substrates of esterases (EC 3.1.1.1). Phospholipases hydrolyze ester linkages in phosphoglycerides with an amphipathic nature instead of neutral lipids (9, 19, 21). In particular, phospholipases A1 and A2 are classified as EC 3.1.1.32 and EC 3.1.1.4 enzymes, respectively. With respect to their similarities of structure and catalytic mechanism, lipases, esterases, and phospholipases A1 and A2 may share activity with each other, because many enzymes have broad substrate specificity or promiscuous functions (6). However, only a few enzymes with both activities have been reported. For example, mammalian lipases, including pancreatic lipase and hepatic lipase, have been known to hydrolyze phospholipids (34). In addition, it was reported that staphylococcal lipases classified in family I.6 can catalyze the hydrolysis of phospholipids (9). Many researchers have studied the amino acid residues contributing to phospholipase activity in lipases by constructing chimeras, using gene replacement, mutation, sequence comparison, or three-dimensional (3D) modeling (26, 29, 30, 33). The crystal structure of a chimeric mutant of pancreatic lipase-related protein 2 from guinea pig (GPLRP2) revealed that the reduced size of an amphipathic helical lid domain contributes to accommodating the polar head of phospholipids (33). Recently, structural analysis of Staphylococcus hyicus lipase (SHL), which has high phospholipase A activity, revealed that its substrate binding cavity contains two large hydrophobic acyl chain binding pockets and a polar third pocket that is capable of binding either a short fatty acid or a phospholipid head group (26).
Metagenomes, i.e., genetic materials recovered directly from the environment without microbial cultivation, have been used successfully for the isolation of novel lipolytic enzymes from various environments, such as the Antarctic, solfataric fields, soil, and deep-sea sediment (8, 10, 18, 31). To date, only two phospholipases from metagenomic libraries, i.e., hot spring and biofilm metagenomic libraries, have been reported, based on sequence similarities (1, 27). Despite their identification, it was unclear whether these enzymes can really hydrolyze phospholipids. We previously reported two novel lipases, LipG and LipEH166, from a metagenomic library obtained from tidal flat sediments in the Saemankum area (35°42′N, 126°33′E) on the western coast of the Republic of Korea (12, 14). Tidal flat regions are attractive environments for the construction of a metagenomic library because they are dynamic physicochemical environments with remarkable and unique microbial diversity resulting from periodic flood tides and high degrees of salinity change as well as temperature fluctuations (11). LipG and LipEH166 constituted new families of bacterial lipolytic enzymes and exhibited high activity and stability under alkaline conditions. In the present study, we describe a novel lipolytic gene from the metagenomic library constructed from the same tidal flat sediments. Although this enzyme was isolated on a lipase screening plate, it was found to exhibit high amino acid similarity to phospholipase A and to display both phospholipase and lipase activities. The novel phospholipase was expressed as a highly soluble form in Escherichia coli and then purified to homogeneity and characterized biochemically.
MATERIALS AND METHODS
Isolation and sequence analysis of lipolytic clone from the metagenomic library.
We extracted metagenomic DNA from the tidal flat sediments of the Saemankum area on the western coast of the Republic of Korea and constructed a library with approximately 386,400 Escherichia coli EPI300-T1 clones harboring recombinant pCC1FOS as described previously (14). Lipolytic clones were screened on an emulsified tricaprylin plate and were observed by a transparent halo around each colony. The recombinant fosmid showing lipolytic activity was finally isolated from an E. coli transformant and used for further analysis of the tidal flat metagenomic enzyme. The complete sequence of the lipolytic fosmid was determined by a shotgun sequencing method and deposited in GenBank. Open reading frames (ORFs) in the sequence were identified by ORF Finder at the National Center for Biotechnology Information (NCBI) website, and the sequence similarities of identified ORFs were analyzed using BLASTX (32). Only hits with E values of <0.01 were considered ORFs. The conserved structural and functional domains in protein sequences were analyzed using the NCBI Conserved Domain Database (CDD) (15). The amino acid sequence of the putative lipolytic enzyme was aligned with those of the homologous proteins by using CLUSTALW and the ESPript method (25). The percent identity was calculated and phylogenetic analysis performed using the MEGALIGN program, implemented in Lasergene software (DNAStar), and TreeView V3.2.
Expression and purification of recombinant lipolytic enzyme.
A putative catalytic domain encoding phospholipase A, mPlaG, was subcloned between the NdeI and XhoI sites in pET-22b(+) (Novagen), resulting in the plasmid pET-MPlaG. E. coli BL21(DE3) harboring pET-MPlaG was cultured at 18°C for 12 h with 0.5 mM isopropyl-β-d-1-thiogalactoside (IPTG) for induction of overexpression. For purification, Ni-nitrilotriacetic acid (Ni-NTA) resin (Qiagen) bound with the crude enzymes was washed with 50 mM Tris-HCl (pH 8.0) containing 5 mM imidazole, and the bound proteins were eluted with 50 mM Tris-HCl (pH 8.0) containing 250 mM imidazole and dialyzed.
Assay of lipolytic activity.
The activity of MPlaG was determined by pH titration and spectrophotometric methods at room temperature. The hydrolyzing activities of MPlaG toward triacylglycerides, olive oil, and phosphatidylcholine were measured by titrating the free fatty acids by use of a pH titrator (model 842 Titrando; Metrohm). After the pH of the substrate emulsion was adjusted to 8.0 by the addition of 10 mM NaOH solution, an appropriate amount of the enzyme solution was added. The release rate of the fatty acid was measured with a pH titrator for 5 min. One unit of lipase activity was defined as the amount of enzyme liberating 1 μmol of fatty acid per minute (16). MPlaG activity against p-nitrophenyl esters was measured by spectrophotometric detection according to a standard assay method. Unless otherwise indicated, p-nitrophenyl caprate (C10) was used as the substrate and 5 mM Ca2+ was added to the reaction solution. The enzyme activity was determined by continuously monitoring the product, p-nitrophenol, at 405 nm with a DU800 spectrophotometer (Beckman). One unit of activity with p-nitrophenyl esters was defined as the amount of enzyme that released 1 μmol of p-nitrophenol per minute (4). Control reactions were carried out without enzymes for every measurement under different conditions in order to subtract the value for the nonenzymatic hydrolysis of substrates.
LC-MS analysis of specificity toward phospholipids.
Positional and chain-length specificities of phospholipolytic MPlaG were determined by using 1-oleoyl-2-palmitoyl-phosphatidylcholine and phosphatidylcholine with various carbon chain lengths (C6, C7, C8, and C14), respectively, as substrates. Purified MPlaG was incubated with 1 mM substrate in 50 mM Tris buffer (pH 8.0) containing 5 mM CaCl2 and 150 mM NaCl for 12 h at 25°C with shaking. Liquid chromatography-mass spectrometry (LC-MS) was performed for analysis of reaction products, using a Finnigan LCQ Advantage Max ion-trap mass spectrometer (Thermo Fisher Scientific) equipped with an electrospray ionization (ESI) source. High-performance liquid chromatography (HPLC) separation was performed with a Kinetex HILIC column (2.6 μm × 2.1 mm × 100 mm; Phenomenex) and a hydrophilic interaction liquid chromatography (HILIC) guard column (4 × 2.0 mm; Phenomenex). Mobile phase A was 10 mM ammonium formate at pH 3.0, adjusted using formic acid, and mobile phase B was acetonitrile. The gradient elution was performed at a flow rate of 0.2 ml/min, as follows: 0 to 10 min, 10% to 40% mobile phase A (linear gradient); and 10 to 20 min, 70% mobile phase A (isocratic) (35). The column temperature was ambient, and the injection volume was 10 μl. The mass spectra were obtained with m/z values ranging from 100 to 1,200 in negative-ion mode, with 3 microscans and a maximum ion injection time of 200 ms.
Effects of pH and temperature on the novel lipolytic enzyme.
GTA buffer (100 mM 3,3-dimethylglutaric acid, 100 mM Tris, and 100 mM 2-amino-2-methyl-1,3-propanediol) was used for the enzyme assay. The pH for maximum activity of MPlaG was determined over a pH range of 6.0 to 12.0, using intervals of 1.0 pH unit. To determine pH stability, the enzyme was incubated at various pHs (4.0 to 12.0, at intervals of 1.0 pH unit) for 3 h, and the residual activity was measured. The optimum temperature of MPlaG was measured in pH 8.0 buffer over the range of 5 to 60°C. The melting temperature (Tm) was determined by differential scanning calorimetry (DSC) using VP-DSC (MicroCal). MPlaG was heated from 25 to 80°C in a DSC aluminum pan at a scan rate of 90°C/h. Before measurements were made, the sample was dialyzed against 50 mM Tris buffer (pH 8.0) with or without Ca2+. The dialysis buffer was used as a reference solution for DSC. A buffer-buffer reference scan was subtracted from each sample scan prior to concentration normalization. Baselines were created in Origin 7.0 (OriginLab).
Effects of metal ions, inhibitors, detergents, and organic solvents on the novel lipolytic enzyme.
The enzyme solution was preincubated with various concentrations of metal ions, enzyme inhibitors, detergents, and organic solvents in 50 mM Tris buffer (pH 8.0) for 3 h at 25°C. An adequate amount of the incubation mixture was withdrawn, and the residual enzyme activities were determined as described above.
Nucleotide sequence accession number.
The complete sequence of the lipolytic fosmid was deposited in the GenBank nucleotide sequence database under accession no. EU285670.
RESULTS
Isolation and annotation of metagenomic fosmid conferring lipolytic activity.
From the metagenomic library of the tidal flat sediments on the Korean west coast, we isolated a fosmid clone from an E. coli transformant displaying lipolytic activity on an emulsified tricaprylin plate. The fosmid clone (GenBank accession no. EU285670) had 28,845 bp of metagenomic DNA, contained a total of 15 ORFs with E values of <0.01 (based on ORF Finder at the NCBI website), and showed database hits with broad identity (22% to 88%) (Table 1). Three of the 15 ORFs, namely, ORFs 8, 13, and 15, were found to have a conserved transposase domain (Table 1). A number of proteins (46%) did not significantly match sequences in common protein databases such as Pfam (7) and COG (24), indicating that the parent organism of the 15 ORFs had a genetic makeup that was different from that of the cultured organisms available in the public databases.
Table 1.
Annotation of ORFs identified on pFosPlaG
| ORF | Length (aa) | % G+C | Most homologous protein | Putative source organism | % Identity/% similarity | E value |
|---|---|---|---|---|---|---|
| 1 | 291 | 36.87 | RNA polymerase sigma factor | Planctomyces maris DSM 8797 | 39/57 | 2e−53 |
| 2 | 565 | 44.94 | Phospholipase A | Grimontia hollisae CIP 101886 | 31/53 | 5e−06 |
| 3 | 334 | 47.56 | NADP-dependent oxidoreductase | Moritella sp. PE36 | 88/96 | 4e−173 |
| 4 | 208 | 44.34 | Hypoxanthine phosphoribosyltransferase | Desulfotalea psychrophila LSv54 | 71/89 | 4e−66 |
| 5 | 90 | 37.36 | Putative regulatory protein | Dictyoglomus thermophilum H-6-12 | 57/80 | 8e−08 |
| 6 | 160 | 33.54 | Hypothetical protein | Pelobacter carbinolicus DSM 2380 | 49/66 | 2e−26 |
| 7 | 328 | 36.58 | Amino acid ABC transporter periplasmic protein | Hahella chejuensis KCTC 2396 | 41/63 | 2e−47 |
| 8 | 274 | 40.48 | Putative transposase | Solibacter usitatus Ellin6076 | 39/58 | 3e−44 |
| 9 | 158 | 46.12 | Hypothetical protein | Methanosarcina barkeri Fusaro | 44/64 | 2e−37 |
| 10 | 107 | 38.58 | Hypothetical protein | Chlorobium chlorochromatii CaD3 | 61/75 | 4e−36 |
| 11 | 1,095 | 35.92 | Transcriptional regulator | Bacillus sp. SG-1 | 22/44 | 5e−57 |
| 12 | 253 | 39.76 | NAD(P)H dehydrogenase | Desulfatibacillum alkenivorans AK-01 | 37/57 | 4e−43 |
| 13 | 114 | 31.59 | Transposase | Pseudomonas aeruginosa | 28/52 | 1e−2 |
| 14 | 435 | 34.79 | Hypothetical exported 24-amino-acid-repeat protein | Fusobacterium nucleatum subsp. vinventii ATCC 49256 | 32/49 | 2e−44 |
| 15 | 308 | 35.38 | Transposase | Marinobacter sp. ELB17 | 25/47 | 2e−11 |
Sequence and phylogenetic analyses of metagenome-derived lipolytic enzyme.
Among all of the ORFs, an ORF encoding a putative phospholipase A of 565 amino acids, with a deduced molecular mass of 61,186 Da, was found. A BLAST search against the sequence databases at GenBank found that only the C-terminal 202 residues showed the highest similarity (31% identity) to phospholipase A (GenBank accession no. ZP_06052044) from Grimontia hollisae CIP 101886. Additionally, the N-terminal 26 residues were predicted to be a signal peptide by SignalP 3.0 (2). The amino acid sequence from Ala27 to Gly157 was repeated after amino acids Thr158 to Gly287 as determined by analysis using the TRUST tandem repeat detection tool (23). These results indicate that the ORF should consist of two domains: an unknown functional domain and a catalytic domain, comprising 287 amino acids of the N-terminal region and 278 amino acids of the C-terminal region, respectively. The putative catalytic domain, starting at Leu288, was designated MPlaG. The Ser435-Asp496-His550 catalytic triad, which is typical of the α/β-hydrolase superfamily, was predicted by multiple sequence alignment with similar phospholipases retrieved from the GenBank database (Fig. 1A) (3). As a catalytic nucleophile, the Ser435 residue was part of a G-X-S-X-G motif considered to be one of the identifying features of α/β-hydrolases (28). In addition, residues in the vicinity of this common motif corresponded well to the characteristic amino acid pattern of phospholipase A1: [LIV]-{KG}-[LIVFY]-[LIVMST]-G-[HYWV]-S-{YAG}-G-[GSTAC] (where brackets indicate alternative amino acids and braces indicate excluded amino acids) (19). Meanwhile, a phylogenetic tree was established from the amino acid sequences of MPlaG and the homologous proteins (Fig. 1B). MPlaG and its phospholipase homologues were compared with lipase and phospholipase families, and they did not belong to any known lipase family and were more phylogenetically related to the bacterial phospholipase A1 family.
Fig 1.

Multiple sequence alignments and phylogenetic analysis of the tidal flat metagenome-derived MPlaG enzyme and closely related proteins. (A) The GenBank accession numbers of the aligned sequences are as follows: ABY56067, MPlaG, a tidal flat metagenome-derived protein; AAD10476, phospholipase A1 from Serratia sp. MK1; AAM13978, phospholipase from Serratia marcescens; YP_001005338, phospholipase A from Yersinia enterocolitica 8081; and YP_001479905, phospholipase A1 from Serratia proteamaculans 568. The black shading represents conserved amino acids. Stars indicate the residues involved in the catalytic triad (Ser-Asp-His). The consensus amino acid pattern around the catalytic serine residue of phospholipase A1 is indicated by underlining. The amino acid sequences of MPlaG and homologous proteins were aligned using the CLUSTALW and ESPript methods. (B) Phylogenetic analysis was performed with MPlaG, phospholipases, and various lipases/esterases in eight families of bacterial lipolytic enzymes. The phylogenetic tree was constructed using the program MEGALIGN. The bar represents the number of amino acid substitution events.
Purification and characterization of metagenome-derived MPlaG.
The heterologously expressed MPlaG enzyme was purified by a single step of Ni-NTA affinity chromatography (see Fig. S1 in the supplemental material). The purified MPlaG enzyme exhibited a specific activity of 169.4 ± 1.8 U mg−1 when p-nitrophenyl palmitate was used as the substrate. Subsequently, purified MPlaG was used for biochemical characterization.
We determined the effects of pH and temperature on the activity of MPlaG and found that MPlaG displayed characteristics of a cold-adapted alkaline enzyme. When the purified enzyme was assayed for the hydrolysis of p-nitrophenyl caprate, it exhibited maximum activities at pH 8.0 (Fig. 2A) and 25°C (Fig. 2B). The activity of MPlaG retained 39% of the maximum at 5°C and drastically decreased at temperatures above the apparent optimum temperature. The effect of pH on the enzymatic stability of MPlaG was also determined by assaying the remaining activity after preincubation of the purified enzyme. MPlaG was remarkably stable (>90% of activity remaining) over a broad pH range (pH 5.0 to 10.0). These results indicate that MPlaG is a cold-adapted alkaline enzyme.
Fig 2.
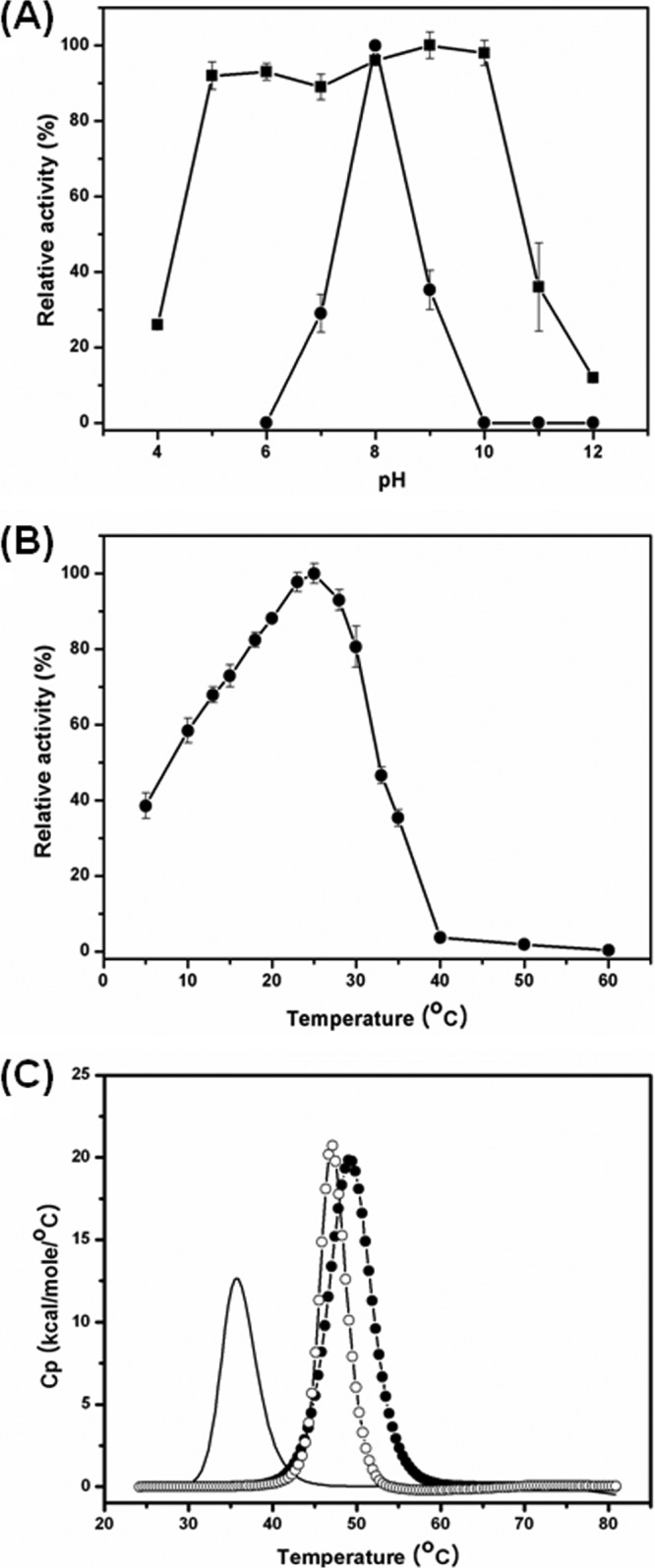
Effects of pH, temperature, and Ca2+ ions on MPlaG activity. Lipase activity was measured by a spectrophotometric method using p-nitrophenyl caprate (C10) and GTA buffer. (A) To determine pH stability, MPlaG was incubated at various pHs (4.0 to 12.0) for 3 h, and the residual activities were measured. Optimum pH and pH stability are indicated by circles and squares, respectively. (B) The optimum temperature of MPlaG within the range of 5 to 60°C was determined in pH 8.0 buffer. All measurements were repeated three times to confirm reproducibility. The values are shown as relative activities, with a maximum value of 100%. (C) The effect of Ca2+ on the temperature of MPlaG thermal unfolding (Tm) was determined by DSC. The Tm without CaCl2 is indicated by a solid line, that with 2 mM CaCl2 is indicated by open circles, and that with 5 mM CaCl2 is indicated by solid circles.
The effects of divalent metal ions on the activity of MPlaG were examined at different concentrations of each ion. Over the range of divalent metal ion concentrations tested, Ca2+ ions drastically increased the activity of MPlaG (10-fold), whereas Cu2+, Zn2+, and Ni2+ ions inhibited the activity and Co2+, Mg2+, and Fe2+ ions somewhat increased the activity (see Table S1 in the supplemental material). In addition, the enzyme was strongly inhibited by EDTA, a chelator of divalent cations: no activity was detected in the presence of 5 mM EDTA. The thermal unfolding of MPlaG was observed by DSC in the absence and presence of Ca2+. The melting temperatures of MPlaG in 2 mM and 5 mM Ca2+ were 47.2°C and 49.2°C, respectively. Thus, 5 mM Ca2+ caused a remarkable 10.7°C shift in melting temperature compared to the melting temperature of 38.5°C without Ca2+ (Fig. 2C). These results indicate that the structural stability of MPlaG is strongly dependent on the presence of Ca2+. In fact, the bacterial production and catalytic activity of several lipases are enhanced by Ca2+ (5, 17). The increased lipase activity in the presence of Ca2+ may be explained mainly by the stabilizing effect of enzyme-bound Ca2+ on the transient enzyme-substrate complex (13). Also, for MPlaG, Ca2+ should have a structural rather than a catalytic role. In order to investigate the organic solvent tolerance of MPlaG, various organic solvents were added to the enzyme solution, at concentrations of 20, 30, 50, and 60% (vol/vol). Over the range of concentrations tested, MPlaG showed a remarkable stability in the presence of most of the water-miscible organic solvents, with more than 80% of the original activity, but not against water-immiscible organic solvents such as butanol and ethyl acetate (see Table S2 in the supplemental material). Additionally, MPlaG activity was inhibited by concentrations of more than 1.0% (vol/vol) of nonionic and ionic detergents (see Table S3 in the supplemental material).
Substrate specificity of MPlaG.
Because MPlaG showed a relatively high amino acid similarity to phospholipase A in the BLAST search, despite being isolated on an emulsified tricaprylin plate, lipase and phospholipase activities were determined preliminarily by applying the purified MPlaG to preformed holes on emulsified tricaprylin and phosphatidylcholine agar plates, respectively. MPlaG formed transparent clear zones around the holes on both plates, indicating that MPlaG hydrolyzes both lipids and phospholipids (data not shown). For the quantitative comparison of phospholipase and lipase activities, the specific activities of MPlaG toward olive oil and phosphatidylcholine from egg yolk were determined by the pH-stat method and were found to be 2,957 ± 144 and 1,735 ± 147 U mg−1, respectively. MPlaG is homologous to phospholipases but displays remarkable lipase activity against olive oil.
To investigate the substrate preference further, the purified enzyme activity was assayed for the hydrolysis of p-nitrophenyl esters, triglycerides, and phosphatidylcholine of different chain lengths (Fig. 3). The results of the pH titration assay against triglycerides showed the highest activity toward tributyrin (C4), and enzymatic activity dramatically decreased with increasing chain length (Fig. 3A). However, in a spectrophotometric assay using p-nitrophenyl esters, MPlaG was most active toward p-nitrophenyl palmitate (C16) (approximately 112-fold), followed by p-nitrophenyl butyrate (C4) (Fig. 3B). Specificity of MPlaG toward phosphatidylcholine molecules with 6-, 7-, 8-, and 14-carbon chain lengths was analyzed by LC-MS (Fig. 3C). Since the mobile phase for LC-MS analysis contained ammonium formate and formic acid, a characteristic mass spectrum showed a deprotonated molecular ion ([M − H]−) and solvent adduct ions such as [M − H + HCOOH]− in the negative-ion mode. The catalytic activity for hydrolysis of phosphatidylcholine to lysophosphatidylcholine was the highest for 1,2-dioctanoyl-phosphatidylcholine (diC8PC) (Fig. 3C). Therefore, MPlaG had diverse chain length specificities according to substrate, showing a high preference for p-nitrophenyl esters with long acyl chains, triglycerides with short acyl chains, and phospholipids with medium acyl chains. The purified enzyme displayed interfacial activity when we measured the hydrolytic activity with increasing concentrations of tributyrin and hydrolyzed triolein (C18:1), which cannot be hydrolyzed by esterases, indicating that MPlaG is not an esterase (Fig. 3A; see Fig. S2 in the supplemental material). Additionally, the site of hydrolysis of MPlaG was determined by LC-MS analysis using 1-oleoyl-2-palmitoyl-phosphatidylcholine (OPPC) (Fig. 4). After incubation of MPlaG with OPPC (molecular weight of 759) at 25°C for 12 h, the LC-MS spectrum revealed that MPlaG degraded OPPC (m/z 804; [M − H + HCOOH]−) to a by-product of m/z 540. If MPlaG cleaved a palmitic acid at the sn-2 position of OPPC, the by-product would be observed at m/z 567 ([M − H + HCOOH-C16:0]−). However, since the MS spectrum detected m/z 540 ([M − H + HCOOH-C18:1]−), corresponding to 2-palmitoyl-lysophosphatidylcholine, MPlaG was identified as a phospholipase A1 that is able to catalyze hydrolysis of the acyl group from the sn-1 position of phospholipids.
Fig 3.

Substrate specificity of MPlaG. The chain length selectivities of MPlaG toward triglycerides (A), p-nitrophenyl esters (B), and phosphatidylcholine (C) were determined by using pH titration, spectrophotometry, and LC-MS, respectively. In panels A and B, the vertical and horizontal axes indicate substrate carbon chain lengths and relative activities (with a maximum value of 100%), respectively. (C) For LC-MS analysis, reaction products obtained from reaction of MPlaG with phosphatidylcholine substrates were separated by HPLC at the indicated retention times: for diC6PC, 8.77 min (m/z 498 of 1,2-dihexanoyl-phosphatidylcholine; 89.6%) and 10.36 min (m/z 400 of 2-hexanoyl-lysophosphatidylcholine; 10.4%); for diC7PC, 8.40 min (m/z 526 of 1,2-diheptanoyl-phosphatidylcholine; 50.5%) and 9.99 min (m/z 414 of 2-heptanoyl-lysophosphatidylcholine; 49.5%); for diC8PC, 8.14 min (m/z 554 of 1,2-dioctanoyl-phosphatidylcholine; 49.6%) and 9.73 min (m/z 428 of 2-octanoyl-lysophosphatidylcholine; 50.4%); and for diC14PC, 7.68 min (m/z 722 of 1,2-dimyristoyl-phosphatidylcholine; 96.5%) and 8.91 min (m/z 512 of 2-myristoyl-lysophosphatidylcholine; 3.5%).
Fig 4.
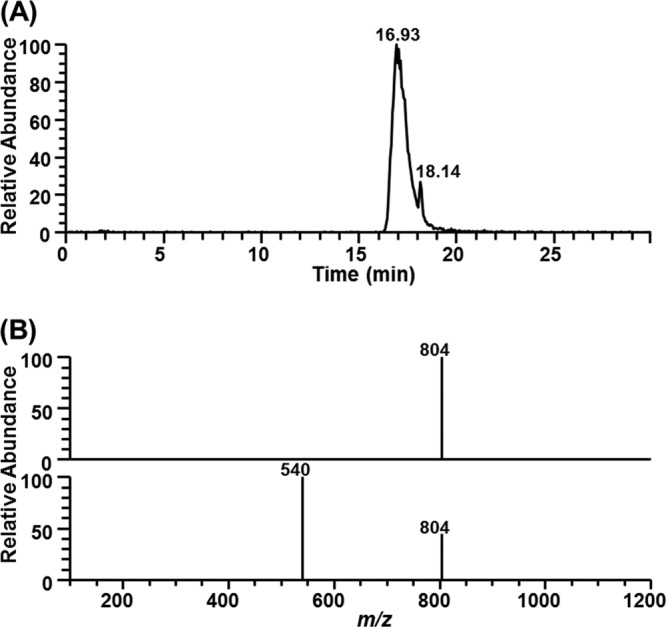
Identification of phospholipase A1 activity of MPlaG. Positional specificity of MPlaG toward OPPC (1-oleoyl-2-palmitoyl-phosphatidylcholine) was determined by LC-MS. (A) The reaction products of MPlaG were separated by HPLC at the indicated retention times, i.e., 16.93 min (m/z 804; OPPC) (upper part of panel B) and 18.14 min (m/z 540; 2-palmitoyl-lysophosphatidylcholine) (lower part of panel B). Gradient elution was performed as follows: 0 to 25 min, 2 to 38% mobile phase A (linear gradient); and 25 to 35 min, 70% mobile phase A (isocratic).
DISCUSSION
Phospholipase A is of particular interest for industrial applications because it yields lysophospholipids, which are excellent emulsifying agents for food, cosmetics, and pharmaceuticals (22). However, relatively few phospholipase A enzymes have been characterized, despite their apparent ubiquity and industrial demands. In this study, MPlaG, a novel lipolytic enzyme with unique properties, was isolated from a metagenomic library obtained from tidal flat sediments. Although the specific activity of MPlaG revealed that lipids rather than phospholipids were preferable substrates for the enzyme, the amino acid similarity to phospholipase A from Grimontia hollisae CIP 101886 and the finding of phospholipase activity of MPlaG indicated that it was a phospholipase rather than a lipase. A phylogenetic tree was established from the amino acid sequences of MPlaG and homologous proteins for comparison with other lipase and phospholipase families (Fig. 1B). Judging from eight lipase families containing staphylococcal lipase family I.6, MPlaG and its phospholipase homologues formed a phylogenetically distinct group that does not belong to the known lipase families and is distant from the staphylococcal lipase family, including SHL (Fig. 1B). As a result, MPlaG is evolutionarily closer to the bacterial phospholipase A1 family, in agreement with the experimental results identifying the phospholipase A1 activity of MPlaG by LC-MS analysis using OPPC as the substrate.
Previously, staphylococcal lipases from Staphylococcus aureus, Staphylococcus epidermidis, Staphylococcus haemolyticus, Staphylococcus xylosus, Staphylococcus hyicus, and Staphylococcus warneri were reported to form a unique lipase family. Among the staphylococcal lipases characterized so far, S. hyicus lipase (SHL) has considerably higher phospholipase than lipase activity, which distinguishes the enzyme not only from other staphylococcal lipases but also from all bacterial lipases. Moreover, SHL shows low chain length specificity and readily hydrolyzes short-, medium-, and long-chain triacylglycerols as well as p-nitrophenyl esters, irrespective of their chain length (20). In addition, SHL is dependent on Ca2+, which is considered to have a function in stabilizing the tertiary structure rather than in the catalytic reaction (26). Although MPlaG and SHL share similar biochemical characteristics, the most important difference is in their primary structures: MPlaG was shown to have relatively low identity (8.2%) with the mature domain of SHL, and even catalytic active site residues were not aligned with each other (data not shown). Thus, based on amino acid sequence similarity, SHL is a lipase and MPlaG is a phospholipase. Furthermore, with respect to substrate specificity, SHL is a phospholipid-preferred lipase, whereas MPlaG is a lipid-preferred phospholipase.
Recently, structure-function relationships of SHL and GPLRP2 with regard to phospholipase activity were elucidated (26, 33). The activity of SHL toward phospholipids comes from a polar acyl binding pocket, whereas the phospholipase activity of GPLRP2 is based on the short lid domain. MPlaG should have a lid domain, considering its interfacial activation. Therefore, MPlaG may possess a substrate binding pocket similar to that of SHL; at least one of three hydrophobic pockets for acyl chains in the active site of MPlaG may be shallower and/or more polar than the others, and another pocket may be more extended and/or flexible, permitting the binding of diverse chains.
In conclusion, the tidal flat metagenome-derived MPlaG enzyme is the first experimentally confirmed phospholipase A1 with lipase activity. The novel MPlaG was identified as having dependence on Ca2+, cold-adapted alkaline properties, and remarkable stability against organic solvents. The sequence of MPlaG has higher homology with phospholipases A than with other lipases and is phylogenetically close to the phospholipase A1 family. However, the experimental results revealed that MPlaG is a phospholipase A1 that has considerably higher lipase than phospholipase activity. When phospholipases evolve from lipases and vice versa, MPlaG may be an intermediate that is closer to phospholipases in the evolutionary relationships between lipases and phospholipases. Finally, further investigations on the substrate specificities of and relationships between lipases and phospholipases through X-ray crystallography and mutagenesis studies are believed to be necessary and will give us more detailed information on this promising enzyme.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jae Kwang Song (Korea Research Institute of Chemical Technology) for useful comments regarding the experiments.
This work was supported by the 21C Frontier Program of Microbial Genomics and Applications (grant 11-2008-00-002-00) from the Ministry of Education, Science and Technology (MEST) of the Republic of Korea.
Published ahead of print 27 April 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Abbai NS, et al. 2011. A deep gold mine metagenome as a source of novel esterases. Afr. J. Biotechnol. 10: 6090–6100 [Google Scholar]
- 2.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340: 783–795 [DOI] [PubMed] [Google Scholar]
- 3.Bugg TD. 2004. Diverse catalytic activities in the αβ-hydrolase family of enzymes: activation of H2O, HCN, H2O2, and O2. Bioorg. Chem. 32: 367–375 [DOI] [PubMed] [Google Scholar]
- 4.Bulow L, Mosbach K. 1987. The expression in E. coli of a polymeric gene coding for an esterase mimic catalyzing the hydrolysis of p-nitrophenyl esters. FEBS Lett. 210: 147–152 [DOI] [PubMed] [Google Scholar]
- 5.Castro-Ochoa LD, Rodríguez-Gómez C, Valerio-Alfaro G, Ros RO. 2005. Screening, purification and characterization of the thermoalkalophilic lipase produced by Bacillus thermoleovorans CCR11. Enzyme Microb. Technol. 37: 648–654 [Google Scholar]
- 6.De Maria L, Oxenbøll VJKM, Svendsen A, Patkar S. 2007. Phospholipases and their industrial applications. Appl. Microbiol. Biotechnol. 74: 290–300 [DOI] [PubMed] [Google Scholar]
- 7.Finn RD, et al. 2008. The Pfam protein families database. Nucleic Acids Res. 36: D281–D288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heath C, Hu XP, Cary SC, Cowan D. 2009. Identification of a novel alkaliphilic esterase active at low temperatures by screening a metagenomic library from Antarctic desert soil. Appl. Environ. Microbiol. 75: 4657–4659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaeger KE, Eggert T. 2002. Lipases for biotechnology. Curr. Opin. Biotechnol. 13: 390–397 [DOI] [PubMed] [Google Scholar]
- 10.Jeon JH, et al. 2009. Cloning and characterization of a new cold-active lipase from a deep-sea sediment metagenome. Appl. Microbiol. Biotechnol. 81: 865–874 [DOI] [PubMed] [Google Scholar]
- 11.Kim BS, Oh HM, Kang H, Chun J. 2005. Archaeal diversity in tidal flat sediment as revealed by 16S rDNA analysis. J. Microbiol. 43: 144–151 [PubMed] [Google Scholar]
- 12.Kim EY, et al. 2009. Novel cold-adapted alkaline lipase from an intertidal flat metagenome and proposal for a new family of bacterial lipases. Appl. Environ. Microbiol. 75: 257–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim MH, Kim HK, Lee JK, Park SY, Oh TK. 2000. Thermostable lipase of Bacillus stearothermophilus: high-level production, purification, and calcium-dependent thermostability. Biosci. Biotechnol. Biochem. 64: 280–286 [DOI] [PubMed] [Google Scholar]
- 14.Lee MH, Lee CH, Oh TK, Song JK, Yoon JH. 2006. Isolation and characterization of a novel lipase from a metagenomic library of tidal flat sediments: evidence for a new family of bacterial lipases. Appl. Environ. Microbiol. 72: 7406–7409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marchler-Bauer A, et al. 2005. CDD: a conserved domain database for protein classification. Nucleic Acids Res. 33: D192–D196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peled N, Krenz MC. 1981. A new assay of microbial lipases with emulsified trioleoyl glycerol. Anal. Biochem. 112: 219–222 [DOI] [PubMed] [Google Scholar]
- 17.Rathi P, Saxena RK, Gupta R. 2001. A novel alkaline lipase from Burkholderia cepacia for detergent formulation. Process Biochem. 37: 187–192 [Google Scholar]
- 18.Rhee JK, Ahn DG, Kim YG, Oh JW. 2005. New thermophilic and thermostable esterase with sequence similarity to the hormone-sensitive lipase family, cloned from a metagenomic library. Appl. Environ. Microbiol. 71: 817–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richmond GS, Smith TK. 2011. Phospholipases a1. Int. J. Mol. Sci. 12: 588–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simons JW, et al. 1996. The lipase from Staphylococcus aureus. Expression in Escherichia coli, large-scale purification and comparison of substrate specificity to Staphylococcus hyicus lipase. Eur. J. Biochem. 242: 760–769 [DOI] [PubMed] [Google Scholar]
- 21.Sitkiewicz I, Stockbauer KE, Musser JM. 2007. Secreted bacterial phospholipase A2 enzymes: better living through phospholipolysis. Trends Microbiol. 15: 63–69 [DOI] [PubMed] [Google Scholar]
- 22.Song JK, Kim MK, Rhee JS. 1999. Cloning and expression of the gene encoding phospholipase A1 from Serratia sp. MK1 in Escherichia coli. J. Biotechnol. 72: 103–114 [DOI] [PubMed] [Google Scholar]
- 23.Szklarczyk R, Heringa J. 2004. Tracking repeats using significance and transitivity. Bioinformatics 20(Suppl 1): i311–i317 [DOI] [PubMed] [Google Scholar]
- 24.Tatusov RL, Galperin MY, Natale DA, Koonin EV. 2000. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28: 33–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tiesinga JJ, van Pouderoyen G, Nardini M, Ransac S, Dijkstra BW. 2007. Structural basis of phospholipase activity of Staphylococcus hyicus lipase. J. Mol. Biol. 371: 447–456 [DOI] [PubMed] [Google Scholar]
- 27.Tirawongsaroj P, et al. 2008. Novel thermophilic and thermostable lipolytic enzymes from a Thailand hot spring metagenomic library. J. Biotechnol. 133: 42–49 [DOI] [PubMed] [Google Scholar]
- 28.Upton C, Buckley JT. 1995. A new family of lipolytic enzymes? Trends Biochem. Sci. 20: 178–179 [DOI] [PubMed] [Google Scholar]
- 29.van Kampen MD, Simons JW, Dekker N, Egmond MR, Verheij HM. 1998. The phospholipase activity of Staphylococcus hyicus lipase strongly depends on a single Ser to Val mutation. Chem. Phys. Lipids 93: 39–45 [DOI] [PubMed] [Google Scholar]
- 30.van Kampen MD, Verheij HM, Egmond MR. 1999. Modifying the substrate specificity of staphylococcal lipases. Biochemistry 38: 9524–9532 [DOI] [PubMed] [Google Scholar]
- 31.Wei P, Bai L, Song W, Hao G. 2009. Characterization of two soil metagenome-derived lipases with high specificity for p-nitrophenyl palmitate. Arch. Microbiol. 191: 233–240 [DOI] [PubMed] [Google Scholar]
- 32.Wheeler DL, et al. 2003. Database resources of the National Center for Biotechnology. Nucleic Acids Res. 31: 28–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Withers-Martinez C, Carriere F, Verger R, Bourgeois D, Cambillau C. 1996. A pancreatic lipase with a phospholipase A1 activity: crystal structure of a chimeric pancreatic lipase-related protein 2 from guinea pig. Structure 4: 1363–1374 [DOI] [PubMed] [Google Scholar]
- 34.Wong H, Schotz MC. 2002. The lipase gene family. J. Lipid Res. 43: 993–999 [DOI] [PubMed] [Google Scholar]
- 35.Zhao YY, Xiong Y, Curtis JM. 2011. Measurement of phospholipids by hydrophilic interaction liquid chromatography coupled to tandem mass spectrometry: the determination of choline containing compounds in foods. J. Chromatogr. A 1218: 5470–5479 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.