
Fig 2.
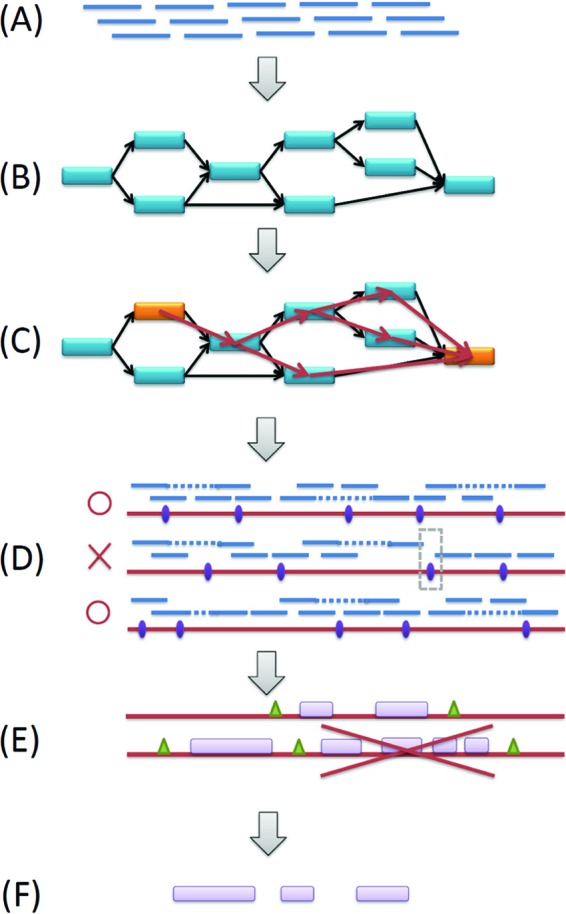
A diagram of the constrained assembly approach. (A) Paired-end and singleton reads from a metagenomic data set. (B) Assembly of all reads using SOAPdenovo, to generate contigs and a de Bruijn graph that connects the contigs. (C) Identification of contigs that consist of integron recombination repeats (shown as orange bars) and search for paths that start and end at a contig with repeats, using a depth-first search algorithm. At any intermediate node, the process will sort the coverage of all contigs connected by its outgoing edges and begin searching from the highest one. The starting and ending contig could be the same contig. (D) Validation of the assembled sequences (the paths) by read mapping and discarding of the paths that are not supported by reads (e.g., the middle sequence in the figure is discarded). (E) Identification of the integron repeats and their exact locations in the assembled sequences. Prediction of genes using FragGeneScan. Output sequences are between two repeats (attC sites) and consist of three or fewer genes. (F) Retrieval of the genes from sequences that pass all criteria.