

Abstract
Background
Pin1-type parvulins are phosphorylation-dependent peptidyl-prolyl cis-trans isomerases. Their functions have been widely reported to be involved in a variety of cellular responses or processes, such as cell division, transcription, and apoptosis, as well as in human diseases including Alzheimer's disease and cancers. TbPin1 was identified as a novel class of Pin1-type parvulins from Trypanosoma brucei, containing a unique PPIase domain, which can catalyze the isomerization of phosphorylated Ser/Thr-Pro peptide bond.
Methodology/Principal Findings
We determined the solution structure of TbPin1 and performed 15N relaxation measurements to analyze its backbone dynamics using multi-dimensional heteronuclear NMR spectroscopy. The average RMSD values of the 20 lowest energy structures are 0.50±0.05 Å for backbone heavy atoms and 0.85±0.08 Å for all heavy atoms. TbPin1 adopts the typical catalytic tertiary structure of Pin1-type parvulins, which comprises a globular fold with a four-stranded anti-parallel β-sheet core surrounded by three α-helices and one 310-helix. The global structure of TbPin1 is relatively rigid except the active site. The 2D EXSY spectra illustrate that TbPin1 possesses a phosphorylation-dependent PPIase activity. The binding sites of TbPin1 for a phosphorylated peptide substrate {SSYFSG[p]TPLEDDSD} were determined by the chemical shift perturbation approach. Residues Ser15, Arg18, Asn19, Val21, Ser22, Val32, Gly66, Ser67, Met83, Asp105 and Gly107 are involved in substantial contact with the substrate.
Conclusions/Significance
The solution structure of TbPin1 and the binding sites of the phosphorylated peptide substrate on TbPin1 were determined. The work is helpful for further understanding the molecular basis of the substrate specificity for Pin1-type parvulin family and enzyme catalysis.
Introduction
The peptidyl-prolyl bond can exist in two distinct cis or trans conformations, due to the unique side chain of proline, which in turn affects an overall protein structure. When the peptide bond switches between cis-trans conformation, protein structures will remodel accordingly, generating functional proteins [1]–[5], although the structural interconversion between the cis and trans conformations is substantially slow and a rate-limiting step. Interestingly, a family of peptidyl-prolyl cis-trans isomerases (PPIases) was identified, which all possessed a PPIase activity and could catalyze the intrinsically slow cis-trans isomerization [2], [5], [6]. So far, the PPIases family is suggested to be divided into three structurally distinct subfamilies: cyclophilins (Cyps), FK506-binding proteins (FKBPs), parvulins [2], [5], [7], [8].
In terms of substrate specificity, the parvulin subfamily can be further divided into the Pin1-type parvulins (phosphorylation-dependent) and the non Pin1-type parvulins (phosphorylation-independent) [2], [8]–[12]. The Pin1-type parvulins specifically catalyze the cis-trans isomerization of either phosphoserine- or phosphothreonine-proline (pSer/pThr-Pro) peptide bond. The phosphorylation-dependent isomerization is unique among all PPIases [13]. The phosphorylation specificity indicates that Pin1 plays an important role in the regulation of proline-directed phosphorylation associated signaling pathways [14], [15] [15]. For example, protein kinases such as MAP kinase (MAPK) and CDK2 specifically phosphorylate the trans conformation of Ser/Thr-Pro peptide bond [16], [17]. Phosphorylation of the substrates would further slow down the cis-trans interconversion rate. However, Pin1-type parvulins could accelerate the interconversion process. Evidences for the biological importance of the Pin1-type parvulins have been elucidated [3], [17]–[24].
Based on the structure, two distinct classes of Pin1-type parvulins have been identified in various organisms. Most of Pin1-type parvulins such as human Pin1 (hPin1), yeast ESS1/PTF1, and Drosophila Dodo, consist of two domains: an N-terminal WW domain and a conserved C-terminal catalytic PPIase domain. The WW domain is a binding module that specifically recognizes pSer/pThr-Pro motifs, while the C-terminal PPIase domain catalyzes the isomerization of pSer/pThr-Pro prolyl-peptide bonds [25]–[28]. However, several Pin1-type parvulins lacking of the WW domain have been reported, such as plant Pin1s [29]. Recently, two new members of the parvulin subfamily, TbPin1 and TbPar42, were identified from Trypanosoma brucei [10], [30]. Like plant Pin1s, TbPin1 lacks the N-terminal WW domain and only contains the catalytic PPIase domain. Instead of a WW domain, TbPar42 contains a forkhead-associated (FHA) domain at its N-terminal and a well conserved PPIase domain at C-terminal. TbPin1 has an ability to compensate for the loss of Ess1 function in yeast but TbPar42 lacks the ability [30], indicating that TbPin1 is a Pin1-type parvulin and TbPar42 is a non Pin1-type parvulin. Furthermore, studies of their subcellular localization in Trypanosoma brucei showed that TbPin1 was uniformly distributed in the cytoplasm, while TbPar42 was localized in the nucleus [30]. On the other hand, previous studies demonstrated that hPin1 was localized in both the nucleus and cytoplasm, and that its nuclear localization was due to the interaction of the WW domain with the target protein [20], [24], [31]. Thus, it is speculated that the exclusion of TbPin1 from the nucleus might be due to its lack of the WW domain. However, studies on plant Pin1s (such as DlPar13 and LjPar1) have shown that despite lacking the WW domain, plant Pin1s could still be localized in the cytoplasm and nucleus [26], [32]. These studies implicated that other structural or physiological conditions might play crucial roles in the specific localization of parvulins, and that TbPin1 potentially have some functions different from its homologues in other species.
The detailed structural interpretations of parvulins provide valuable information for addressing the functions of parvulins. So far, the three-dimensional structure of TbPin1 has not been interpreted and characterized. In the present work, we determined the solution structure and dynamics of TbPin1, performed the PPIase activity analysis and chemical shift perturbation for TbPin1 using NMR spectroscopy.
Results
Sequence alignment of TbPin1 with selected members of the parvulin family
To understand the primary sequence relationship, the amino acid sequences of TbPin1 (Swiss-Prot ID: Q57YG1) and the PPIase domain of TbPar42 (Q57XM6) were aligned with those of selected Pin1-type parvulins including Pin1At (Q9SL42) from A. thaliana; hPin1 (Q13526) from H. sapiens and CaEss1 (G1UA02) from C. albicans, and some non Pin1-type parvulins including EcPar10 (P0A9L5) from E. coli; CsPinA (P60747) from C. symbiosum; hPar10 (Q13526) from H. sapiens; PrsA-PPIase domain (P60747) from S. aureus. Furthermore, the available structures of the parvulins were also aligned onto the structure of TbPin1 using DaliLite [33]. Sequence alignment shows that the catalytic PPIase domain is well conserved. The subfamilies of parvulins differ in length and composition of the β1/α1 loop (Figure 1). In Pin1-type parvulins, the loop is considered the phosphate binding loop containing several positively charged residues (Lys63, Arg68 and Arg69 in hPin1) [25]. It is thereby expected that the loop could induce the preference for substrates with a negatively charged residue especially the phospho-Ser/Thr [25], [34]. In non Pin1-type parvulins, the β1/α1 loop is mostly missed or shorter than that in Pin1-type parvulins [34]–[38], such as EcPar10, CsPinA, hPar14 and PrsA-PPIase domain (Figure 1). However, the loop in TbPar42 is longer than those in other parvulins, which also contains a high number of positively charged residues (Lys276, Arg280 and Arg281) similar to those in Pin1-type parvulins. Thus, the role of the β1/α1 loop both in Pin1-type parvulins and in non Pin1-type parvulins is in need of further investigation.
Figure 1. Sequence alignment of TbPin1 with other parvulins.

The following structures and protein sequences were obtained from PDB and SwissProt: TbPin1 (Q57YG1); TbPar42 (Q57XM6); 1J6Y, Pin1At (Q9SL42); 1PIN, hPin1 (Q13526); 1YW5, CaEss1 (G1UA02); 1JNT, EcPar10 (P0A9L5); 2RQS, CsPinA (O74049); 3UI4, hPar14 (Q9Y237); 2JZV, PrsA (P60747). The sequences of parvulins were aligned with that of TbPin1 and the available tertiary structures of parvulins were also aligned onto the structure of TbPin1 using DaliLite [33]. DSSP information for helix (H) and strand (E) conformations from the PDB files is given in light gray below the protein sequences except TbPar42. Aligned residues are written as capital letters and the most frequent residues are colored in each column. Residues considered crucial for the PPIase activity are highlighted in yellow.
Chemical Shift Assignments of TbPin1
Sequence-specific assignments of backbone 1H, 13C and 15N resonances of TbPin1 were obtained based on a suite of 3D heteronuclear NMR spectra including HNCACB, CBCA(CO)NH, HNCA, HN(CO)CA, HNCO, HN(CA)CO spectra. Side-chain assignments of 1H and 13C resonances of TbPin1 were achieved on the basis of 3D HBHA(CO)NH, H(CCH)(CO)NHTOCSY, (H)CC(CO)NHTOCSY, HCCH-TOCSY, CCHTOCSY and 15N-edited TOCSY spectra. More than 95.9% of the backbone assignments were obtained and about 85.7% of the side chain assignments were extracted by this procedure with the exception of signals from aromatic side chains and three residual residues (Gly-Ser-His) from the N-terminal extension. All assigned 1H, 13C and 15N chemical shifts of TbPin1 were deposited in the Biological Magnetic Resonances Bank (BMRB ID: 17918), and the assigned 1H-15N HSQC spectrum is shown in Figure 2.
Figure 2. Assigned 1H-15N HSQC spectrum of TbPin1 in 20 mM sodium phosphate buffer at pH 7.0.

The spectrum was recorded at 298 K on a Varian Unity Inova 600 MHz spectrometer. Resonance assignments of backbone amide groups are indicated by the residue type and number. The unlabeled resonances are the side chain amides. T25 and G72 are shown in negative resonances corresponding to the resonances aliased in the 15N dimension. Positive resonances are colored red and negative resonances are colored green.
NMR structures of TbPin1 in solution
Using the Aria2.2/CNS1.2 software [39], [40], we calculated the solution structure of TbPin1 with 1918 distance restraints (711 intra-residual, 453 sequential, 195 short-range, 103 medium-range and 456 long-range NOEs) derived from 3D 15N-edited and 13C-edited NOESY-HSQC spectra, and 187 backbone torsion angle restraints generated by TALOS+ [41]. A family of 200 structures was calculated according to the simulated annealing protocol. Then, 20 lowest energy structures were selected and further refined in water. The structural statistics of the final 20 lowest energy conformers are summarized in Table 1. There are no distance restraint violations greater than 0.5 Å and no torsion angle restraint violations above 5°in the 20 lowest energy structures (Figure 3A). Ramachandran plot analysis of the 20 lowest energy structures indicates 99.1% of residues are located in the allowed regions. The average RMSD values are 0.50±0.05 Å for backbone heavy atoms and 0.85±0.08 Å for all heavy atoms. The atomic coordinates of TbPin1 were deposited in the Protein Data Bank (PDB: 2LJ4). The three-dimensional structure of TbPin1 is a typical Pin1-type parvulin-fold consisting of a four-stranded anti-parallel β-sheet core (β1, residues 4–12; β2, 73–78; β3, 101–103; β4, 108–113), three α-helices (α1, 34–50; α2, 55–62; α3, 84–90) and one 310-helix (η1, 66–70) surrounding the core (Figure 3B).
Table 1. Structural statistic for the 20 lowest energy structures of TbPin1.
| Restraints for calculations | |
| Total NOE restraints used | 1918 |
| Intraresidue (|i–j| = 0) | 711 |
| Sequential (|i–j| = 1) | 453 |
| Short-range (2≤|i–j|≤3) | 195 |
| Medium-range (4≤ |i–j|≤5) | 103 |
| Long-range (|i–j|>5) | 456 |
| Hydrogen bond restraints | 35 |
| Dihedral angle restraints (Φ and Ψ) a | 187 |
Figure 3. Solution structure of TbPin1.

(A) Superimposed backbone traces of the 20 lowest energy structures in the structure ensemble; (B) Ribbon diagram of the mean structure showing the secondary structure elements. The structure was determined in 20 mM sodium phosphate buffer at pH 7.0. Structure visualization is generated using MOLMOL.
In TbPin1, hydrophobic residues (Leu74, Met82 and Phe86) and two histidine residues (His9 and His109) forming the hydrophobic core are located on the concave side of the β-sheet cluster. The histidine side chains adopt the very similar orientation observed in other parvulin structures. However, the imidazole moieties show different ring-flipping states in comparison with those in hPin1 (PDB: 1PIN, 1F8A) and the orientations reported in hPar14 and PrsA-PPIase domain [34], [42]. It may be due to that the limited resolution disallows an unequivocal assignment.
Superposition of backbone heavy atoms of PPIase domains of hPin1 (PDB: 1PIN), CaEss1 (PDB: 1YW5), AtPin1 (PDB: 1J6Y), EcPar10 (PDB: 1JNT) and PrsA (PDB: 2JZV) with TbPin1 show RMSD values of 1.6 Å, 1.8 Å, 2.6 Å, 2.4 Å, 1.7 Å, respectively (Figure 4). They display a well conserved PPIase domain. In the solution structure of TbPin1, the β1/α1 loop (residues 14–33) exhibits a folded conformation, which has also been observed in hPin1 (PDB: 1PIN), Pin1At (PDB: 1J6Y) and CaEss1 (PDB: 1YW5) (Figure 4). In the crystal structure of hPin1 (PDB: 1PIN), a sulfate ion was found to be bound with the β1/α1 loop [25] (Figure 4A). In the solution structure of Pin1At, sodium sulfate salt was employed to stabilize the β1/α1 loop [43], and a buffer contained phosphate was used to crystallize CaEss1 [27]. Moreover, Daum et al. suggested the phosphate group in the buffer could be bound to the phosphate binding pocket [44]. On the other hand, the β1/α1 loop in the crystal structure of hPin1 (PDB: 1F8A) shows an unfolded conformation (Figure 4B), in which neither phosphate nor sulfate was present [45]. Therefore, a phosphate group would be bound to the phosphate binding pocket to induce the folded conformation of the β1/α1 loop in TbPin1, which was dissolved in 20 mM sodium phosphate buffer. In addition, structural alignment of the two non Pin1-type parvulins (PrsA-PPIase domain and EcPar10) with TbPin1 demonstrates that the β1/α1 loop of PrsA-PPIase domain is shorter than that of TbPin1 (Figure 4E), and the loop of EcPar10 is totally absent (Figure 4F). Thus, this loop might be important for the phosphorylation-dependent cis-trans isomerization, and potentially serves for distinguishing Pin1-type parvulins from non Pin1-type parvulins.
Figure 4. Structural comparison of TbPin1 (2LJ4) with other parvulins.

(A) hPin1 in complex with the Ala-Pro dipeptide and a sulfate ion (1PIN); (B) hPin1 without a sulfate ion (1F8A); (C) CaEss1 (1YW5); (D) Pin1At (1J6Y); (E) PrsA-PPIase domain (2JZV); (F) EcPar10 (1JNT). The structures of TbPin1, hPin1, CaEss1, Pin1At, EcPar10 and PrsA-PPIase domain are colored green, cyan, blue, pink, lightblue and yellow, respectively. The β1/α1 loop of TbPin1 is displayed in red. The sulfate ion is used to mimic to the phosphate group.
Enzymes activity analysis
Based on a phosphorylated peptide {SSYFSG[p]TPLEDDSD} derived from the substrate of Pin1At, Agamous-like 24 (AGL24) [22], we performed isomerase activity assays in vitro for TbPin1 using 2D 1H-1H NMR spectra including EXSY (exchange spectroscopy) and TOCSY (total correlation spectroscopy) spectra. The pSer/pThr-Pro motif displays two distinct sets of 1H signals in the 2D TOCSY and 2D EXSY spectra. No cross-peaks between the cis and trans isomers could be observed in the 2D EXSY spectrum due to the slow exchange rate between the cis and trans conformations [22], [46], [47]. Thus, in the absence of TbPin1, no cross peaks were observed in the EXSY spectrum of the phosphorylated peptide (Figure 5A), indicating that the exchange between the cis and trans conformations was too slow to be detected on the NMR timescale. By contrast, in the presence of TbPin1, the proline isomerization rate of the phosphorylated peptide was greatly enhanced, and cross-peaks resulting from the conformational exchange were observed in the EXSY spectrum (Figure 5B). In the presence of the TbPin1-C65A mutant, no cross-peaks were detected (Figure 5C), hence the PPIase activity to the peptide was shut down by the mutation. Furthermore, as shown in Figure 5D, no cross-peaks were observed in the presence of the FHA-truncated TbPar42 (TbPPIase) mutant. In addition, no exchange peaks were observed for the non-phosphorylated peptide SSYFSGTPLEDDSD in the presence of and absence of TbPin1, or TbPPIase (Figure S1 in supporting information). These results suggest that TbPin1 is a Pin1-type parvulin possessing phosphorylation-dependent isomerase activity. Shut down the activity by mutation of Cys65 to Ala further confirms that the conserved Cys65 residue plays a crucial role in the PPIase activity of TbPin1, as observed for other Pin1-type PPIases [25], [29].
Figure 5. TbPin1 catalyzes the cis-trans isomerization of a phosphorylated peptide.

A selected region of the 2D-EXSY spectrum of the phosphorylated peptide SSYFSG[p]TPLEDDSD is displayed (A) in the absence of TbPin1; (B) in the presence of TbPin1; (C) in the presence of the TbPin1-C65A mutant; (D) in the presence of the FHA-truncated TbPar42 mutant (TbPPIase). A mixing time of 300 ms was used. The cis and trans T7-HN and L9-HN are shown in the spectra, which were assigned based on the 2D-TOCSY spectrum of the peptide. Diagonal peaks from cis and trans conformers are indicated by cc and tt, respectively, whereas exchange peaks resulting from isomerization are labeled with ct and tc.
Determination of substrate binding sites on TbPin1
Chemical shift perturbation experiments were used to determine the binding sites of the phosphorylated peptide {SSYFSG[p]TPLEDDSD} on TbPin1. 15N-labeled TbPin1 was titrated stepwise with the peptide to a final molar ratio of 1∶8. 2D 1H-15N HSQC spectra were acquired following each addition of the peptide. To some extent, the peptide titration caused non-significant chemical shift perturbations in the HSQC spectra of TbPin1. The dissociate constant Kd was estimated to be about millimolar. Figure 6A shows the HSQC spectrum of unliganded TbPin1 protein (red peaks) overlaid with the final HSQC spectrum in which TbPin1 was saturated with the peptide (blue peaks). To identify the residues involved in the interaction of TbPin1 with the peptide, the magnitudes of the chemical shift changes caused by the peptide binding versus residue number are shown in Figure 6B. Residues on TbPin1 exhibiting significant amide chemical shift changes (>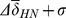 ) are mapped to the tertiary structure of TbPin1 (Figure 6C). Approximately 11 peaks are observed with significant chemical shift perturbations. These residues are concentrated to the catalytic loop (Ser15, Arg18, Asn19, Val21, Ser22 and Val32), η1 (Gly66 and Ser67), and the linker regions connecting β2 and α3 (Met83), β3 and β4 (Asp105 and Gly107).
) are mapped to the tertiary structure of TbPin1 (Figure 6C). Approximately 11 peaks are observed with significant chemical shift perturbations. These residues are concentrated to the catalytic loop (Ser15, Arg18, Asn19, Val21, Ser22 and Val32), η1 (Gly66 and Ser67), and the linker regions connecting β2 and α3 (Met83), β3 and β4 (Asp105 and Gly107).
Figure 6. Chemical shift perturbation analysis reveals the substrate binding pocket on TbPin1.

(A) Overlay of the 1H-15N HSQC spectra of TbPin1 free (red) and in complex with the unlabeled phosphorylated peptide substrate (blue). The molar ratio of TbPin1 to the phosphorylated peptide is 1∶8. (B) Diagram of amide chemical shift changes (ΔδHN) of TbPin1 versus residue number at a molar ratio of 1∶8. The average amide chemical shift change (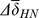 ) and the mean standard deviation (
) and the mean standard deviation (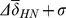 ) are indicated with solid and dashed lines, respectively. Residues with chemical shift changes larger than
) are indicated with solid and dashed lines, respectively. Residues with chemical shift changes larger than 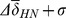 (dashed line) are considered to be involved in substantial contact with the substrate. (C) Mapping the substantial contact residues on the tertiary structure of TbPin1. Residues with significant chemical shift change are highlighted in stick style.
(dashed line) are considered to be involved in substantial contact with the substrate. (C) Mapping the substantial contact residues on the tertiary structure of TbPin1. Residues with significant chemical shift change are highlighted in stick style.
Dynamics in the free enzyme
To analyze the global and local backbone dynamics of TbPin1, we performed 15N relaxation experiments. As a whole, 105 assigned residues were used except the unobservable resonances and partially overlapped peaks such as Phe14 and Met82. The relaxation rates R1, R2 and heteronuclear NOEs versus residue number are shown in Figure 7. The R1 values do not change markedly with the sequence, ranging from 1.4 to 1.9 s−1. Different from the R1 distribution, the R2 values are relatively variable with residue number, ranging from 6.0 to 10.2 s−1. The residue Ser106 shows the largest R2 value over 10.1 s−1, and Asn19, Ser22 also display distinctly large R2 value as high as 10.0 s−1. Almost all of the NOE values are between 0.7 and 0.9 except for Glu3, Lys4 and Val21 (< 0.6), indicating that the overall structure of TbPin1 is highly rigid.
Figure 7. Relaxation rates R1, R2 and {1H}-15N heteronuclear NOEs of TbPin1.
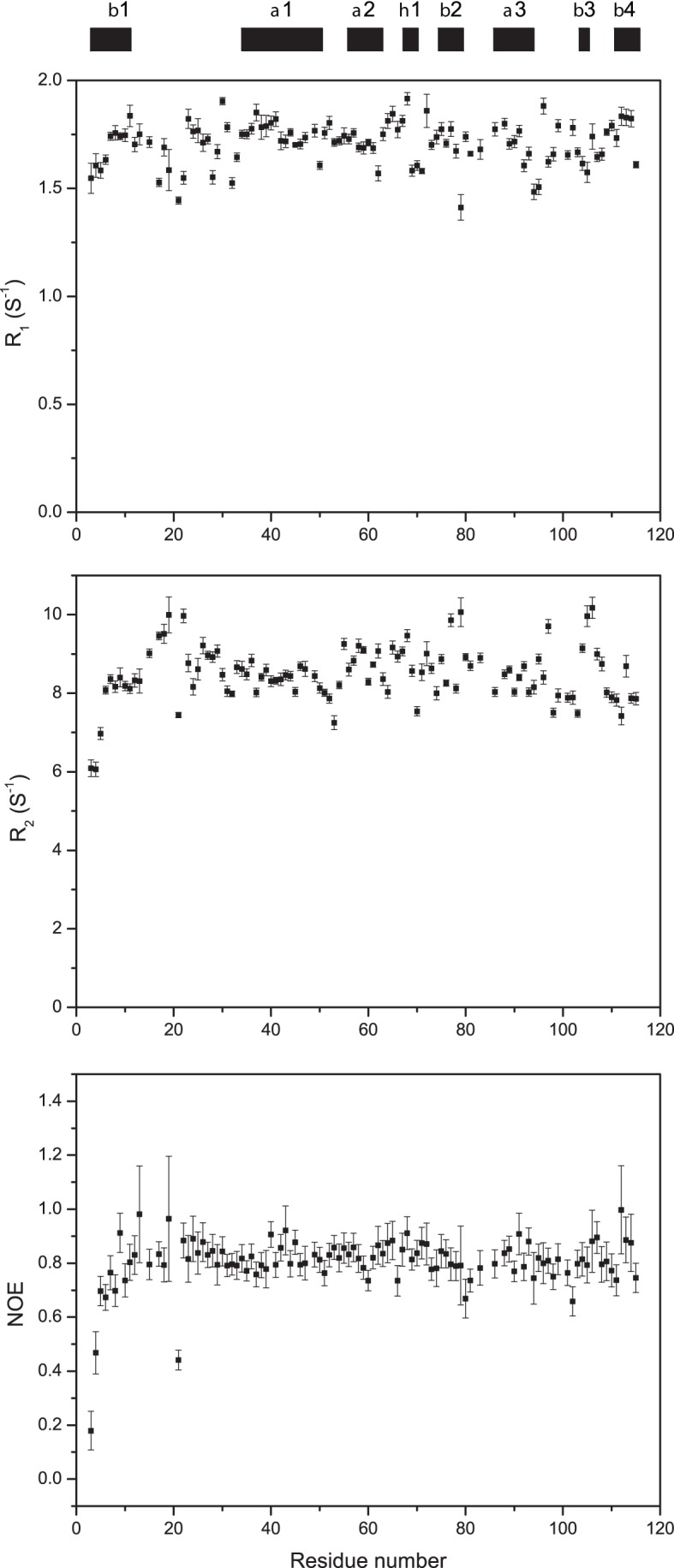
The regular secondary structure elements are indicated on the top. NMR spectra were recorded at 298 K on a Varian Unity Inova 600 MHz spectrometer. Proteins were dissolved in 20 mM phosphate sodium buffer containing 1 mM DTT, 0.1 mM NaN3, 10% D2O, pH 7.0.
Analysis of 15N relaxation data
We used both the TENSOR2 program [48] and Mathematica notebooks from Spyracopoulos' lab [49] to analyze the backbone 15N relaxation data for TbPin1. Residues were eliminated due to NOE <0.65 and filtered due to short T2 values based on the criterion {[(<T2>–T2n)/<T2>–(<T1>–T1n)/<T1>] >1.5 SD}, where T2n is the T2 value of residue n, and <T2> is the average T2 value, SD is the standard deviation of (<T2>–T2n)/<T2>–(<T1>–T1n)/<T1> [50], [51]. Thus, 78 out of 105 residues were used to determine the rotational diffusion parameters of TbPin1. The inertia tensor of the TbPin1 protein was calculated from the PDB coordinates. Its principal value ratios (Ix: Iy: Iz) were 1.00:0.89:0.56, allowing the approximation of the protein molecule as a prolate ellipsoid. The D∥: D⊥ ratio of the rotational diffusion tensor of TbPin1 was calculated to be 1.17±0.01, suggesting that the axially symmetric model was suitable for the data fitting. In the model-free approach, mobility was characterized by the order parameter S2, which could be interpreted as the amplitude of the inter-motion on a nanosecond time scale. The calculated S2 values versus residue number are mapped into the tertiary structure of TbPin1 with a color pattern (Figure 8). Residues with S2 values ranging from 0.7 to 0.9 are located on most of secondary structure elements, indicating that TbPin1 adopts a much rigid structure. Only Cys65, Ser67, Tyr68, Gly72 and Lys84 show relatively high S2 value (S2>0.9), indicating that this region (Figure 8B) is very restricted. The other two dynamics parameters, i.e., correlation time (τe) for ps-ns timescale internal motion, and conformational exchange rate (Rex) for µs-ms timescale internal motion are shown in Figure 8. Residues with Rex >1.0 S−1 are also mostly located on the region of the peptide binding pocket, including the catalytic loop (Ser15, Arg18, Asn19, Ser22 and Ser28), the α2 helix (Arg62), the C-terminal end of β2 (Phe77 and Ser79), the linker regions connecting α3 and β3 (Ile95 and Asp97), β3 and β4 (Thr104, Asp105, Ser106) (Figure 8). This result indicates that TbPin1 is well structured except the binding pocket of the phosphorylated peptide which exhibits significant conformational exchange.
Figure 8. Model-free analysis of TbPin1.

(A, C and E) The plots of the order parameters S2, the effective internal correlation time τe, and the conformational exchange rate Rex versus residue number of TbPin1, respectively; (B, D and F) Residues with S2, τe (blue) and Rex (orange) values are mapped onto the solution structure of TbPin1, respectively. (B) Gray, S2 unavailable due to the absence of NMR data or failure in NMR data fitting; red, S2<0.6; orange, 0.6≤S2<0.7; yellow, 0.7≤S2<0.8; green, 0.8≤S2<0.9; and blue, 0.9≤S2<1.0. The ribbon graph was generated by PyMOL.
Discussion
In this work, we have determined the solution structure of the Pin1-type parvulin TbPin1 from Trypanosoma brucei. TbPin1 has the unique PPIase domain with a lack of the WW domain, which shares high homologies with other members of Pin1-type parvulins, and exhibits phosphorylation-dependent PPIase activity (Figure 1 and 5).
Recent studies of hPin1 and other parvulins have provided new insights into the functional status of active site residues [34], [42], [52], [53]. In the Pin1-type parvulin family, the basic cluster (Lys63, Arg68 and Arg69 in hPin1) has been considered essential for phospho-specific recognition [25], [54]. However, Behrsin et al. used a unigenic evolution strategy to reveal that only 19 residues were perfectly conserved in the hPin1 sequence, and the two residues crucial for the phosphate binding, Arg68 and Arg69, were not always conserved through unigenic evolution [52]. They proposed that only one basic residue (Arg) at either position 68 or 69 was essential for the function of hPin1. In line with this finding, alignment of TbPin1 with its homologues shows that the two Arg residues at the positions are not all conserved. Arg69 in hPin1 is replaced by Asn19 in TbPin1, in the case of CaEss1 the equivalent basic residue is Lys83 (Figure 1). Interestingly, TbPar42 shows absolutely conserved residues Lys276, Arg280, Arg281 in the corresponding position. Thereby, TbPar42 would have a phosphate-binding loop or prefer the substrates with a negatively charged residue preceding the proline. However, the enzyme activity assays illustrated that, TbPin1 displayed the phosphorylation-dependent isomerization ability for the substrate, but the FHA-truncated TbPar42 did not show the isomerase activity in the same experimental condition (Figure 5). Maybe some specific phosphorylated peptides or a peptide containing Glu- or Asp-Pro motif could serve as the substrate for TbPar42 [13], [34]. The results from the enzyme activity assays are consistent with the previous report that TbPin1 had the ability to rescue the lethal mitotic phenotype of a temperature-sensitive mutation in Saccharomyces cerevisiae while TbPar42 lacked the ability [30], suggesting that TbPin1 is a Pin1-type parvulin and TbPar42 is a non Pin1-type parvulin. TbPar42 shows well conserved residues to Pin1-type parvulins, but why it lacks the Pin1-like isomerase activity still remains to be elucidated.
The active site residues were originally defined by the crystal structure of hPin1 in complex with an Ala-Pro dipeptide [25], in which the dipeptide is used as a pseudo substrate for hPin1. In the present work, we identified the active site residues of TbPin1 for a phosphorylated peptide substrate using the chemical shift perturbation approach. NMR titration of the phosphorylated peptide to TbPin1 demonstrated that residues Ser15, Arg18, Asn19, Val21, Ser22, Val32, Gly66, Ser67, Met83, Asp105 and Gly107 experienced significant chemical shift changes (Figure 6). These residues may be involved in substantial contact with the phosphorylated peptide substrate. Most of these residue side chains point to the peptide binding pocket. For instance, in comparison with the corresponding residue Gln131 in hPin1, the Met83 side chain points in an orientation to the hydrophobic pocket (Figure S2). Dynamics data derived from the 15N relaxation experiments indicated that these residues except Gly66, Ser67 possessed significant conformational exchange rates Rex on the µs-ms timescale (Figure 8). The result suggests that the substrate binding pocket is flexible, similarly with the previous result reported for the archaeal parvulin CsPinA [36].
On the other hand, the crucial residue Cys65 did not exhibit marked chemical shift perturbation during the peptide titration process, even though the PPIase activity analysis demonstrated that the mutation of Cys65 to Ala abolished the enzyme activity of TbPin1. It is well known that chemical shift perturbations monitored by 2D 1H-15N HSQC spectrum are sensitive to the binding of backbone amide groups, but usually not sensitive to that of side-chain groups. This reason might account for why the residues which were considered crucial for enzyme catalysis (including His9, Lys13, Cys65, Leu74, Met82, Phe86, Ser106 and His109 in TbPin1, equivalent to His59, Lys63, Cys113, Leu122, Met130, Phe134, Ser154 and His157 in hPin1) did not experience significant chemical shift perturbations [13]. Similarly, His12, Lys16, Cys70, Leu79, and His114 in PinAt (equivalent to His9, Lys13, Cys65, Leu74 and His109 in TbPin1) also showed non-significant chemical shift perturbations in 2D 1H-15N HSQC spectrum [43]. However, in 2D 1H-13C HSQC spectrum, the two conserved histidine residues (i.e. His9 and His86 in CsPinA, His146 and His239 in S.aureus PrsA-PPIase) displayed significant chemical shift perturbations [34], [36]. It was postulated that a hydrogen bonding between the two histidine residues were required for the catalysis [34], [42]. Therefore, both His9 and His109 in TbPin1 might also be involved in the catalysis.
Although TbPin1 exhibited evident phosphorylation-dependent PPIase activity for the phosphorylated peptide, chemical shift perturbation analysis illustrated that TbPin1 interacted weekly with the phosphorylated peptide. The weak binding affinity of TbPin1 for the substrate is supported by the previous studies conducted on other parvulins [34], [55], [56], which might be due to the fast conformational exchange between the free and complex forms.
In conclusion, the solution structure of TbPin1 from Trypanosoma brucei was determined and characterized using NMR spectroscopy. TbPin1 only contains a unique PPIase domain and adopts the typical catalytic tertiary structure (β-α2-η-β-α-β2 fold) of Pin1-type parvulins. The overall structure of TbPin1 is highly rigid except that the substrate binding pocket is flexible. TbPin1 catalyzes the cis/trans conformational change of the phosphorylated peptide {SSYFSG[p]TPLEDDSD}, and residues including Ser15, Arg18, Asn19, Val21, Ser22, Val32, Gly66, Ser67, Met83, Asp105 and Gly107 are involved in the substantial contact with the phosphorylated substrate. Mutation of Cys65 to Ala confirms that the conserved Cys is crucial for the enzyme catalysis in Pin1-type parvulins. These results shed light on the molecular mechanism of enzyme catalysis for TbPin1 and other Pin1-type parvulins.
Materials and Methods
Sample preparation
The amplified gene fragments coding for TbPin1 and TbPPIase (truncation of FHA domain of TbPar42) were cloned into the vector pET28b via NdeI and SalI restriction sites as mentioned previously [30]. A single point mutant on the pET28b-TbPin1 plasmid (C65A) was constructed using the site-directed mutagenesis. The recombinant proteins were expressed at 37°C in E.coli strain BL21 (DE3) cells and induced with 0.4 mM IPTG for 4 hours in LB media until an OD600 of ∼1.0. For 15N-labeled or 15N,13C-labeled TbPin1 protein samples, cells were grown in a M9 minimal medium containing 15N-NH4Cl (Cambridge Isotopes Laboratories) or 15N-NH4Cl and 13C6-Glucose (Cambridge Isotopes Laboratories) as sole nitrogen or/and carbon sources, respectively. Cells were resuspended in a lysis buffer containing 20 mM sodium phosphate (pH 7.4), 500 mM NaCl, 10 mM β-mercaptoethanol, 1% (V/V) Tween 20 and sonicated at 4°C. Following sonication, soluble supernatants were incubated with a Ni-NTA resin (Qiagen) for 2 hours and then washed with a lysis buffer without Tween 20 and a wash buffer containing 20 mM sodium phosphate (pH 7.4), 500 mM NaCl, 20 mM imidazole. The recombinant proteins were eluted with a wash buffer supplemented with 500 mM imidazole. Eluted proteins were digested with thrombin (Sigma) overnight at 4°C and followed by FPLC gel-filtration on a Superdex 75 10/300 GL column (GE Healthcare). Three additional residues (Gly-Ser-His) were remained at the N terminus of TbPin1 after thrombin cleavage and were not included in later assignment and structure calculation. The resulting samples were concentrated to 0.8∼1 mM and then exchanged into 20 mM sodium phosphate buffer (pH 7.0), 1 mM DTT, 0.01 mM NaN3.
Both phosphopeptide {SSYFSG[p]TPLEDDSD} and non-phosphorylated peptide SSYFSGTPLEDDSD were purchased from ChinaPeptides Corporation (Shanghai, China). Concentrated peptide stocks were prepared in 20 mM sodium phosphate buffer (pH 7.0).
NMR Spectroscopy and Resonance Assignment
All NMR spectra were recorded at 298 K on a Varian Unity Inova 600 MHz spectrometer equipped with three RF channels and a triple-resonance pulse-field gradient probe. Sequence-specific assignments of backbone 1H, 13C, and 15N resonances were performed by using a suit of 3D heteronuclear spectra including HNCACB, CBCA(CO)NH, HNCA, HN(CO)CA, HNCO, HN(CA)CO, and HAHB(CO)NH spectra. Side-chain assignments were obtained by using 3D H(CCH)(CO)NHTOCSY, (H)CC(CO)NHTOCSY, HCCH-TOCSY, CCHTOCSY and 15N-edited TOCSY spectra. Secondary structures were predicted from secondary chemical shifts of 1Hα, 13Cα, 13Cβ and 13CO with the Chemical Shift Index (CSI) approach [57]. A mixing time of 100 ms was used for 3D 15N- and 13C-edited NOESY experiments. All NMR data were processed with NMRPipe [58] and analyzed by using Sparky (T. D. Goddard and D. G. Kneller, University of California, USA) and CcpNmr Analysis [59].
Structure Calculation
Conformational restraints were used for structure calculation of TbPin1, including 1H-1H distance restraints derived from 3D 15N- and 13C-edited NOESY spectra, backbone dihedral angle restraints generated by the TALOS+ software [41], and hydrogen bonds restraints derived from H-D exchange experiments. Structures were calculated using the ARIA2.2/CNS1.2 software [39], [40]. A family of 200 structures was calculated according to the simulated annealing protocol. Twenty lowest energy structures were selected and further refined in water. Quality of the ensemble was assessed by the PROCHECK program [60], [61]. Figures were generated with MOLMOL [62] or PyMOL (DeLano Scientific LLC).
Enzyme activity analysis
NMR experiments were performed on peptide samples in a NMR buffer containing 20 mM sodium phosphate, 90% H2O and 10% D2O (pH 7.0) with or without TbPin1, the TbPin1-C65A mutant or the FHA-truncated TbPar42 mutant. The final concentration of the phosphorylated peptide {SSYFSG[p]TPLEDDSD} or non-phosphorylated peptide SSYFSGTPLEDDSD was 2.4 mM, and the final concentration of the protein was 0.03 mM. 2D 1H-1H NMR spectra were recorded with spectral widths of 8000×8000 Hz in t1 × t2 dimensions. EXSY spectra were acquired at a mixing time of 200 or 300 ms with 16 scans, while TOCSY spectra were recorded at a mixing time of 75 ms with 16 scans [22], [30], [46].
Chemical shift perturbation
15N-labeled TbPin1 was dissolved in the NMR buffer described above. The phosphorylated peptide was titrated stepwise up to a final molar ratio of 1∶8, with each step monitored by recording 2D 1H-15N HSQC spectrum. The reciprocal titration of 15N-labeled TbPin1 with the phosphorylated peptide was performed accordingly with the initial TbPin1 concentration of 0.4 mM. The chemical shift changes of amide 1H and 15N chemical shifts were calculated by using Eq.1.
| (1) |
15N Relaxation Measurements
All 15N relaxation data were recorded at 298 K on Varian Unity Inova 600 spectrometer. The standard pulse sequences with minimal water suppression were used to record the 2D spectra of T1, T2, and {1H}-15N NOE. In the direct (1H) dimension, the carrier frequency was set on the water resonance with a spectral width of 10,000 Hz; while in the indirect (15N) dimension, the spectral width was 1400 Hz. A recycle delay time was 2 s. T1 was measured by using a series of spectra recorded with 10 relaxation delays: 10.83, 54.17, 108.34, 216.68, 325.02, 541.70, 866.72, 1191.74, 1570.93, 1950.12 ms. T2 measurement was carried out with 10 relaxation delays: 15.62, 31.23, 46.85, 62.46, 78.08, 93.70, 109.31, 124.93, 140.54 and 156.16 ms. The relaxation rates and experimental errors were calculated by a mono-exponential curve fitting of the corresponding signals in a series of 2D spectra using the CCPN software [59]. The steady-state {1H}-15N NOE enhancements were determined from the ratio of peak heights in spectra recorded with or without proton saturation. The saturated spectra were recorded with a 2 s relaxation delay followed by a 3 s period of proton saturation. The spectra without proton saturation were acquired with a relaxation delay of 5 s. The experimental errors in NOE values can be assessed as follows [63]:
| (2) |
where I and σ denote the peak intensity and the level of experimental noise, respectively. In the saturation and non-saturation experiments NOE can be determined as follow [63]:
| (3) |
Analysis of 15N relaxation data
Several methods have been developed for analyzing protein dynamics [51], [64], [65]. Here, we used the TENSOR2 program [48] and a suit of Mathematica notebooks provided by Spyracopoulos's lab [49] to analyze 15N relaxation data. TENSOR2 was used to determine rotational diffusion tensor from the coordinate PDB file and 15N relaxation data. TENSOR2 can also be used to perform the model-free analysis from relaxation rate R1, R2 and {1H}-15N NOE. Spyracopoulos et al. have developed a suit of Mathematica notebooks to analyze backbone 15N NMR relaxation data, such as spectral density, diffusion tensor, model-free analysis. These notebooks can be downloaded from www.bionmr.ulabert.ca/~lspy. The tensors and internal mobility parameters were estimated using Monte-Carlo sampling methods and F-tests [49].
Supporting Information
TbPin1 does not catalyze the cis-trans isomerization of the non-phosphorylated peptide. A selected region of the 2D EXSY spectrum of the non-phosphorylated peptide SSYFSGTPLEDDSD is displayed (A) in the absence of TbPin1; (B) in the presence of TbPin1; (C) in the presence of the FHA-truncated TbPar42 mutant (TbPPIase). A mixing time of 300 ms was used. The cis and trans T7-HN and L9-HN are shown in the spectra, which were assigned based on the 2D-TOCSY spectrum of the peptide. No exchange cross peaks were observed in the spectra.
(EPS)
Comparison of the binding sites between TbPin1 and hPin1. (A) Binding sites of the phosphorylated peptide SSYFSG[p]TPLEDDSD on TbPin1 identified by using the chemical shift perturbation approach. (B) Binding sites of an Ala-Pro dipeptide and a sulfate ion on hPin1 which were defined by the crystal structure of hPin1 in complex with the dipeptide (PDB: 1PIN). These residues are highlighted in stick style and displayed in cyan and orange, respectively. The dipeptide and Q131 in hPin1 are colored magentas, slate, respectively. Q131 which is not considered to be involved in the dipeptide binding shows an orientation far away from the hydrophobic pocket constituted by H59, L122, M130, F134 and H157. However, the corresponding residue M83 in TbPin1 points in an orientation to the hydrophobic pocket, indicating that M83 is involved in the binding of the phosphorylated peptide.
(TIF)
Acknowledgments
The authors would like to thank all the members of the Zhao's and Lin's laboratories for technical helps and advices.
Funding Statement
This work was supported by the Chinese National Science Foundation (Nos. 20972130, 91129713, 31170717), the Major State Basic Research Development Program of China (973 Program, No. 2012CB821600) and the Program of Shanghai Subject Chief Scientist (No. 09XD1405100). Y-CL was supported by a grant (06/1/21/19/473) from the Biomedical Research Council, the Agency for Science, Research and Technology, Singapore. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Velazquez HA, Hamelberg D (2011) Conformational selection in the recognition of phosphorylated substrates by the catalytic domain of human Pin1. Biochemistry 50: 9605–9615. [DOI] [PubMed] [Google Scholar]
- 2. Lu KP, Finn G, Lee TH, Nicholson LK (2007) Prolyl cis-trans isomerization as a molecular timer. Nat Chem Biol 3: 619–629. [DOI] [PubMed] [Google Scholar]
- 3. Stukenberg PT, Kirschner MW (2001) Pin1 acts catalytically to promote a conformational change in Cdc25. Mol Cell 7: 1071–1083. [DOI] [PubMed] [Google Scholar]
- 4. Park ST, Aldape RA, Futer O, DeCenzo MT, Livingston DJ (1992) PPIase catalysis by human FK506-binding protein proceeds through a conformational twist mechanism. J Biol Chem 267: 3316–3324. [PubMed] [Google Scholar]
- 5. Galat A, Metcalfe SM (1995) Peptidylproline cis/trans isomerases. Prog Biophys Mol Biol 63: 67–118. [DOI] [PubMed] [Google Scholar]
- 6. Fischer G, Bang H, Mech C (1984) Determination of enzymatic catalysis for the cis-trans-isomerization of peptide binding in proline-containing peptides. Biomed Biochim Acta 43: 1101–1111. [PubMed] [Google Scholar]
- 7. Gothel SF, Marahiel MA (1999) Peptidyl-prolyl cis-trans isomerases, a superfamily of ubiquitous folding catalysts. Cell Mol Life Sci 55: 423–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fanghanel J, Fischer G (2004) Insights into the catalytic mechanism of peptidyl prolyl cis/trans isomerases. Front Biosci 9: 3453–3478. [DOI] [PubMed] [Google Scholar]
- 9. Lu KP, Zhou XZ (2007) The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol 8: 904–916. [DOI] [PubMed] [Google Scholar]
- 10. Erben ED, Daum S, Tellez-Inon MT (2007) The Trypanosoma cruzi PIN1 gene encodes a parvulin peptidyl-prolyl cis/trans isomerase able to replace the essential ESS1 in Saccharomyces cerevisiae. Mol Biochem Parasitol 153: 186–193. [DOI] [PubMed] [Google Scholar]
- 11. Lippens G, Landrieu I, Smet C (2007) Molecular mechanisms of the phospho-dependent prolyl cis/trans isomerase Pin1. FEBS J 274: 5211–5222. [DOI] [PubMed] [Google Scholar]
- 12. Mueller JW, Bayer P (2008) Small family with key contacts: par14 and par17 parvulin proteins, relatives of pin1, now emerge in biomedical research. Perspect Medicin Chem 2: 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, et al. (1997) Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science 278: 1957–1960. [DOI] [PubMed] [Google Scholar]
- 14. Zhou XZ, Lu PJ, Wulf G, Lu KP (1999) Phosphorylation-dependent prolyl isomerization: a novel signaling regulatory mechanism. Cell Mol Life Sci 56: 788–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liou YC, Zhou XZ, Lu KP (2011) Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem Sci 36: 501–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou XZ, Kops O, Werner A, Lu PJ, Shen M, et al. (2000) Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol Cell 6: 873–883. [DOI] [PubMed] [Google Scholar]
- 17. Wulf G, Finn G, Suizu F, Lu KP (2005) Phosphorylation-specific prolyl isomerization: is there an underlying theme? Nat Cell Biol 7: 435–441. [DOI] [PubMed] [Google Scholar]
- 18. Lim J, Lu KP (2005) Pinning down phosphorylated tau and tauopathies. Biochim Biophys Acta 1739: 311–322. [DOI] [PubMed] [Google Scholar]
- 19. Smet C, Sambo AV, Wieruszeski JM, Leroy A, Landrieu I, et al. (2004) The peptidyl prolyl cis/trans-isomerase Pin1 recognizes the phospho-Thr212-Pro213 site on Tau. Biochemistry 43: 2032–2040. [DOI] [PubMed] [Google Scholar]
- 20. Ryo A, Nakamura M, Wulf G, Liou YC, Lu KP (2001) Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat Cell Biol 3: 793–801. [DOI] [PubMed] [Google Scholar]
- 21. Zheng H, You H, Zhou XZ, Murray SA, Uchida T, et al. (2002) The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 419: 849–853. [DOI] [PubMed] [Google Scholar]
- 22. Wang Y, Liu C, Yang D, Yu H, Liou YC (2010) Pin1At encoding a peptidyl-prolyl cis/trans isomerase regulates flowering time in Arabidopsis. Mol Cell 37: 112–122. [DOI] [PubMed] [Google Scholar]
- 23. Bialik S, Kimchi A (2011) Pin-pointing a new DAP kinase function: the peptidyl-proly isomerase Pin1 Is Negatively Regulated by DAP kinase-mediated phosphorylation. Mol Cell 42: 139–141. [DOI] [PubMed] [Google Scholar]
- 24. Liou YC, Sun A, Ryo A, Zhou XZ, Yu ZX, et al. (2003) Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 424: 556–561. [DOI] [PubMed] [Google Scholar]
- 25. Ranganathan R, Lu KP, Hunter T, Noel JP (1997) Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell 89: 875–886. [DOI] [PubMed] [Google Scholar]
- 26. Metzner M, Stoller G, Rucknagel KP, Lu KP, Fischer G, et al. (2001) Functional replacement of the essential ESS1 in yeast by the plant parvulin DlPar13. J Biol Chem 276: 13524–13529. [DOI] [PubMed] [Google Scholar]
- 27. Li Z, Li H, Devasahayam G, Gemmill T, Chaturvedi V, et al. (2005) The structure of the Candida albicans Ess1 prolyl isomerase reveals a well-ordered linker that restricts domain mobility. Biochemistry 44: 6180–6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hsu T, McRackan D, Vincent TS, Gert de Couet H (2001) Drosophila Pin1 prolyl isomerase Dodo is a MAP kinase signal responder during oogenesis. Nat Cell Biol 3: 538–543. [DOI] [PubMed] [Google Scholar]
- 29. Yao JL, Kops O, Lu PJ, Lu KP (2001) Functional conservation of phosphorylation-specific prolyl isomerases in plants. J Biol Chem 276: 13517–13523. [DOI] [PubMed] [Google Scholar]
- 30. Goh JY, Lai CY, Tan LC, Yang D, He CY, et al. (2010) Functional characterization of two novel parvulins in Trypanosoma brucei. FEBS Lett 584: 2901–2908. [DOI] [PubMed] [Google Scholar]
- 31. Lu PJ, Zhou XZ, Liou YC, Noel JP, Lu KP (2002) Critical role of WW domain phosphorylation in regulating phosphoserine binding activity and Pin1 function. J Biol Chem 277: 2381–2384. [DOI] [PubMed] [Google Scholar]
- 32. Kouri ED, Labrou NE, Garbis SD, Kalliampakou KI, Stedel C, et al. (2009) Molecular and biochemical characterization of the parvulin-type PPIases in Lotus japonicus. Plant Physiol 150: 1160–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Holm L, Rosenstrom P (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res 38: W545–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heikkinen O, Seppala R, Tossavainen H, Heikkinen S, Koskela H, et al. (2009) Solution structure of the parvulin-type PPIase domain of Staphylococcus aureus PrsA – implications for the catalytic mechanism of parvulins. BMC Struct Biol 9: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kuhlewein A, Voll G, Hernandez Alvarez B, Kessler H, Fischer G, et al. (2004) Solution structure of Escherichia coli Par10: The prototypic member of the Parvulin family of peptidyl-prolyl cis/trans isomerases. Protein Sci 13: 2378–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jaremko L, Jaremko M, Elfaki I, Mueller JW, Ejchart A, et al. (2011) Structure and dynamics of the first archaeal parvulin reveal a new functionally important loop in parvulin-type prolyl isomerases. J Biol Chem 286: 6554–6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bitto E, McKay DB (2002) Crystallographic structure of SurA, a molecular chaperone that facilitates folding of outer membrane porins. Structure 10: 1489–1498. [DOI] [PubMed] [Google Scholar]
- 38. Sekerina E, Rahfeld JU, Muller J, Fanghanel J, Rascher C, et al. (2000) NMR solution structure of hPar14 reveals similarity to the peptidyl prolyl cis/trans isomerase domain of the mitotic regulator hPin1 but indicates a different functionality of the protein. J Mol Biol 301: 1003–1017. [DOI] [PubMed] [Google Scholar]
- 39. Rieping W, Habeck M, Bardiaux B, Bernard A, Malliavin TE, et al. (2007) ARIA2: automated NOE assignment and data integration in NMR structure calculation. Bioinformatics 23: 381–382. [DOI] [PubMed] [Google Scholar]
- 40. Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, et al. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 54: 905–921. [DOI] [PubMed] [Google Scholar]
- 41. Shen Y, Delaglio F, Cornilescu G, Bax A (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR 44: 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mueller JW, Link NM, Matena A, Hoppstock L, Ruppel A, et al. (2011) Crystallographic proof for an extended hydrogen-bonding network in small prolyl isomerases. J Am Chem Soc 133: 20096–20099. [DOI] [PubMed] [Google Scholar]
- 43. Landrieu I, Wieruszeski JM, Wintjens R, Inze D, Lippens G (2002) Solution structure of the single-domain prolyl cis/trans isomerase PIN1At from Arabidopsis thaliana. J Mol Biol 320: 321–332. [DOI] [PubMed] [Google Scholar]
- 44. Daum S, Fanghanel J, Wildemann D, Schiene-Fischer C (2006) Thermodynamics of phosphopeptide binding to the human peptidyl prolyl cis/trans isomerase Pin1. Biochemistry 45: 12125–12135. [DOI] [PubMed] [Google Scholar]
- 45. Verdecia MA, Bowman ME, Lu KP, Hunter T, Noel JP (2000) Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat Struct Biol 7: 639–643. [DOI] [PubMed] [Google Scholar]
- 46. Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, et al. (2006) The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature 440: 528–534. [DOI] [PubMed] [Google Scholar]
- 47. Zhou W, Yang Q, Low CB, Karthik BC, Wang Y, et al. (2009) Pin1 catalyzes conformational changes of Thr-187 in p27Kip1 and mediates its stability through a polyubiquitination process. J Biol Chem 284: 23980–23988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dosset P, Hus JC, Blackledge M, Marion D (2000) Efficient analysis of macromolecular rotational diffusion from heteronuclear relaxation data. J Biomol NMR 16: 23–28. [DOI] [PubMed] [Google Scholar]
- 49. Spyracopoulos L (2006) A suite of Mathematica notebooks for the analysis of protein main chain 15N NMR relaxation data. J Biomol NMR 36: 215–224. [DOI] [PubMed] [Google Scholar]
- 50. Tjandra N, Feller SE, Pastor RW, Bax A (1995) Rotational diffusion anisotropy of human ubiquitin from 15N NMR relaxation. J Am Chem SOC 117: 12562–12566. [Google Scholar]
- 51. Palmer AG 3rd (2004) NMR characterization of the dynamics of biomacromolecules. Chem Rev 104: 3623–3640. [DOI] [PubMed] [Google Scholar]
- 52. Behrsin CD, Bailey ML, Bateman KS, Hamilton KS, Wahl LM, et al. (2007) Functionally important residues in the peptidyl-prolyl isomerase Pin1 revealed by unigenic evolution. J Mol Biol 365: 1143–1162. [DOI] [PubMed] [Google Scholar]
- 53. Bailey ML, Shilton BH, Brandl CJ, Litchfield DW (2008) The dual histidine motif in the active site of Pin1 has a structural rather than catalytic role. Biochemistry 47: 11481–11489. [DOI] [PubMed] [Google Scholar]
- 54. Terada T, Shirouzu M, Fukumori Y, Fujimori F, Ito Y, et al. (2001) Solution structure of the human parvulin-like peptidyl prolyl cis/trans isomerase, hPar14. J Mol Biol 305: 917–926. [DOI] [PubMed] [Google Scholar]
- 55. Bayer E, Goettsch S, Mueller JW, Griewel B, Guiberman E, et al. (2003) Structural analysis of the mitotic regulator hPin1 in solution: insights into domain architecture and substrate binding. J Biol Chem 278: 26183–26193. [DOI] [PubMed] [Google Scholar]
- 56. Labeikovsky W, Eisenmesser EZ, Bosco DA, Kern D (2007) Structure and dynamics of pin1 during catalysis by NMR. J Mol Biol 367: 1370–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wishart DS, Sykes BD, Richards FM (1992) The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 31: 1647–1651. [DOI] [PubMed] [Google Scholar]
- 58. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, et al. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- 59. Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, et al. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59: 687–696. [DOI] [PubMed] [Google Scholar]
- 60. Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8: 477–486. [DOI] [PubMed] [Google Scholar]
- 61. Doreleijers JF, Rullmann JA, Kaptein R (1998) Quality assessment of NMR structures: a statistical survey. J Mol Biol 281: 149–164. [DOI] [PubMed] [Google Scholar]
- 62.Koradi R, Billeter M, Wuthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14: 51–55, 29–32. [DOI] [PubMed]
- 63.Zerbe O (2003) BioNMR in drug research; Mannhold R, Kubinyi H, Folkers G, editors. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA. 283–308 p.
- 64. Ishima R (2012) Recent Developments in (15)N NMR Relaxation Studies that Probe Protein Backbone Dynamics. Top Curr Chem 326: 99–122. [DOI] [PubMed] [Google Scholar]
- 65. Jarymowycz VA, Stone MJ (2006) Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem Rev 106: 1624–1671. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TbPin1 does not catalyze the cis-trans isomerization of the non-phosphorylated peptide. A selected region of the 2D EXSY spectrum of the non-phosphorylated peptide SSYFSGTPLEDDSD is displayed (A) in the absence of TbPin1; (B) in the presence of TbPin1; (C) in the presence of the FHA-truncated TbPar42 mutant (TbPPIase). A mixing time of 300 ms was used. The cis and trans T7-HN and L9-HN are shown in the spectra, which were assigned based on the 2D-TOCSY spectrum of the peptide. No exchange cross peaks were observed in the spectra.
(EPS)
Comparison of the binding sites between TbPin1 and hPin1. (A) Binding sites of the phosphorylated peptide SSYFSG[p]TPLEDDSD on TbPin1 identified by using the chemical shift perturbation approach. (B) Binding sites of an Ala-Pro dipeptide and a sulfate ion on hPin1 which were defined by the crystal structure of hPin1 in complex with the dipeptide (PDB: 1PIN). These residues are highlighted in stick style and displayed in cyan and orange, respectively. The dipeptide and Q131 in hPin1 are colored magentas, slate, respectively. Q131 which is not considered to be involved in the dipeptide binding shows an orientation far away from the hydrophobic pocket constituted by H59, L122, M130, F134 and H157. However, the corresponding residue M83 in TbPin1 points in an orientation to the hydrophobic pocket, indicating that M83 is involved in the binding of the phosphorylated peptide.
(TIF)