

Introduction
In the DDC feeding mouse model where liver cells proliferate, Mallory-Denk bodies (MDBs) form and later, after DDC withdrawal, hepatocellular carcinomas (HCCs) develop (1). Similarly, patients who abuse alcohol develop alcoholic liver disease (ALD), MDBs form (2) and later, after alcohol abstinence, the patients develop HCCs (2). Also MDBs form in many of the HCCs, both in the mouse model and in ALD. Because of this, it has been suggested that MDBs are a preneoplastic change formed in balloon hepatocytes which transform into cancer cells (3-6). But there may be other links to the preneoplastic process in ALD-induced HCCs such as the role that macrophages play in the TLR4 pathway response to LPS (4) or the transformation of stem cells seen in both cirrhosis and the associated HCC in ALD (7). In this review the role played by the following is discussed: (I) cell cycle arrest, (II) TLR signaling macrophages and stem cell transformation to form cancer stem cells, (III) ballooned hepatocytes that form Mallory-Denk bodies as progenitor pre-cancer cells in the pathogenesis of the ALD/HCC transformation.
Cell arrest
Alcohol-induced cell cycle arrest plays a role in the ALD-HCC transformation. It also plays a major role in alcoholic hepatitis (AH) as determined in liver biopsies from AH patients. Our hypothesis is based on the observation that the expression of both PCNA and cyclin D1 is increased in almost all of the hepatocytic nuclei in liver biopsies taken from AH patients. The stain for Ki-67 was positive in only a very few hepatocytes in the same biopsies. Both p21 and p27 positive nuclei were very numerous in these liver biopsies of patients with AH or NASH (7) (Figure 1). This indicates that p21 and p27 inhibition of the cell cycle at both the G1/S growth phase and the G2 phase (8,9) was the reason. Because of the cell cycle arrest, regeneration of liver cells is impeded and apoptosis, genome instability and oncogenic effects result (9). P53 dependent and independent mechanisms of p21 and p27 induction exist. Stress from liver injury increases the expression of p53 and mitochondrial stress, both increasing p21 expression, which leads to cell cycle arrest (10,11). It has been reported that p21, but not Ki-67 expression, is increased in the liver cell nuclei of patients with AH, but not in NASH (12,13). This means that the cell cycle progression is arrested and regeneration of the liver is prevented in AH. A similar phenomenon occurs in decompensated cirrhosis where oxidative stress induces p21 up regulation (14-16). Rats fed ethanol chronically have up regulation of p21 and p27 in liver cell nuclei and this explains how ethanol inhibited liver regeneration after partial hepatectomy (15).
Figure 1.

Liver biopsy from a patient with alcoholic hepatitis showing (A) an immunostain of numerous p27 positive nuclei (700×); (B) MDBs also stain positive (arrows) (1,050×)
The increase in PCNA positive nuclei in AH has been reported previously (12,13). The mechanisms by which p21 regulates cell cycle progression are complex. Inhibition of cyclin/CDK kinase activity by p21 induces cell cycle arrest (17). P21 can directly inhibit PCNA-dependent DNA replication (16,18). In response to mitogen, p21 is induced during the G1 phase and plays a role in normal cell cycle progression (19,20). Activated p53 binds DNA and activates WAF-1/Cdip-1 encoding for p21, which binds to the G1-S/CDK2 and S/CDK complexes (molecules that are important for the G1/S transition) inhibiting their activation. When p21 (WAF 1) is complexed with CDK2 the cell cannot continue to the next stage of the cell cycle.
PCNA positive nuclei are markedly increased in hepatocytes in AH (7,21). PCNA is important for both DNA synthesis and DNA repair (22,23). PCNA becomes post-translationally modified by ubiquitin (24). Polyubiquitin-mediated degradation of cell cycle proteins such as p21 is bound to PCNA by the E3 ligase CRL4 (Cdt2 ubiquitination and the 26s proteasome). This promotes several DNA repair processes when p21 is degraded by the proteasome. PCNA is then freed for the repair process of the DNA (25). If the U3 ligase/proteasome digestion mechanism fails to degrade p21, the cell cycle progression is arrested. This may turn out to be the mechanism involved in HCC formation in ALD, since chronic ethanol feeding leads to inhibition of the 26S proteasome activity in the liver (26). Chronic infection can also induce p21 levels in the liver where the balance of the liver cell proliferation/growth arrest leads to changes in the levels of Gadd 45B, PCNA, cyclin D1, Gadd 45r, p53 and activated caspase 3 (27).
P21 and p27 are up regulated in cirrhosis and HCCs (28) and up regulated by deacetylase inhibitors such as vorinostat (SAHA) used in chemotherapy (29). The implication is that histone acetyltransferases regulate p21 and p27 expression such as HADC1 (30). HADC1 is over expressed in the nuclei of hepatocytes forming Mallory Denk bodies in alcoholic hepatitis (31). P27 has oncogenic effects (32). Therefore, p21 and p27 may play important roles in the pathogenesis of HCCs in ALD patients, probably because of the DNA damage that develops during cell cycle arrest caused by p21 and p27 over expression.
The role of macrophages TLR4 signaling and stem cell transformation to form cancer stem cells in the pathogenesis of ALD-HCC transformation
Liver cell injury in AH is in part, due to macrophage generated proinflammatory cytokines and sinusoidal obstruction. The function of some macrophages (Kupffer cells) causes injury to hepatocytes by way of innate immune injury in response to endotoxin. This was found in rodent models of early alcoholic liver disease and possibly in AH in humans (33). However, these changes are increased in response to acute alcohol ingestion. They are responses that are reversible when ethanol ingestion is stopped in experimental alcohol fed rodent models. The question is: What has happened to the macrophages in chronic alcohol ingestion in humans who have AH? Plasticity and functional polarization are hallmarks of different types of macrophages i.e. M1i, M2a, M2b, and M2c which might be involved in AH.
This differential modulation of the macrophage chemokine system integrates polarized macrophages in pathways of resistance to or promotion of immune-regulation, tissue repair and remodeling (34). The T cell response to chemokines and cytokines differs when M1 and M2 macrophages are compared. M1 has a Th1 response to IFNα and LPS. M2a, b and c give a Th2 response of immune-regulation, matrix deposition and remodeling. M2a is a response to IL-4 and 13, M2b is a response to TLR/IL-1R agonists, and M2c responds to 1L-10 and suppresses immune responses to tissue remodeling (34,35). The type of macrophages in the sinusoids determines the inflammatory process in AH. We have done preliminary studies on the type of macrophages that occupies the sinusoids in liver biopsies of AH. We did IHC stains for CD-68 and CD163 to determine the degree of macrophage infiltrate in the sinusoids in AH (Figure 2A). We were surprised to find that the sinusoids were diffusely filled with macrophages (obstructed) all of which stained heavily for CD163 and not so heavily for CD68. The CD163 (M2c) plays an immuno-regulation role (34). The soluble form of CD163 can be measured in the serum to assess the degree of macrophage activation since CD163 is an activated macrophage marker (35). To assess the sinusoidal macrophages morphologically, we performed electron microscopy (Figure 2B, C). The morphology was that of two types of macrophages. The first type was smaller and filled with phagocytic bodies (secondary lysosomes). The second type was much larger and less common and contained lysosomes and rough ER (Figure 2).
Figure 2.

CD-163 positive macrophages fill all the sinusoids in a liver biopsy from an AH patient ×612 (A); EM of the same liver as A showing 2 types of macrophages in the sinusoids, phagocytic on the left (B arrow) and (C) secretory on the right ×1973
The marked increase in the activity of CD163 positive macrophages involves a cascade of intracellular signals which lead to the secretion of IL6 and CSF1. CD163 positive macrophages are positive for the CD14 and CD16 subunits. CD-163 expression is down regulated by proinflammatory mediators like LPS, IFNg and TNFα. IL-6 and IL-10 strongly up regulate CD-163 (36). Thus, up regulation of CD-163 as noted in the livers of AH implies that the positive staining macrophages are functionally anti-inflammatory (36).
The link between the activated macrophage in the sinusoids in the liver of patients with AH and the development of HCC is through chronic activation of TLR4 in response to a “leaky gut” increase in LPS into the portal vascular system (4). The link to HCC pathogenesis was first developed using a model of alcohol-fed NSSA Tg mice with a diet supplement of LPS. The combination, over time led to synergistic liver damage and liver tumor formation due to alcohol-induced endotoxemia (37). In this mouse model, Nanog, a stem cell/progenitor cell marker, was up regulated by TLR4 activation. CD133/Nanog positive cells were found in the mouse liver tumors that formed (38). These observations supported the concept that the synergism between alcohol abuse and HCV leads to liver tumorigenesis through TLR signaling up regulation of the Nanog expressing stem cells, causing them to transform into cancer stem cells in HCC formation (TISCs). Nanog is up regulated by TLR4 activation. CD133/Nanog positive cells are consequently found in the HCCs of affected Tg mice (39) (Figures 3, 4, 5). CD133, a marker for cancer stem cells, is regulated epigenetically by TGFβ (40). In fact there is compelling evidence that TGFβ signals the expansion of progenitor liver stem cells, which lead to HCC formation and stimulate the progression of the HCCs (41-43). It’s a paradox that the cytostatic, tumor suppressor, TGFβ becomes a tumor promoter, which stimulates the transition from stem cells to progenitor cells to cancer stem cells (39,42,43). Yap1 and Igf2bp3 that are Nanog-dependent genes inhibit TGFβ signaling in TISCs (39). Yap1 and Igf2bp positive cells are present in the livers of ALD and associated HCCs (Figures 3, 4, 5). Taken together, TLR4 expression may be a universal proto-oncogene responsible for the genesis of TLR4-Nanog dependent TISCs (39).
Figure 3.

Immunostain (IHC) of liver showing an HCC. A. Shows a positive stained cell for YAP1 (green); B. Same cell stained positive for 1GF2bdr3 (red); and C. tricolor combining A and B (×654)
Figure 4.

Liver from a patient with alcoholic liver disease showing alcoholic hepatitis and cirrhosis, immunostained for Oct 3-4 (A green), ubiquitin (B red) and (C tricolor) combining A and B. Note the co localization of Oct 3-4 and ubiquitin in the nucleus (×654)
Figure 5.

Liver from a patient with alcoholic liver disease showing cirrhosis and HCC. The photos are of a fibrous septa in the cirrhosis. A. Shows numerous Nanog (green) stem cells; B. One cell staining positive for SOX2 (red) (arrow); C. is tricolor combining A and B (×436)
The role of chronic inflammation of the liver in the development of liver cancer has long been suspected (44). Transcription factors such as TLR4, JNK, NFκB, STAT3, IL-6, IL-1α and EGF receptor are involved in inflammation associated HCC development (44,45). TLR4 and TLR2 signaling activated by inflammation up regulate NFκB and JNK cytokine expression. In experimental alcoholic liver disease TLR4 signaling in mice fed ethanol is increased through a MyD88 independent pathway (46). However, in rats fed ethanol by intragastric tube, where high blood alcohol levels are achieved, TLR4 expression increased as well as MyD88 protein levels indicating that the MyD88 signaling pathway was activated (47). When S-adenosylmethionine was fed with ethanol the up regulation of TLR signaling was prevented indicating that the changes in TLR expression were the result of epigenetic mechanisms. Chronic alcohol feeding also up regulated CD34, FOS, IRF-1, Jun, TLR1, 2, 3, 6 and 7 and Traf6. IL-6, IL10 and IFNγ were also up regulated. Both IL-6 and IL-10 are cytokines that are up regulated by Kupffer cells (M2) in ALD (48). TL-6 activates STAT3. STAT3 acts as a proinflammatory signal (34). The activation of the TLR signaling pathway leads to the up activation of NFκB which stimulates cytokine expression in chronic liver diseases, including ALD and this triggers, over time, the formation of HCC (49).
The role of ballooned hepatocytes that form Mallory-Denk bodies (MDB) as progenitor precancer cells
Balloon cell differentiation (BCD) with (MDB) occurs in chronic hepatitis and cirrhosis due to diverse causes such as alcoholic hepatitis (5). Their occurrence associated with HCC is well established (3). In an experimental mouse model where BCD/MDBs develop in large numbers similar to alcoholic hepatitis, liver tumors develop many months after the withdrawal of the carcinogen DCC. This is similar to the development of the HCCs that develop years after alcohol abstinence in ALD patients (1). In the mouse model BCD/MDBs are associated with the development of preneoplastic changes (48). MDB forming hepatocytes express the same preneoplastic hepatocyte phenotype in both mice (50) and humans (4). The basic morphology of the MDB forming BCD is the same in the human liver and the liver in the mouse model of MDB formation (7) (Figure 6).
Figure 6.

Liver biopsy stained for H&E (A) ×700 and CAM5.2 (B) ×1,050 for keratin 8 and 18. Balloon cells that formed in alcoholic hepatitis are shown where they have formed MDBs. Note that the balloon cells are devoid of keratin except for the MDBs which stain intensely. (C) ×1,875 and (D) ×7,500 electron micrographs of an hepatocyte balloon degeneration cell which had formed an MDB (arrow)
The first change that occurs when the balloon cell degeneration occurs is the disappearance of the keratin 18/8 cytoskeleton and rounding up of the cell. The balloon cell then differs from the normal polyhedral-shaped cell of neighboring hepatocytes (5). Electron microscopy of balloon cells (Figure 6B, C) shows micro-vesicular fat, reduced numbers of mitochondria, reduced glycogen and loss of the normal organelle arrangement due to the loss of the keratin filament structure. The most dramatic change is in the nucleus, which is large, with euchromatin and vesicular with a prominent nucleolus. When the balloon cell nucleus was immunostained for H3K27me3 the fluorescent intensity was low compared to the surrounding normal liver cell nuclei as shown by morphometric comparison (7). Similarly, pEZH2 was increased in the balloon cells that had formed (7). PEZH2 was increased in the liver when measured by Western blot. These observations supported the working hypothesis that the balloon cell change is due to epigenetic alteration of gene expression where the nuclear DNA methylation was reduced and gene expression was up regulated globally (1).
The working hypothesis is that balloon cells are phenotypically changed due to a failure of the H3K27me3/EZH2 to repress gene expression (51). The hallmark of the balloon cell/MDB forming cell is the loss of keratin intermediate filaments which normally span from the plasma membrane to the nuclear membrane (52). Keratin protein regulates protein synthesis and epithelial cell growth in keratinocytes (53). When MDBs form in the balloon cells in AH, the bile canaliculi disappear and organelles become randomly arranged. In an electron microscopic autoradiography study of synthesis of keratin filament protein using radio labeled S35 methionine as a marker, we showed that the nascent keratin proteins went to MDBs preferentially compared to the normally formed intermediate filaments (54).
Most relevant to the role of the BCD/MDB cells linking them to the formation of HCCs is the fact that HCCs often form MDBs in large numbers in humans and in the mouse model (7). In the mouse model the BCD/MDB cells (FAT10+cells) have a growth advantage compared to the normal neighboring cells in response to liver cell injury (1). They show an increased expression of α-fetoprotein, have a decreased expression of DNA repair enzyme glycosylase OGG1, have decreased levels of DNA 5’methyl cytosine, decreased nuclear levels of DNA methyltransferase enzyme DNMT36 and have a large increase in the expression of the mouse form of FAT10 (UBD). Fat10 is over expressed in human HCCs (1,55,56). The markers for the MDB associated preneoplastic phenotype, which indicate that the BCD/MDB cells are preneoplastic; include A2 macroglobin, gamma glutamyl transpeptidase, GSTmu2, fatty acid synthase, glypican-3, p38 and AKT, as well as AFP (1). The BCD cell as well as the MDBs stain positive with an antibody to SOX2 (Figure 7) a marker for hepatic stem cells, suggesting that these cells are stem cell/progenitor cells which have the potential to transform into cancer stem cells, which drive the formation of HCCs (57).
Figure 7.
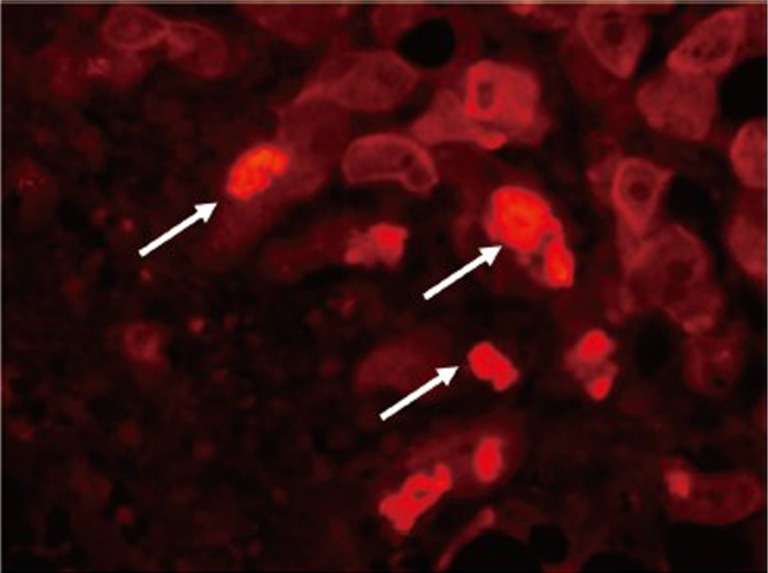
Liver from a patient with alcoholic hepatitis immunostained for SOX2 (red). Note that numerous MDBs stained positive for SOX2 (arrows) (2,224×)
Acknowledgements
The authors thank Adriana Flores for typing the manuscript. Supported by a grant from NIH/NIAAA 6772 and the Morphology Core from grant P50-011999.
Disclosure: The authors declare no conflict of interest.
References
- 1.Oliva J, Bardag-Gorce F, French BA, et al. Fat10 is an epigenetic marker for liver preneoplasia in a drug-primed mouse model of tumorigenesis. Exp Mol Pathol 2008;84:102-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chedid A, Mendenhall CL, Gartside P, et al. Prognostic factors in alcoholic liver disease. VA Cooperative Study Group. Am J Gastroenterol 1991;86:210-6 [PubMed] [Google Scholar]
- 3.Nakanuma Y, Ohta G.Is mallory body formation a preneoplastic change? A study of 181 cases of liver bearing hepatocellular carcinoma and 82 cases of cirrhosis. Cancer 1985;55:2400-4 [DOI] [PubMed] [Google Scholar]
- 4.French SW, Oliva J, French BA, et al. Alcohol, nutrition and liver cancer: role of Toll-like receptor signaling. World J Gastroenterol 2010;16:1344-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.French SW, Nash J, Shitabata P, et al. Pathology of alcoholic liver disease. VA Cooperative Study Group 119. Semin Liver Dis 1993;13:154-69 [DOI] [PubMed] [Google Scholar]
- 6.French SW, Eidus LB, Freeman J. Nonalcoholic fatty hepatitis: An important clinical condition. Canadian J Gastroenterol 1989;3:189-97 [Google Scholar]
- 7.French BA, Oliva J, Bardag-Gorce F, et al. Mallory-Denk bodies form when EZH2/H3K27me3 fails to methylate DNA in the nuclei of human and mice liver cells. Exp Mol Pathol 2012;92:318-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abbas T, Dutta A.p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 2009;9:400-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cazzalini O, Scovassi AI, Savio M, et al. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat Res 2010;704:12-20 [DOI] [PubMed] [Google Scholar]
- 10.Li CH, Tzeng SL, Cheng YW, et al. Chloramphenicol-induced mitochondrial stress increases p21 expression and prevents cell apoptosis through a p21-dependent pathway. J Biol Chem 2005;280:26193-9 [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Jenkins CW, Nichols MA, et al. Cell cycle expression and p53 regulation of the cyclin-dependent kinase inhibitor p21. Oncogene 1994;9:2261-8 [PubMed] [Google Scholar]
- 12.Fang JW, Bird GL, Nakamura T, et al. Hepatocyte proliferation as an indicator of outcome in acute alcoholic hepatitis. Lancet 1994;343:820-3 [DOI] [PubMed] [Google Scholar]
- 13.Crary GS, Albrecht JH. Expression of cyclin-dependent kinase inhibitor p21 in human liver. Hepatology 1998;28:738-43 [DOI] [PubMed] [Google Scholar]
- 14.Lunz JG, 3rd, Tsuji H, Nozaki I, et al. An inhibitor of cyclin-dependent kinase, stress-induced p21Waf-1/Cip-1, mediates hepatocyte mito-inhibition during the evolution of cirrhosis. Hepatology 2005;41:1262-71 [DOI] [PubMed] [Google Scholar]
- 15.Koteish A, Yang S, Lin H, et al. Ethanol induces redox-sensitive cell-cycle inhibitors and inhibits liver regeneration after partial hepatectomy. Alcohol Clin Exp Res 2002;26:1710-8 [DOI] [PubMed] [Google Scholar]
- 16.Luo Y, Hurwitz J, Massagué J.Cell-cycle inhibition by independent CDK and PCNA binding domains in p21Cip1. Nature 1995;375:159-61 [DOI] [PubMed] [Google Scholar]
- 17.Morgan DO. Principles of CDK regulation. Nature 1995;374:131-4 [DOI] [PubMed] [Google Scholar]
- 18.Waga S, Hannon GJ, Beach D, et al. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature 1994;369:574-8 [DOI] [PubMed] [Google Scholar]
- 19.Macleod KF, Sherry N, Hannon G, et al. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev 1995;9:935-44 [DOI] [PubMed] [Google Scholar]
- 20.Gray-Bablin J, Rao S, Keyomarsi K.Lovastatin induction of cyclin-dependent kinase inhibitors in human breast cells occurs in a cell cycle-independent fashion. Cancer Res 1997;57:604-9 [PubMed] [Google Scholar]
- 21.French SW. Epigenetic effects of alcohol-induced hepatocellular carcinoma. Alcohol Res Health. 2012 [Epub ahead of print] [Google Scholar]
- 22.Essers J, Theil AF, Baldeyron C, et al. Nuclear dynamics of PCNA in DNA replication and repair. Mol Cell Biol 2005;25:9350-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shivji KK, Kenny MK, Wood RD. Proliferating cell nuclear antigen is required for DNA excision repair. Cell 1992;69:367-74 [DOI] [PubMed] [Google Scholar]
- 24.Hoege C, Pfander B, Moldovan GL, et al. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002;419:135-41 [DOI] [PubMed] [Google Scholar]
- 25.Abbas T, Sivaprasad U, Terai K, et al. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev 2008;22:2496-506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fataccioli V, Andraud E, Gentil M, et al. Effects of chronic ethanol administration on rat liver proteasome activities: relationship with oxidative stress. Hepatology 1999;29:14-20 [DOI] [PubMed] [Google Scholar]
- 27.Zhang C, Wang J, Lü G, et al. Hepatocyte proliferation/growth arrest balance in the liver of mice during E. multilocularis infection: a coordinated 3-stage course. PLoS One 2012;7:e30127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitselou A, Karapiperides D, Nesseris I, et al. Altered expression of cell cycle and apoptotic proteins in human liver pathologies. Anticancer Res 2010;30:4493-501 [PubMed] [Google Scholar]
- 29.Dokmanovic M, Clarke C, Marke PA. Histone deacetylase inhibitors: Overview and perspectives. Mol Cancer Res 2007;5:981-9 [DOI] [PubMed] [Google Scholar]
- 30.Gui CY, Ngo L, Xu WS, et al. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci U S A 2004;101:1241-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.French SW, Bardag-Gorce F, Li J, et al. Mallory-Denk body pathogenesis revisited. World J Hepatol 2010;2:295-301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Serres MP, Zlotek-Zlotkiewicz E, Concha C, et al. Cytoplasmic p27 is oncogenic and cooperates with Ras both in vivo and in vitro. Oncogene 2011;30:2846-58 [DOI] [PubMed] [Google Scholar]
- 33.Miller AM, Horiguchi N, Jeong WI, et al. Molecular mechanisms of alcoholic liver disease: innate immunity and cytokines. Alcohol Clin Exp Res 2011;35:787-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mellins ED, Macaubas C, Grom AA. Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions. Nat Rev Rheumatol 2011;7:416-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bleesing J, Prada A, Siegel DM, et al. The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor alpha-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum 2007;56:965-71 [DOI] [PubMed] [Google Scholar]
- 36.Buechler C, Ritter M, Orsó E, et al. Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J Leukoc Biol 2000;67:97-103 [PubMed] [Google Scholar]
- 37.Machida K, Chen CL, Liu JC, et al. Cancer stem cells generated by alcohol, diabetes, and hepatitis C virus. J Gastroenterol Hepatol 2012;27:19-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morgan TR, Mandayam S, Jamal MM. Alcohol and hepatocellular carcinoma. Gastroenterology 2004;127:S87-96 [DOI] [PubMed] [Google Scholar]
- 39.Machida K, Tsukamoto H, Mkrtchyan H, et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc Natl Acad Sci U S A 2009;106:1548-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.You H, Ding W, Rountree CB. Epigenetic regulation of cancer stem cell marker CD133 by transforming growth factor-beta. Hepatology 2010;51:1635-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thenappan A, Li Y, Kitisin K, et al. Role of transforming growth factor beta signaling and expansion of progenitor cells in regenerating liver. Hepatology 2010;51:1373-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dooley S, Weng H, Mertens PR. Hypotheses on the role of transforming growth factor-beta in the onset and progression of hepatocellular carcinoma. Dig Dis 2009;27:93-101 [DOI] [PubMed] [Google Scholar]
- 43.Ikegami T.Transforming growth factor-beta signaling and liver cancer stem cell. Hepatol Res 2009;39:847-9 [DOI] [PubMed] [Google Scholar]
- 44.Berasain C, Castillo J, Perugorria MJ, et al. Inflammation and liver cancer: new molecular links. Ann N Y Acad Sci 2009;1155:206-21 [DOI] [PubMed] [Google Scholar]
- 45.Maeda S. NF-κB, JNK and TLR signaling pathways in hepatocarcinogenesis. Gastroenterol Res Pract 2010;2010:367694. [DOI] [PMC free article] [PubMed]
- 46.Szabo G, Mandrekar P, Petrasek J, et al. The unfolding web of innate immune dysregulation in alcoholic liver injury. Alcohol Clin Exp Res 2011;35:782-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oliva J, Bardag-Gorce F, Li J, et al. S-adenosylmethionine prevents the up regulation of Toll-like receptor (TLR) signaling caused by chronic ethanol feeding in rats. Exp Mol Pathol 2011;90:239-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tazawa J, Irie T, French SW. Mallory body formation runs parallel to gamma-glutamyl transferase induction in hepatocytes of griseofulvin-fed mice. Hepatology 1983;3:989-1001 [DOI] [PubMed] [Google Scholar]
- 49.Luedde T, Schwabe RF. NF.-κB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2011;8:108-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nan L, Bardag-Gorce F, Wu Y, et al. Mallory body forming cells express the preneoplastic hepatocyte phenotype. Exp Mol Pathol 2006;80:109-18 [DOI] [PubMed] [Google Scholar]
- 51.Margueron R, Li G, Sarma K, et al. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 2008;32:503-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Katsuma Y, Swierenga SH, Marceau N, et al. Connections of intermediate filaments with the nuclear lamina and the cell periphery. Biol Cell 1987;59:193-203 [DOI] [PubMed] [Google Scholar]
- 53.Kim S, Wong P, Coulombe PA. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature 2006;441:362-5 [DOI] [PubMed] [Google Scholar]
- 54.Kachi K, Cadrin M, French SW. Synthesis of Mallory body, intermediate filament, and microfilament proteins in liver cell primary cultures. An electron microscopic autoradiography assay. Lab Invest 1993;68:71-81 [PubMed] [Google Scholar]
- 55.Canaan A, Yu X, Booth CJ, et al. FAT10/diubiquitin-like protein-deficient mice exhibit minimal phenotypic differences. Mol Cell Biol 2006;26:5180-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee CG, Ren J, Cheong IS, et al. Expression of the FAT10 gene is highly upregulated in hepatocellular carcinoma and other gastrointestinal and gynecological cancers. Oncogene 2003;22:2592-603 [DOI] [PubMed] [Google Scholar]
- 57.Ghodsizadeh A, Taei A, Totonchi M, et al. Generation of liver disease-specific induced pluripotent stem cells along with efficient differentiation to functional hepatocyte-like cells. Stem Cell Rev 2010;6:622-32 [DOI] [PubMed] [Google Scholar]