

Abstract
Disruption of the BRCA2 tumor suppressor is associated with structural and numerical chromosomal defects. The numerical abnormalities in BRCA2 deficient cells may partly result from aberrations in cell division caused by disruption of BRCA2 during cytokinesis. Here we show that BRCA2 is a component of the midbody that is recruited through an interaction with Filamin A actin-binding protein. At the midbody BRCA2 influences the recruitment of endosomal sorting complex required for transport (ESCRT) associated proteins, Alix and Tsg101, and formation of CEP55-Alix and CEP55-Tsg101 complexes during abscission. Disruption of these BRCA2 interactions by cancer associated mutations results in increased cytokinetic defects but has no effect on BRCA2-dependent homologous recombination repair of DNA damage. These findings identify a specific role for BRCA2 in the regulation of midbody structure and function, separate from DNA damage repair, that may explain in part the whole-chromosomal instability in BRCA2-deficient tumors.
Keywords: Cytokinesis, BRCA2, Midbody, Filamin A
Introduction
Germline mutations in the BRCA2 breast and ovarian cancer susceptibility gene result in an increased lifetime risk of breast and other cancers. Tumors forming in these patients and in brca2-/- mouse models exhibit significant structural and numerical chromosomal defects. Since BRCA2 is directly involved in homologous recombination repair of DNA double strand breaks and interstrand crosslinks, the observed structural chromosomal alterations are thought to derive from the absence of RAD51-mediated BRCA2 DNA repair activity. In contrast, whole-chromosomal defects detected in BRCA2 mutant tumors and deficient cells have been associated with aberrations in both chromosome segregation and cell division (Daniels et al., 2004). One explanation for these chromosomal defects is the presence of BRCA2 at the centrosome throughout the cell cycle and the involvement of BRCA2 in centrosome duplication (Wu et al., 2005). Specifically, depletion or inactivation of BRCA2 results in centrosome amplification, which can lead to unequal separation of chromosomes during the metaphase to anaphase transition (Ganem et al., 2009). BRCA2 has also been found to localize to the central spindle and midbody during telophase and cytokinesis, and depletion or inactivation of BRCA2 has been associated with multinucleation (Daniels et al., 2004; Jonsdottir et al., 2009; Ryser et al., 2009). While the influence of BRCA2 on cytokinesis is not well defined, loss of BRCA2 activity during this phase of the cell cycle has been implicated in disruption of myosin II organization at the cleavage furrow and the intercellular bridge. Similarly, disruption of the interaction between BRCA2 and HMG20b, a kinesin-like coiled coil protein implicated in G2-M transition has been associated with defects in the completion of cell division (Lee et al., 2011). Furthermore, it has been suggested that BRCA2 complexes with Aurora B, an important regulator of midbody function, during cytokinesis (Ryser et al., 2009). In contrast one study based on BRCA2 depletion by siRNA has suggested that BRCA2 is not localized to the midbody and has no influence on cytokinesis (Lekomtsev et al., 2010).
Here we confirm that BRCA2 localizes to the central spindle and midbody during telophase and cytokinesis, and we demonstrate that absence of BRCA2 from the midbody disrupts localization of several components of the central spindle and the midbody and impairs cytokinesis. We establish that an interaction between BRCA2 and Filamin A is required for the recruitment of BRCA2 to the central spindle and the midbody. In addition, we provide evidence suggesting that BRCA2 influences CEP55-dependent midbody localization of endosomal sorting complex required for transport (ESCRT) complexes that are required for abscission during the terminal stage of cytokinesis. Furthermore, we identify breast cancer associated mutations in the Filamin A interacting domain of BRCA2 that exclude BRCA2 from the midbody and mutations in the N-terminal CEP55, Alix and Tsg101 interacting domains of BRCA2 that disrupt these interactions and reduce ESCRT complex recruitment to midbody. Thus, disorganization of the midbody caused by the absence or disruption of BRCA2 may account, in part, for the numerical chromosomal instability observed in BRCA2-deficient tumors.
Results
BRCA2 is a component of the midbody
To verify that BRCA2 localizes to the spindle midzone during telophase and the intercellular bridge and midbody during late abscission (Daniels et al., 2004) we studied the localization of endogenous BRCA2 in HeLa cells throughout mitosis by immunofluorescence (IF) microscopy. BRCA2 was detected at the centrosome, the spindle midzone during telophase (Figure 1A), and at the midbody during abscission and cytokinesis (Figure 1A, high magnification), co-incident with MgcRacGAP, a marker of the central spindle and the midbody (Figure 1B). The specificity of antibodies for BRCA2 was verified by an inability to detect BRCA2 at the centrosome, midzone, or midbody by immunofluorescence in the presence of BRCA2 siRNAs. In addition, quantification of BRCA2 intensity at the midbody and normalization by adjacent α-tubulin intensity in 293T cells treated with a BRCA2 siRNA pool (Dharmacon), two individual BRCA2 siRNAs (Qiagen) and a BRCA2 siRNA scrambled control showed that both antibodies (Ab-1 and Ab-2) detected much reduced levels of BRCA2 following BRCA2 depletion by siRNAs (Figure S1A). The Ab-1 antibody also detected BRCA2 at the midbody, centrosomes and central spindle during mitosis in pancreatic tumor cells from brca2+/+ but not brca2-/- mice (Figure 1C and S1B). Western blot analysis of the cell extracts derived from brca2-/- and brca2+/+ mice confirmed the absence of BRCA2 from brca2-/- cells (Figure S1C). In addition, time-lapse microscopy of cells expressing EGFP-tagged BRCA2 (WT-BRCA2) and mCherry tagged α-tubulin identified BRCA2 at centrosomes, the spindle midzone, and the midbody until dissolution of the intercellular bridge and separation of daughter cells (Figure S1D and Movie-S1).
Figure 1. Influence of midbody-associated BRCA2 on cytokinesis.
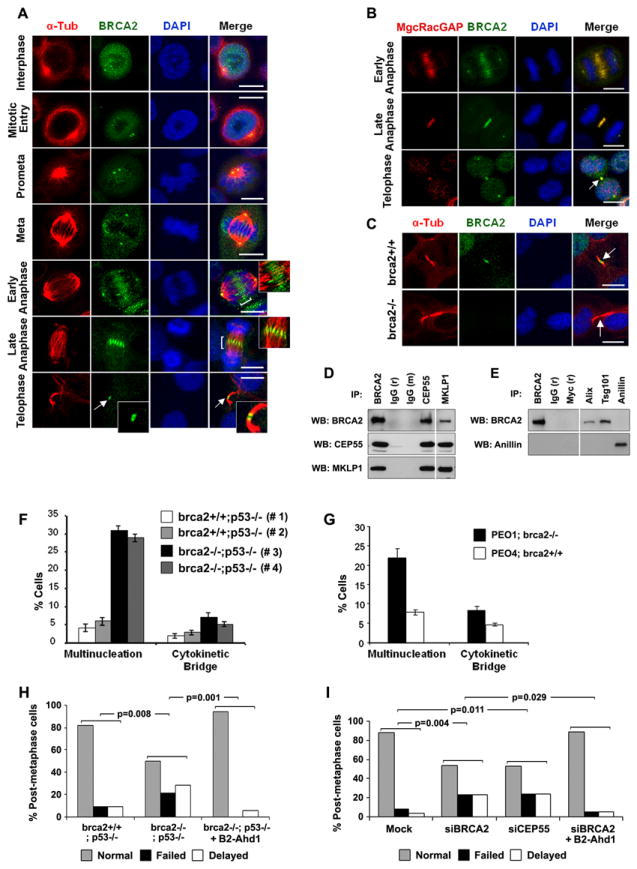
(A) Cellular localization of BRCA2 in mitotic HeLa cells was evaluated by immunofluorescence (IF) staining for BRCA2 and α-tubulin. (B) IF of HeLa cells for BRCA2 and MgcRacGAP during anaphase and telophase. (C) IF of brca2+/+ and brca2-/- cells for Brca2. Arrows indicate the midbody. Bars (A-C) 10μm. (D, E) Immunoblotting (WB) of defined immunoprecipitates (IP) from mitotic HeLa cells for BRCA2, CEP55 and MKLP1 (D), and for BRCA2 and Anillin (E). (F-G) Quantification of multinucleation and unresolved cytokinetic bridges in brca2+/+ (#1, #2) and brca2-/- (#3, #4) tumor cells (F) and PEO1 and PEO4 tumor cells (G). Error bars represent standard error of the mean (SE). (H) Quantitation (%) of post-metaphase brca2+/+ and brca2-/- cells and B2-Adh1 reconstituted brca2-/- cells exhibiting normal, failed or delayed (for > 1hr) cytokinesis. (I) Quantitation (%) of post-metaphase HeLa cells treated with BRCA2, CEP55 siRNA, or BRCA2 siRNA and B2-Adh1 as in (H). The significance (p-values) of differences between ratios of combined failed/delayed cytokinesis to normal cytokinesis under various conditions are shown. (See also Figures S1, S2 and supplemental movies 1-4).
To evaluate possible interactions between BRCA2 and central spindle and midbody proteins we immunoprecipitated FLAG-tagged BRCA2 from 293T cells and western blotted for a number of midbody markers. We verified that BRCA2 formed a complex with Aurora B (Figure S1E) (Ryser et al., 2009), and showed that BRCA2 interacted with PRC1 (Figure S1E) and CEP55 (Figure 1D). Characterization of immune complexes from mitotic HeLa cells confirmed associations between endogenous BRCA2 and components of the central spindle and midbody including MKLP1, CEP55 and Aurora B (Figure 1D and Figure S1F), which localize to the midbody and are involved in cytokinesis (Morita et al., 2007). We also verified a previously described interaction between BRCA2 and Filamin A (Yuan and Shen, 2001), a component of the actomyosin complex, during cytokinesis (Figure S1G). However, we could not detect any interaction between BRCA2 and Anillin (Figure 1E), which colocalizes with myosin II at the cellular cortex during cleavage furrow formation.
Inactivation or depletion of BRCA2 induces a failure of cytokinesis
Given the interaction of BRCA2 with components of the central spindle and midbody, we further evaluated the influence of BRCA2 on cytokinesis. Consistent with previous reports of a failure of cytokinesis in BRCA2 deficient cells (Daniels et al., 2004; Jonsdottir et al., 2009; Ryser et al., 2009) brca2-/- murine pancreatic tumor cells displayed an elevated frequency of unresolved cytokinetic bridges and multinucleated cells compared to brca2+/+ cells (Figure 1F and Figure S1H). Similar effects of brca2 deficiency were observed in mouse embryonic fibroblast (MEF) lines derived from the brca2-/- and brca2+/+ mice, and in human BRCA2 deficient PEO1 ovarian cancer cells, relative to spontaneously reverted wildtype BRCA2 PEO4 controls (Sakai et al., 2009; Sakai et al., 2008) (Figure 1G). To demonstrate a direct effect of BRCA2 deficiency on cytokinesis we performed live cell microscopy of H2B-mRFP tagged cells derived from the brca2-/- and brca2+/+ mice. We focused on post-metaphase mitotic cells and categorized cells by normal, delay (> 1hr to progress from early anaphase to complete abscission), or failure (inability to complete abscission) of cytokinesis (Movies S2-S4). Brca2-/- cells displayed significantly greater delay or failure of cytokinesis than brca2+/+ cells (p=0.008) (Figure 1H). In addition, acute depletion of BRCA2 from HeLa cells by siRNA (Figure S1I) increased delay and failure of cytokinesis relative to controls (p=0.004), similarly to the depletion of CEP55 (p=0.01), a known mediator of cytokinesis (Zhao et al., 2006) (Figure 1I and Figure S1J).
Next, we evaluated the ability of BRCA2 to rescue the cytokinetic defects in BRCA2 deficient cells. Initially we generated a BRCA2 mini-gene (B2-Ahd1) that eliminated the central Rad51-binding domains and part of the DNA binding domain of BRCA2 but retained the N- and C- termini (Wang et al., 2011; Yuan and Shen, 2001) (Figure S1K), such that effects on cytokinesis could be studied independently of influences on DNA damage repair. B2-Ahd1 expressed a ~200kd protein that appropriately localized to the centrosome and midbody of 293T cells (Figure S1, L and M), interacted with midbody proteins CEP55 and Filamin A (Figure S1N), and exhibited resistance to BRCA2 siRNA depletion (Figure S1M) due to the absence of the siRNA binding site. Importantly, B2-Ahd1 reduced the frequency of cytokinesis defects in brca2-/- cells similarly to wildtype BRCA2 (Figure S1O). In addition, live cell imaging of post-metaphase mitotic brca2-/- cells (p=0.001) and HeLa cells treated with BRCA2 siRNA (p=0.03) showed that expression of B2-Ahd1 significantly reduced the cytokinetic defects associated with BRCA2 absence or depletion (Figure 1, H and I).
Failure of cytokinesis in BRCA2 deficient cells is not associated with chromatin/DNA bridges
Given these data we postulated that the failure of cytokinesis associated with BRCA2 deficiency results from disruption of a specific BRCA2 function at the midbody. However, since unresolved DNA and chromatin bridges during anaphase and telophase can induce cytokinesis defects (Steigemann et al., 2009), DNA segregation abnormalities might contribute to the cytokinetic defects in BRCA2 deficient cells. Here we adopted Lap2β (Steigemann et al., 2009) as a marker of unresolved DNA and chromatin bridges during late abscission, after verifying a significant increase in the frequency of Lap2β staining chromatin bridges in HeLa cells treated with etoposide (p=0.03) to induce segregation defects. No significant difference in the frequency of Lap2β staining was observed in murine brca2-/- cells relative to brca2+/+ cells (p=0.86) (Figure S2, A and B), in PEO1 BRCA2 deficient cells relative to isogenic wildtype BRCA2 PEO4 cells (Sakai et al., 2008) (p=0.81), and in HeLa cells with and without BRCA2 siRNA treatment (p=0.93) (Figure S2, A and B). Because of the known interaction between BRCA2 and Filamin A, we also evaluated but found no difference in the frequency of chromatin bridges in human Filamin A deficient cells and wild type controls (p=0.95) (Figure S2, A and B). In contrast, elevated levels of unresolved cytokinetic bridges were observed in all BRCA2 deficient cells compared to cells expressing wildtype BRCA2.
Since ultrafine DNA bridges (UFBs) are an established source of cytokinesis defects in certain cells types (Vinciguerra et al., 2010) we also compared the frequencies of UFBs in BRCA2 depleted cells, control cells and FANCA depleted cells. While immunostaining of Bloom’s syndrome protein (BLM) revealed a high frequency (~52%) of UFBs in FANCA depleted cells, only 22% of BRCA2 depleted cells and 16% of control cells displayed UFBs (Figure S2C-E). Overall these results indicate that unresolved UFBs and chromatin bridges do not account for much of the influence of BRCA2 inactivation/ depletion on cytokinesis.
BRCA2 is required for localization of microtubule associated proteins to the midbody
Further characterization of the influence of BRCA2 on the midbody by immunofluorescence analysis revealed that BRCA2 deficiency had no effect on the localization of Plk1, Aurora B (Figure S3A), Eg5, CENP-E, Anillin, F-actin (data not shown), Filamin A and CEP55 (Figure 2, A and B) to the central spindle and midbody during telophase and cytokinesis. In contrast, and consistent with previous observations, myosin II was depleted and mislocalized at the cleavage furrow and the spindle midzone (Figure 2, A and B) (Daniels et al., 2004). In addition, the staining for MKLP1 and MKLP2 during late abscission was disordered and no longer specific to the midbody ring in brca2-/- cells (Figure 2, A and B). Defective localization of MKLP1 at the midbody ring was highly significantly associated with BRCA2 inactivation (p<0.001) (Figure 2C). Similarly, PRC1 inappropriately stained the entire cytokinetic bridge during abscission indicating an inability of PRC1 to efficiently localize to the midbody in the absence of BRCA2 (Figure 2B) (Zhao et al., 2006). In contrast, the absence of BRCA2 had no effect on the localization of MKLP1, MKLP2 and PRC1 to the central spindle (Figure S3B). The mislocalization of these markers from the midbody, but not from the central spindle, suggested a specific role for BRCA2 in maintaining the structure of the midbody.
Figure 2. BRCA2-dependent localization of midbody markers.
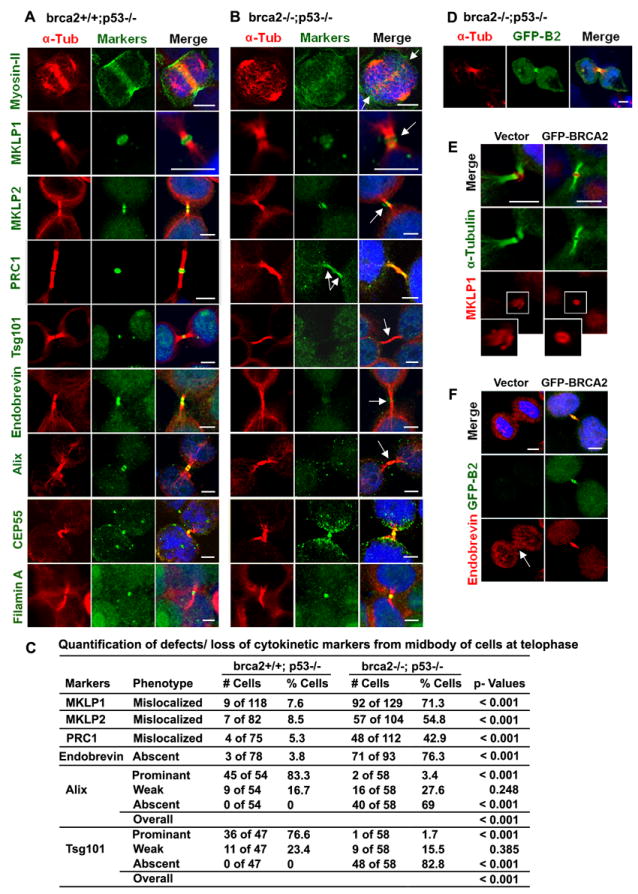
(A-B) Localization of midbody markers in brca2-/- and brca2+/+ cells by immunofluorescence (IF) staining with antibodies against markers (green) and α-tubulin (red). (C) Quantification of proportions of cells from (A,B) displaying defects in localization of markers at the midbody. p-values for differences in proportions of cells exhibiting defined defects were obtained by Poisson regression analysis. (D) Reconstitution of GFP-BRCA2 to the centrosomes and midbody of brca2-/- mouse tumor cells. (E, F) IF for MKLP1 (red in E) or Endobrevin (red in F) and α-tubulin (green) in brca2-/- mouse tumor cells after reconstitution with GFP-BRCA2 (green). Arrows (B and F) indicate mislocalized markers in brca2-/- cells; Bars (A-B and D-F), 5μm. (See also Figure S3).
The final stage of cytokinesis involves ESCRT-associated membrane abscission that is mediated, in part, by CEP55 interactions with Alix and Tsg101 (Carlton and Martin-Serrano, 2007; Morita et al., 2007). Staining for these markers in brca2-/- and brca2+/+ cells identified significant differences in the levels of both Alix (p<0.001) and Tsg101 (p<0.001), when categorized as prominent, weak and absent staining at the midbody (Figure 2, A-C). Furthermore, the v-SNARE complex component, Endobrevin, that is involved in membrane fission during terminal cytokinesis, was also frequently reduced or absent from the midbody of brca2-/- cells (Figure 2, A and B). Although CEP55 localization to the midbody appeared normal, the mislocalization of Alix, Tsg101, and Endobrevin was consistent with the effect of CEP55 knockdown on cytokinesis (Zhao et al., 2006), where the midbody ring is disrupted, both Tsg101 and Endobrevin are absent from the midbody (Zhao et al., 2006), and the organization of midbody microtubules is altered (Matuliene and Kuriyama, 2002). However, BRCA2 deficiency differs from CEP55 deficiency in that Plk1 and Aurora B are absent from the central spindle and midbody in CEP55 depleted cells (Zhao et al., 2006), but are present in BRCA2 deficient cells (Figure S3A).
To confirm the mislocalization of midbody markers in the absence of functional BRCA2 protein, we also assessed the ability of reconstituted BRCA2 (Figure S3C) to correct the defective localization of midbody markers in brca2-/- cells. GFP-tagged BRCA2 correctly localized to the centrosome and midbody during telophase (Figure 2D), relocalized MKLP1 to the midbody ring (Figure 2E) and relocalized Endobrevin to the cytokinetic bridge and the midbody (Figure 2F).
Filamin A mediates BRCA2 localization to the midbody
To identify the domain of BRCA2 responsible for localization to the midbody, the ability of nine overlapping FLAG-tagged BRCA2 fragments to localize to this structure was evaluated by immunofluorescence staining (Figure 3A). The BRCA2 F8 fragment containing amino acids 2516-3030 displayed prominent staining at the spindle midzone and midbody (Figure S3D) and BRCA2 fragments F2 and F3 displayed weak staining at the midbody (Figure S3E). Consistent with the F8 findings, time lapse microscopy of 293T cells expressing a GFP-tagged C-terminal BRCA2 fragment (amino acids 2126-3418) detected the BRCA2 fragment at the central spindle following initiation of furrow ingression and at the midbody during abscission (Movie S5). Importantly, the F8 region of BRCA2 contains part of the BRCA2 DNA binding domain (Yang et al., 2002) and a binding domain (amino acids 2973 to 3001) for Filamin A (Yuan and Shen, 2001) and BCCIPα (Liu et al., 2001).
Figure 3. Characterization of the Filamin A binding domain of BRCA2.
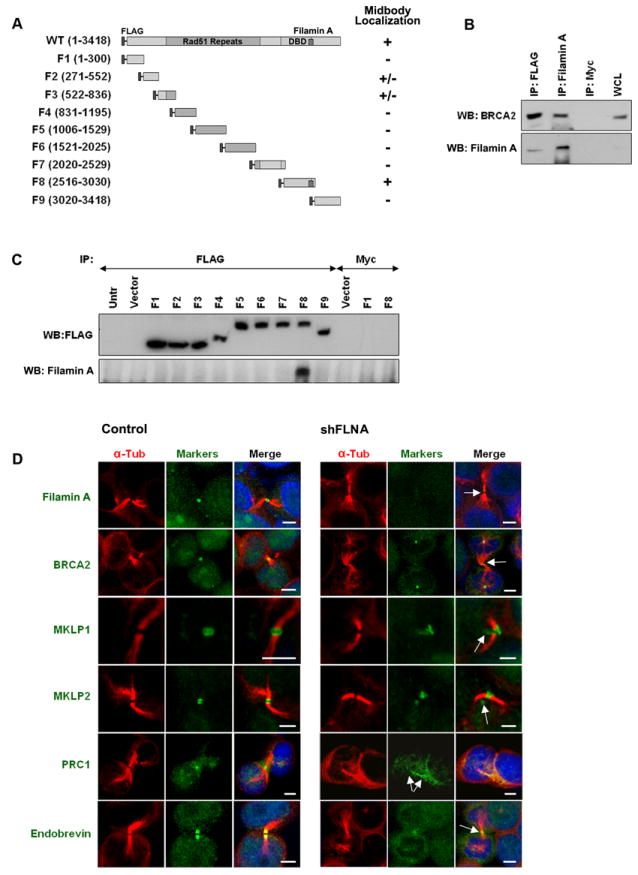
(A) Schematic representations of BRCA2 fragments and the ability (+), inability (-), or partial ability (+/-) to localize to the midbody. Amino acids positions are shown in parentheses. (B) Interaction between BRCA2 and Filamin A. Immunoblotting (WB) for BRCA2 and Filamin A after the indicated IPs from 293T cells expressing FLAG-BRCA2. (C) Identification of a BRCA2 Filamin A interaction domain by immunoblot (WB) analysis for FLAG and Filamin A in anti-FLAG IPs from 293T cells expressing FLAG-tagged fragments of BRCA2. (D) Localization by IF of midbody markers (green) and α-tubulin (red) in 293T cells treated with lentiviral control or FLNA (Filamin A) shRNAs. Bars (D), 5μm. (See also Figure S4 and supplemental movie 5).
The Filamin A actin binding protein is associated with the cleavage furrow (Nunnally et al., 1980), acts as a scaffold for cytoskeletal and cytokinetic proteins (Playford et al., 2006), is present at the spindle midzone and the midbody during telophase (Figure 2A), and interacts with both full-length BRCA2 (Figure 3B and Figure S1G) and BRCA2-F8 (Figure 3C). To determine whether BRCA2 is dependent on Filamin A for localization to the central spindle and midbody, we depleted Filamin A from 293T cells and from brca2+/+ tumor cells using anti-Filamin A lentiviral shRNAs. In the absence of Filamin A, BRCA2 was substantially depleted from the spindle midzone and midbody but was retained at centrosomes (Figure 3D and Figure S4A). In addition, MKLP1 and MKLP2 displayed disordered localization at the midbody ring (Figure 3D), PRC1 stained the entire intercellular bridge (Figure 3D), and the levels of Endobrevin at the midbody were substantially reduced (Figure 3D and Figure S4A). To exclude the possibility of off-target shRNA effects, midbody localization of BRCA2, PRC1, Endobrevin, MgcRacGAP and Plk1 were assessed in melanoma cells lacking Filamin A (M2/FLNA-/-) or stably reconstituted with Filamin A (A7/FLNA+/+) (Cunningham et al., 1992). In the absence of Filamin A the levels of BRCA2 and Endobrevin at the central spindle and midbody were substantially reduced (Figure S4B) and PRC1 no longer localized to the midbody (Figure S4B), whereas, MgcRacGAP and Plk1 localization to the midbody was unaffected (Figure S4C). Furthermore, FLNA-/- cells displayed elevated levels of multinucleation and unresolved cytokinetic bridges similarly to brca2-/- cells (Figure S4, D and E).
Mutations in the Filamin A binding domain of BRCA2 disrupt localization to the midbody
Next we explored the influence of missense mutations in the Filamin A binding domain of BRCA2 on the interaction with Filamin A, localization of BRCA2 to the central spindle and midbody, and the integrity of cytokinesis. We selected 12 mutations from the Filamin A binding site (residues 2970-3020) of BRCA2 for evaluation, including five mutations (L2972W/R2973C/E3002D/E3002K/S3020C) from the Breast Cancer Information Core (BIC) website which lists cancer-associated missense mutations in the BRCA2 gene identified through clinical genetic screening of high-risk breast cancer families; six mutations (L2987A/W2990A/L3000A/G3003A/R3005A/S3016A) in residues fully conserved between pufferfish (Fugu rubripes) and human; and one mutation in a non-conserved residue (K3017A) as a control (Figure 4A and Figure S5A). Full-length FLAG-tagged BRCA2 expression constructs encoding these missense mutant proteins (Farrugia et al., 2008; Wu et al., 2005) were immunoprecipitated from 293T cells and western blotted for BRCA2 and Filamin A to assess the ability of each mutation to disrupt the BRCA2-Filamin A interaction. Of the 12 mutations, E3002K disrupted the interaction with Filamin A, R3005A substantially reduced the interaction, and E3002D partially reduced the interaction (Figure 4A and Figure S5A). Confocal microscopy showed that the E3002K and R3005A mutants, failed to localize efficiently to the midbody during abscission (Figure 4B). In contrast, other mutants that did not influence the interaction with Filamin A, such as L2987A and W2990A, localized normally to the midbody (Figure 4B). Live cell imaging of 293T cells expressing GFP-tagged full-length E3002K BRCA2 also confirmed that this mutant was unable to localize to the central spindle or the midbody (Figure S5B), although the mutant did localize to centrosomes (Figure S5B and Movie S6).
Figure 4. Influence of BRCA2 missense mutations on Filamin A binding and localization of midbody markers.
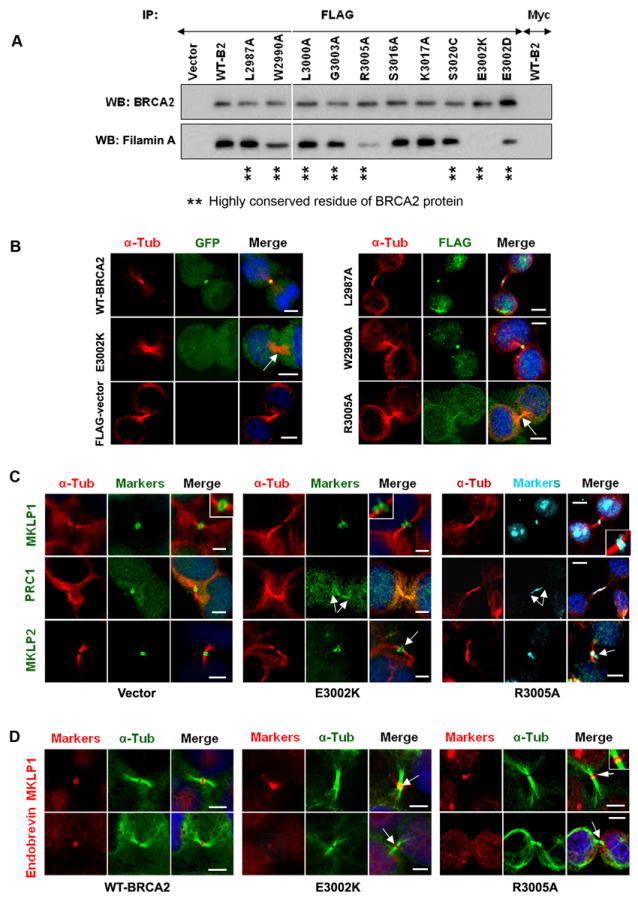
(A) Disruption of the Filamin A interaction with BRCA2 by BRCA2 mutations shown by immunoblot (WB) for BRCA2 and Filamin A in anti-FLAG IPs from 293T cells expressing FLAG-tagged wildtype (WT-B2) or missense mutants of BRCA2. (B) Absence of BRCA2 mutants from the midbody shown by direct visualization of GFP-tagged BRCA2 and by IF for FLAG (green) and α-tubulin (red) in 293T cells expressing FLAG-tagged WT or mutant BRCA2. (C) Disruption of midbody marker localization by E3002K and R3005A shown by IF of 293T cells expressing mutant BRCA2 for midbody markers (green) and α-tubulin (red). (D) Reconstitution of midbody components by E3002K and R3005A in brca2-/- cells shown by IF of brca2-/- cells expressing mutant BRCA2 for α-tubulin (green) and MKLP1 or Endobrevin (green) in. Arrows indicate absence of BRCA2 protein (B) and disruption of midbody markers (C and D). Bars (B-D), 5μm. (See also Figure S5 and supplemental movie 6).
We then used the E3002K and R3005A mutants to assess the effect of disrupting the Filamin A-BRCA2 interaction on the localization of central spindle and midbody markers. Neither mutant excluded Filamin A from the central spindle or the midbody. However, both mutants disordered localization of MKLP1 and MKLP2 at the midbody ring and mislocalized PRC1 along the intercellular bridge during abscission (Figure 4C), but did not alter the localization of Plk1 and CEP55 to the midbody (Figure S5C). The mutants also had no influence on the localization of these markers to the central spindle (data not shown). In additional studies in brca2-/- cells, E3002K and R3005A did not rescue MKLP1 localization to the midbody ring or reconstitute Endobrevin at the midbody during abscission (Figure 4D). Together these findings are consistent with a direct influence of BRCA2 on the midbody during late abscission that is dependent on recruitment of BRCA2 to the midbody through an interaction with Filamin A.
Disruption of the Filamin A interacting domain in BRCA2 causes a failure of cytokinesis
To confirm that disruption of the BRCA2-Filamin A interaction resulted in reduced efficiency of cytokinesis we evaluated the ability of the BRCA2 missense mutants to induce multinucleation and unresolved cytokinetic bridges in 293T cells. Immunofluorescence analysis determined that the E3002K and R3005A mutations, which disrupted the BRCA2-Filamin A interaction, induced substantially elevated frequencies of unresolved cytokinetic bridges and multinucleated cells, whereas other mutations had no effect (Figure 5A). Likewise, E3002D induced a moderate increase in multinucleation and cytokinetic bridges, consistent with the mild effect of this mutation on Filamin A binding (Figure 4A and Figure 5A). Similar dominant negative effects on cytokinesis were observed when expressing these mutants in brca2+/+ cells (Figure 5B). In addition, in contrast to wildtype BRCA2 reconstitution of BRCA2 deficient cells, the E3002K and R3005A mutants failed to reduce the frequency of cytokinetic bridges and multinucleation (Figure 5C). Furthermore, quantitation of chromosome numbers in metaphase spreads of brca2-/- and brca2+/+ cancer cell lines showed that the E3002K and R3005A mutants increased whole-chromosomal instability in brca2+/+ cells and failed to reduce instability in brca2-/- cells similarly to wildtype BRCA2 (Figure 5D), consistent with a delay or failure of cytokinesis.
Figure 5. Association of BRCA2 mutations with cytokinetic but not DNA repair defects.
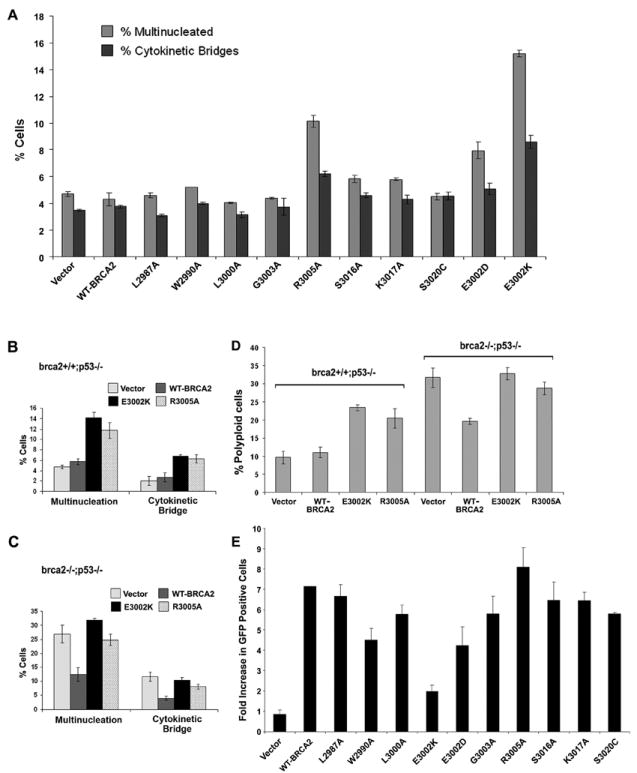
(A) Percent of 293T cells (n=200) with multiple nuclei (multinucleated) and unresolved cytokinetic bridges 72 hrs after expression of wildtype (WT) and missense mutants of BRCA2. (B,C) Percent of brca2+/+ (n=200) (B) or brca2-/- (n=200) (C) cells with multiple nuclei and unresolved cytokinetic bridges 72 hr after expression of E3002K and R3005A mutants. (D) Percent polyploidy of brca2+/+ and brca2-/- cells in metaphase spreads (n=50), 72 hr after expression of wildtype (WT) E3002K or R3005A BRCA2. Error bars (A-D) show SE. (E) Rescue of homologous directed repair activity by BRCA2 mutants in brca2 deficient DR-GFP V-C8 cells. Fold increase in GFP positive cells relative to vector identified by FACS analysis following co-transfection with I-Sce1 and WT or mutant BRCA2 constructs.
Since numerical chromosomal alterations can also result from multipolar mitoses and merotelic attachment of microtubules following centrosome amplification (Ganem et al., 2009), we also evaluated the influence of the E3002K and R3005A variants on centrosome number. Immunofluorescence staining for centrosomal γ-tubulin in brca2+/+ and brca2-/- cells expressing the FLAG-tagged E3002K or R3005A mutants showed that both mutants maintained a normal centrosome complement in brca2+/+ cells and partially rescued centrosome amplification in brca2-/- cells, similarly to wildtype BRCA2 (Farrugia et al., 2008) (Figure S5D). Thus, the failure of cytokinesis and chromosomal instability induced by disruption of the BRCA2-Filamin A interaction is independent of an effect on centrosome duplication.
BRCA2 function at the midbody is independent of a role in DNA repair
Since the Filamin A binding and midbody localization domain of BRCA2 overlaps with the homologous recombination (HR) DNA repair associated DNA binding domain, we assessed whether disruption of Filamin A binding also disrupted the homology directed repair (HDR) activity of BRCA2. All mutations that had no effect on Filamin A binding to BRCA2 reestablished wildtype HDR activity in BRCA2 deficient V-C8 cells (Figure 5E). However, the E3002K mutant that disrupted binding and increased the frequency of cytokinetic failure (Figure 5A) failed to rescue HDR activity, consistent with disruption of the BRCA2 DNA binding domain. Similarly, the E3002D and W2990A mutants that partially reduced Filamin A binding failed to completely rescue HDR activity (Figure 5E). In contrast, the BRCA2 R3005A mutation that disrupted Filamin A binding and promoted failure of cytokinesis successfully corrected HDR activity similarly to wildtype BRCA2 (Figure 5E). Thus, we conclude that the R3005A mutation in the Filamin A interacting domain of BRCA2, which overlaps with the DNA binding domain, can distinguish between the influence of BRCA2 on cytokinesis and on DNA repair.
BRCA2 regulates the presence of CEP55 signaling complexes at the midbody
While recruitment of BRCA2 to the midbody is important for completion of cytokinesis, the mechanisms by which BRCA2 regulates these processes are unclear. We previously showed that BRCA2 interacts with CEP55 (Figure 1D), a midbody component that has been implicated in the formation of the midbody ring (Zhao et al., 2006) and localization of ESCRT complexes to the midbody through interactions with Alix and Tsg101 (Carlton and Martin-Serrano, 2007; Morita et al., 2007). Immunoprecipitation and western blotting were subsequently used to confirm interactions between ectopically expressed BRCA2 and CEP55 (Figure S6A) and to show that BRCA2 also formed complexes with Alix and Tsg101 in cells aligned in mitosis (Figure S6B). Next we established that recombinant GST-tagged BRCA2 fragment F2 (amino acids 421-982) directly bound to recombinant His-tagged full-length CEP55 (Figure 6A) and to a fragment of CEP55 (amino acids 160-217) containing the ESCRT- and Alix-binding region (EABR) (Figure 6A). Given this interaction between BRCA2 and the CEP55 EABR domain, we explored the influence of BRCA2 on the formation of CEP55-Alix and CEP55-Tsg101 complexes. CEP55 interactions with Alix and Tsg101 were substantially reduced in brca2-/- cells relative to brca2+/+ tumor cells, and in BRCA2 depleted 293T cells relative to controls (Figure 6B), suggesting a role for BRCA2 in the formation of these complexes.
Figure 6. Influence of BRCA2 on CEP55-Alix and CEP55-Tsg101 complex formation.
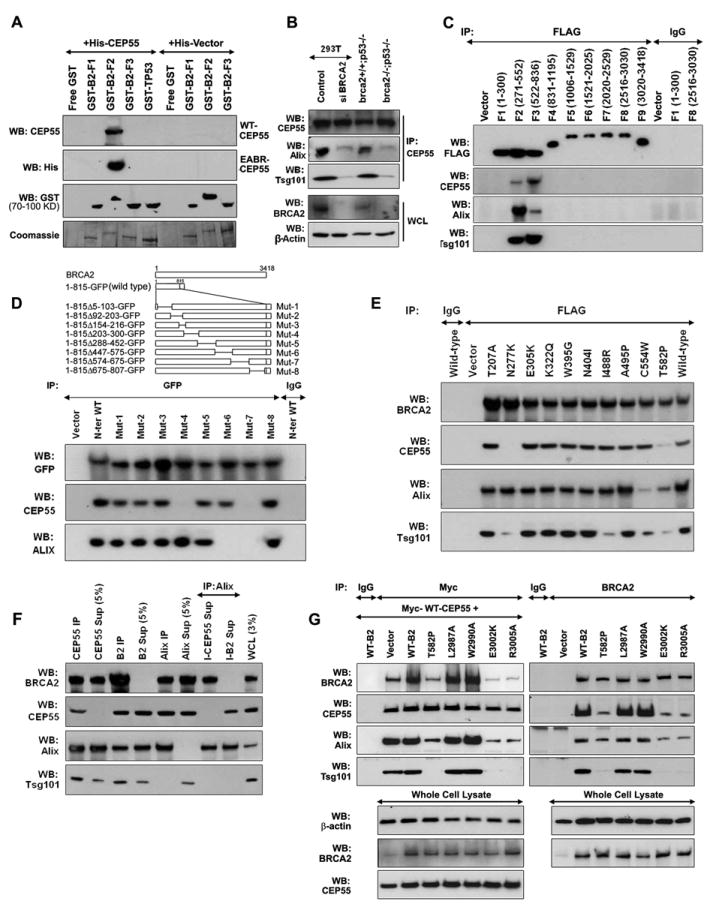
(A) In vitro binding of His-tagged CEP55 and His-tagged EABR-CEP55 to GST-tagged fragments of BRCA2 shown by WB for CEP55, His-tag and GST; coomassie stained gels show bacterially expressed BRCA2 fragments. GST-TP53 is a negative control. (B) Influence of BRCA2 loss on CEP55-Alix and CEP55-Tsg101 complexes. Immunoblot (WB) for CEP55, Alix, and Tsg101 in endogenous CEP55 immunoprecipitates (IPs) from brca2+/+ and brca2-/- cells and 293T cells treated with control or BRCA2 siRNA. Whole cell lysates (WCL) are shown as controls. (C) Identification of the CEP55, Alix and Tsg101 interacting domains of BRCA2 by WB of anti-FLAG IPs from 293T cells expressing FLAG-tagged fragments of BRCA2. (D) Fine-mapping of Alix and CEP55 interacting domains of BRCA2; GFP, CEP55 and Alix WB of anti-GFP IPs from late mitotic 293T cells expressing EGFP-tagged WT and internal deletion mutants (shown in schematic) of a BRCA2 N-terminal fragment (aa 1-815). (E) Effect of BRCA2 missense mutations on the BRCA2 interactions with CEP55, Alix and Tsg101; BRCA2, CEP55, Alix and Tsg101 WB of anti-FLAG IPs from late mitotic 293T cells expressing FLAG-tagged full-length WT and missense mutants of BRCA2 (as indicated). (F) WB (as shown) of IPs, IP supernatants and anti-Alix secondary-IP of CEP55 and BRCA2 (B2) immunodepleted supernatants from Hela cells synchronized in late mitosis. Sup: supernatant; I-CEP55: CEP55 immunodepleted supernatant; I-BRCA2: BRCA2 immunodepleted supernatant. (G) Disruption of CEP55-Alix and CEP55-Tsg101 complexes by BRCA2 mutations. WB (as shown) of Myc or BRCA2 IPs from synchronized 293T cells, 48 hr after transfection with vector, wildtype (WT-B2) or indicated mutant BRCA2 constructs in combination with Myc-tagged CEP55. (See also Figure S6).
Fine mapping of the regions of BRCA2 involved in the interactions was undertaken to gain further insight into the composition of the complexes. Immunoprecipitation of BRCA2 fragments showed that CEP55, Alix and Tsg101 all interacted with BRCA2-F2 (amino acids 271-552) and BRCA2-F3 (amino acids 522-836) (Figure 6C). Further mapping of the interactions of CEP55 and Alix with an N-terminal BRCA2 fragment (amino acids 1-815) containing eight different ~100 amino acid in-frame deletions in 293T cells synchronized in late mitosis (Figure 6D) found that amino acids 574-675 (Mut-7) of BRCA2 were required for interactions with both CEP55 and Alix. In addition, amino acids 203-300 (Mut-4) were necessary for the interaction with Alix, whereas amino acids 447-575 (Mut-6) were only necessary for the CEP55 interaction (Figure 6D), suggesting complex interactions between these proteins possibly involving independent binding sites. In keeping with these results, we found that Alix had different binding sites for CEP55 and BRCA2 in cells synchronized during mitosis (Figure S6C). Specifically, amino acids 101-296 of Alix (Del-2 and Del-3) were required for the Alix interaction with BRCA2, while amino acids 709-800 containing a previously defined CEP55 binding site (Lee et al., 2008), as well as amino acids 101-296, were required for the Alix-CEP55 interaction (Figure S6C). Since BRCA2 can interact with Alix in the absence of amino acids 709-800, it appeared that BRCA2 forms complexes with Alix during mitosis independently of the Alix interaction with CEP55.
To confirm these results we evaluated the influence of a series of breast cancer associated missense mutations from the N-terminus of BRCA2 on BRCA2 interactions with CEP55, Alix and Tsg101 (Figure 6E). The mutations were selected because they altered amino acids displaying evolutionary conservation from sea urchin to human and were considered likely to alter protein function. T582P disrupted interaction with all three partners (Figure 6E). The N277K mutant interacted with Alix but failed to interact with CEP55, consistent with the inability of Mut-4 (amino acids 203-300) to interact with CEP55 (Figure 6E). In contrast, C554W reduced the BRCA2 interaction with Alix and Tsg101 but had no influence of the interaction with CEP55 (Figure 6E), consistent with the effect of Mut-6 (amino acids 447-575) (Figure 6D). Separately, I488R only impaired the Tsg101 interaction with BRCA2. These findings suggested that BRCA2 has the ability to independently interact with CEP55, Alix and Tsg101 during mitosis. To further evaluate this possibility we conducted a series of immunodepletion studies. Specifically, we depleted CEP55 and BRCA2 by immunoprecipitation from Hela cells synchronized in late mitosis (Figure 6F, lanes 1-6) and assessed the ability of Alix to co-immunoprecipitate with BRCA2, CEP55 and Tsg101 in the cleared supernatants (Figure 6F, lanes 7 and 8). Immunoblot analysis revealed that Alix formed a complex with BRCA2 when CEP55 was depleted (Figure 6F, lane 7) and that Alix interacted with CEP55 when BRCA2 was depleted (Figure 6F, lane 8), suggesting that once these proteins have been recruited to the midbody, removal of certain components (by immunodepletion) does not influence complex formation. In contrast, depletion of CEP55 by siRNAs inhibits formation of BRCA2-Alix and BRCA2-Tsg101 complexes (Figure S6D) and BRCA2 depletion by siRNA inhibits formation of CEP55-Alix and CEP55-Tsg101 complexes (Figure 6B) because Alix and Tsg101 cannot be recruited to the disrupted midbody (Zhao et al., 2006). These findings suggest that BRCA2 can interact with Alix independently of CEP55 or Tsg101 and vice versa at the midbody during cytokinesis.
BRCA2 interaction influences midbody localization of Alix and Tsg101
Since BRCA2 co-localizes with CEP55, Alix and Tsg101 at the midbody (Figure 2A) we evaluated whether localization of BRCA2 to the midbody influenced BRCA2 and CEP55 complexes involving Alix and Tsg101. In addition to T582P, which disrupts the BRCA2 interaction with CEP55, we tested the effect of E3002K and R3005A mutations that impaired Filamin A dependent midbody localization of BRCA2 on formation of these complexes. Similarly to the N-terminal T582P mutant, the BRCA2 E3002K and R3005A mutants substantially inhibited BRCA2 interaction with CEP55, Alix, and Tsg101 and also CEP55 interaction with Alix and Tsg101 (Figure 6G). Interestingly, this suggests that the BRCA2-Alix interaction observed in the absence of CEP55 (Figure 6F) is dependent on localization of BRCA2 to the midbody.
Next, we used immunofluorescence staining of 293T cells expressing BRCA2 mutants to test whether the interactions of BRCA2 with CEP55, Alix and Tsg101 influenced the midbody localization of these proteins. The E3002K and R3005A mutants from the Filamin A interacting domain, that failed to localize to the midbody, substantially reduced the levels of Alix and Tsg101 at the midbody (Figure 7A-C). Similarly, the C554W and T582P mutants that perturbed the BRCA2 interactions with Alix and Tsg101 diminished the levels of Alix and Tsg101 at the midbody, but did not influence localization of BRCA2 (Figure 7A) or Plk1, MKLP1 or PRC1 to the midbody (Figure S7A, B). In addition, the N277K mutation that only influenced the BRCA2 interaction with CEP55 and Tsg101 (Figure 6E), reduced localization of Tsg101 to the midbody but had no effect on Alix localization (Figure 7A). In all cases, no effect on CEP55 localization to the midbody was observed, consistent with our finding that depletion of BRCA2 did not influence the midbody localization of CEP55 (Figure 2A,B) and that depletion of CEP55 had no effect on BRCA2 localization to the midbody (Figure S7C, D). Together these results indicate that individual interactions of BRCA2 with CEP55, Alix and Tsg101 during cytokinesis are important for recruitment of ESCRT-associated and ESCRT complexes to the midbody.
Figure 7. Effect of BRCA2 mutation on Alix and Tsg101 localization and cytokinesis.
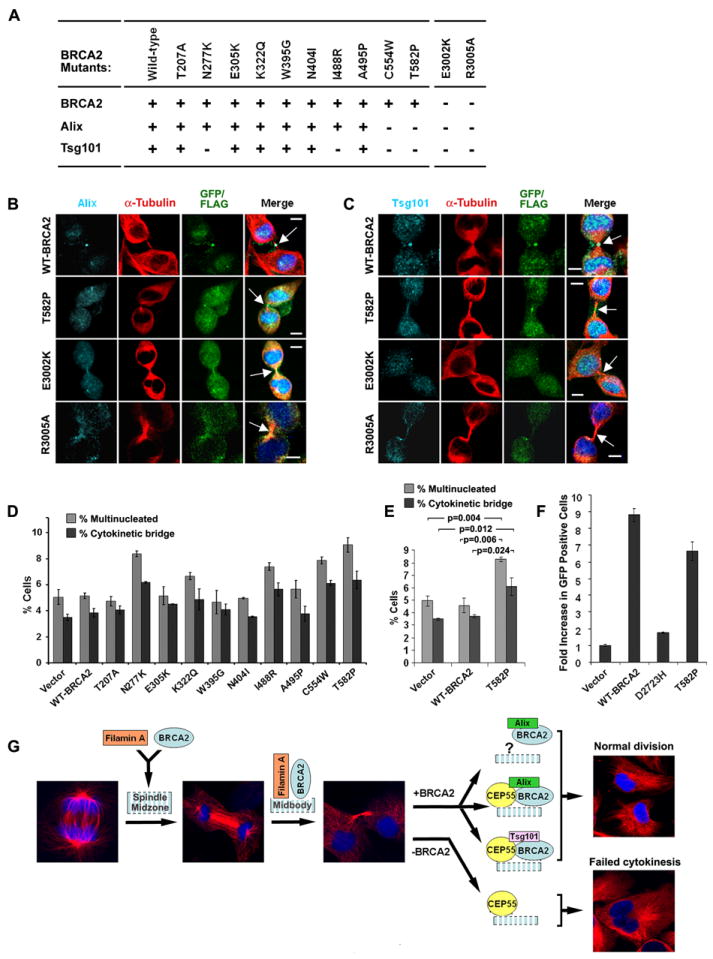
(A) Summary of midbody localization of BRCA2, Alix and Tsg101 in response to BRCA2 missense mutants. “+” normal and “−” reduced localization. (B-C) Disruption of Alix (B) and Tsg101 (C) midbody localization by BRCA2 mutations shown by immunofluorescence (IF) staining for Alix and Tsg101 in 293T cells expressing FLAG or GFP-tagged WT and mutant BRCA2. Arrows indicate midbodies. Bars (B, C), 5μm. (D-E) Bar graph of percent 293T cells with multiple nuclei and unresolved cytokinetic bridges 72 hr after transfection with the indicated BRCA2 constructs; p-values (E) indicate the statistical significance of the differences. (F) Rescue of homology directed repair activity of BRCA2 mutants in brca2 deficient DR-GFP V-C8 cells. GFP positive cells were identified by FACS analysis following co-transfection with I-Sce1 and WT or mutant BRCA2 vectors. Error bars (in D-F) show standard error of the mean. (G) Model of BRCA2 activity at the midbody. Stages of progression from anaphase through completion of cytokinesis are shown by α-tubulin and DAPI staining. BRCA2 is recruited to the midbody by Filamin A during furrow ingression and cytokinetic bridge formation, and interacts with CEP55 to assist the formation of CEP55-Alix and CEP55-Tsg101 complexes. Impairment of BRCA2 function reduces the availability of these complexes at the midbody leading to abnormal or failed cytokinesis. (See also Figure S7).
BRCA2 interaction with CEP55, Alix and Tsg101 regulates cytokinesis
Previously we showed that missense mutations in the Filamin A interacting domain of BRCA2 reduced the efficiency of BRCA2 localization to the midbody and caused an increase in cytokinesis defects (Figure 4B and 5A-D). Here we evaluated the influence of the missense mutations in the N-terminal domain of BRCA2 that disrupted interactions with CEP55, Alix and Tsg101 on the integrity of cytokinesis (Figure 7D). The N277K, I488R and C554W mutants, which disrupted complex formation and localization of Alix or Tsg101 to the midbody, all increased the frequency of multinucleation and delayed cytokinetic bridge resolution relative to wildtype BRCA2 (Figure 7D). Likewise the T582P mutant that reduced the interactions with CEP55, Alix and Tsg101caused a significant difference in the frequency of multinucleation (p=0.006) and unresolved cytokinetic bridges (p=0.024) relative to wildtype BRCA2 (WT-BRCA2) (Figure 7D, E). Importantly, T582P did not influence BRCA2 homology directed repair activity (Figure 7F). Furthermore, like wildtype and the E3002K mutant, the T582P mutant partially rescued localization of Myosin II to the cortex of BRCA2 deficient PEO1 cells during furrow ingression (Figure S7E) and did not disrupt Myosin II localization to the cleavage furrow or central spindle in 293T cells (Figure S7E). Thus, disruption of the BRCA2 interaction with CEP55 or the downstream components Alix and Tsg101 appears to directly influence the terminal stage of cytokinesis but has no effect on HDR activity or Myosin II associated furrow formation.
Discussion
Several lines of evidence suggest that BRCA2 has a role in regulation of cytokinesis. Both normal and malignant BRCA2 deficient cells exhibit numerical chromosomal abnormalities often in the form of polyploidy, and both depletion and inactivation of BRCA2 is associated with an accumulation of 4N cells (Daniels et al., 2004; Jonsdottir et al., 2009), consistent with a failure of cytokinesis. Here we confirm that BRCA2 accumulates at the midbody, regulates midbody structure and function, and regulates the efficiency of cytokinesis, independently of DNA damage repair. Specifically, we detected both endogenous BRCA2 and FLAG-tagged versions of BRCA2 in HeLa cells, and demonstrated reconstitution of BRCA2 to the midbody in brca2-/- murine pancreatic tumor cells and BRCA2 deficient PEO1 human ovarian cancer cells. We also used reconstitution and live cell confocal imaging experiments to establish that BRCA2 is involved in regulation of cytokinesis. Furthermore, we established that the recruitment of BRCA2 to the central spindle and the midbody is dependent on an interaction with Filamin A, a component of actomyosin complexes (Figure 7G). Our data suggest that inactivation or loss of BRCA2 from the midbody disrupts recruitment of specific markers to the midbody. Moreover, we show that BRCA2 regulates the recruitment of Alix and Tsg101 to the midbody and the formation of CEP55-Alix and CEP55-Tsg101 complexes that are required for recruitment of ESCRT complexes and subsequent cell separation (Figure 7G). Overall, although the effect of BRCA2 depletion is not as severe as depletion of essential structural components of the midbody such as MKLP1 or CEP55 (Matuliene and Kuriyama, 2002; Morita et al., 2007), BRCA2 appears to play an important role in regulation of final abscission by mediating the assembly of a number of midbody factors and signaling components (Figure 2A, B). These results are consistent with previous studies suggesting that BRCA2 influences the midbody (Daniels et al., 2004; Jonsdottir et al., 2009) but contrast with results from a recent study by Lekomtsev et al. which suggested that BRCA2 did not localize to the cytokinetic bridge and had no influence on cytokinesis (Lekomtsev et al., 2010). The absence of data supporting the influence of BRCA2 on the midbody and cytokinesis may have resulted from the use of a limited set of antibodies and siRNAs. Importantly, the Lekomsev et al. findings also contrast with those from a recent study showing that HMG20b, a kinesin-like coiled coil protein, interacts with BRCA2 BRC5 and influences the completion of cell division, but does not influence BRCA2-Rad51 interactions or BRCA2 DNA repair activity (Lee et al., 2011). Filamin A, an actin binding protein that can act as a scaffold for numerous proteins has been detected at the midbody (Feng and Walsh, 2004; Nunnally et al., 1980) and has been implicated in regulation of cytokinesis (Playford et al., 2006). Here, we demonstrate that disruption of the Filamin A-BRCA2 interaction is sufficient to exclude BRCA2 from the midbody, to disrupt the localization of midbody markers, and to substantially increase the proportion of cells that exhibit delay or failure of cytokinesis. In addition, we show that depletion of BRCA2 or disruption of the BRCA2 interaction with Filamin A has no effect on Filamin A localization at the midbody, suggesting that BRCA2 functions downstream of Filamin A during cytokinesis. Taken together these data strongly implicate Filamin A-dependent recruitment of BRCA2 to the central spindle and midbody in the maintenance of cytokinesis (Figure 7G).
In this study we report that BRCA2 is required for normal positioning of a number of important regulators of cytokinesis, including PRC1, MKLP1 and MKLP2, at the midbody during abscission. In particular, in the absence of BRCA2, the PRC1 marker of the central spindle and midbody (Kurasawa et al., 2004) was dispersed along the microtubules on the cytokinetic bridge during late abscission. PRC1 is required for recruitment of MKLP1 and MgcRacGAP (centralspindlin) to the central spindle and depletion of PRC1 results in the absence of centralspindlin from the central spindle and failure of abscission (Mollinari et al., 2005). However, PRC mislocalization due to BRCA2 deficiency did not displace MgcRacGAP or MKLP1 from the central spindle. Instead the effect on PRC1 appears to be restricted to efficient recruitment and/or retention of PRC1 at the midbody after successful recruitment of MgcRacGAP and MKLP1 to the centralspindle. This finding is reminiscent of the dispersion of PRC1 along the cytokinetic bridge and the failure of abscission observed after CEP55 depletion (Fabbro et al., 2005; Zhao et al., 2006). Furthermore, while the MKLP1 and MKLP2 microtubule associated proteins were efficiently recruited to the central spindle and midbody during abscission in BRCA2 deficient cells, these kinesins displayed disordered positioning at the midbody ring (Figure 2A and B), a tubulin enriched structure decorated by the centralspindlin complex, MKLP2, and CEP55 that forms in late telophase in the phase-dense Flemming body (Gromley et al., 2005). This structure is thought to function as a docking site for machinery involved in membrane fusion during late abscission, including SNARE and ESCRT complexes (Prekeris and Gould, 2008). Since we show that BRCA2 interacts with CEP55, influences MKLP1 and MKLP2 positioning, and disrupts recruitment of ESCRT complexes to the midbody, the suggestion is that BRCA2 is an important regulator of the structure and function of the midbody ring.
Dimeric CEP55 localizes to the central spindle and midbody during cytokinesis, possibly through direct binding to MKLP1, where it influences the establishment and proper function of the midbody structure (Lee et al., 2008). In addition, CEP55 is required for membrane fusion at the terminal stage of cytokinesis through direct recruitment of the ESCRT-1 component, Tsg101, and the ESCRT-interacting protein Alix to the midbody ring (Carlton and Martin-Serrano, 2007). Disruption of the BRCA2-CEP55 interaction resulted in reduced CEP55-Alix and CEP55-Tsg101 complex formation and reduced levels of Alix, Tsg101, and Endobrevin at the midbody, consistent with the effects of CEP55 or BRCA2 depletion (Carlton and Martin-Serrano, 2007; Zhao et al., 2006). However, in contrast to BRCA2 deficiency, absence of CEP55 displaced Plk1, Aurora B and MKLP2 from the midbody, consistent with reports of complete absence of the Flemming body from the midbody following CEP55 depletion (Zhao et al., 2006). Since CEP55 depletion had a greater effect on the midbody than exclusion of BRCA2 from the midbody or disruption of CEP55-BRCA2 complexes, we speculate that BRCA2 regulates a subset of CEP55 signaling pathways in the midbody, including CEP55 interactions with membrane sorting complexes.
It has been shown that CEP55 interactions with Alix and Tsg101 through the EABR domain can be regulated by other binding partners, such as Tex14, which competes with Alix and Tsg101 for binding to CEP55 at the midbody and prevents completion of cytokinesis in germ cells (Iwamori et al.). Given that BRCA2 also interacts with CEP55 through the EABR domain and is required for cytokinesis, we evaluated whether BRCA2 influences CEP55 complex formation and functions as a scaffold for assembly of protein complexes at the midbody. Depletion of CEP55 by siRNAs disrupted the interaction between BRCA2 and Alix in mitotic cells. However, we successfully identified Alix-BRCA2 complexes after CEP55 immunodepletion from cells aligned in mitosis (Figure 6F), when BRCA2 and Alix were already present at the midbody. Similarly, depletion of BRCA2 disrupted formation of CEP55-Alix complexes, whereas immunodepletion of BRCA2 during cytokinesis after recruitment of Alix to the midbody had no effect on CEP55-Alix complex formation. We also showed that BRCA2 interacted with Alix through a CEP55-independent binding site (Figure 6, D and E) and that disruption of BRCA2 binding sites in Alix (amino acids 101-209 and 216-296) interfered with the interaction between Alix and CEP55, despite the continued presence of a CEP55 binding site (amino acid region 709-800) in the mutant Alix protein. Together these data suggest that the presence of BRCA2 at the midbody is required for efficient recruitment of Alix, Tsg101 and ESCRT complexes to the midbody, consistent with a scaffolding activity. While it is intriguing to speculate that CEP55, Alix and Tsg101 all interact with BRCA2 in the midbody and that BRCA2 promotes the interaction of Alix and Tsg101 with CEP55 through the CEP55 EABR domain, this model does not appear to be consistent with recent studies showing direct binding of Alix and Tsg101 to CEP55 in vitro in the absence of BRCA2 (Lee et al., 2008). However, it is possible that BRCA2 recruits and delivers Alix and Tsg101 to CEP55 in vivo by binding to CEP55 EABR and facilitates loading of Alix and Tsg101 onto CEP55 by exchange of binding to the EABR site. This recruitment and loading activity would not be observed in an in vitro binding model in the presence of excess protein. Alternatively, BRCA2 may protect CEP55 from Tex14 binding at the EABR domain or may displace Tex14 from the EABR domain, thereby facilitating completion of abscission. Further studies are needed to resolve the mechanism by which BRCA2 influences CEP55-associated recruitment of ESCRT complexes to the midbody (Figure 7G).
The contribution of BRCA2 to homologous recombination repair of DNA double strand breaks has been well documented (Wu et al., 2005; Yang et al., 2002). Given recent evidence that unresolved chromatin in cytokinetic bridges can result in delays and failure of abscission and cytokinesis (Steigemann et al., 2009), the possibility existed that impaired DNA repair activity in the absence of BRCA2 also accounted for the observed cytokinetic defects. Here we provide evidence that regulation of cytokinesis by BRCA2 is independent of the established DNA repair activity of the protein by showing that Lap2β stained unresolved DNA strands or BLM stained UFBs were not present at increased frequency in BRCA2 deficient or mutant cells relative to wildtype controls. Thus, the failure of cytokinesis due to inactive BRCA2 is at least in part independent of defects in DNA decatenation or sister chromatid separation. In addition, we showed that reconstitution of brca2-/- cells with a BRCA2 mini-gene, deficient in HDR activity due to the absence of a large part of the BRCA2 DNA-binding domain, efficiently reduced the frequency of cytokinesis defects (Figure S1O), suggesting an HDR-independent role for BRCA2 in regulation of cytokinesis. Furthermore, we used missense mutations in the Filamin A interacting domain of BRCA2 to distinguish between the ability of BRCA2 to mediate DNA repair and cytokinesis. We found that the E3002K mutation that disrupted the BRCA2 interaction with Filamin A and induced a failure of cytokinesis also inhibited the homologous recombination activity of BRCA2 (Farrugia et al., 2008; Wu et al., 2005). Importantly the E3002K mutation may disrupt predicted hydrogen bonding and van der Waal’s interactions at the interface between the OB1 and OB2 domains in the crystal structure of rat BRCA2 leading to structural alterations and inactivation of all BRCA2 functions involving this region of BRCA2. In contrast, the R3005A mutation, which also disrupted Filamin A binding and cytokinesis, had no effect on homologous recombination. This is consistent with the crystal structure based finding that Filamin A interacting residues in BRCA2 are distinct from the major DNA binding and DSS1 binding residues in the same region (Yang et al., 2002). Thus, R3005A may only influence Filamin A binding whereas E3002K may induce a more significant conformational change in this region of BRCA2 resulting in disruption of both Filamin A and DNA binding. Similarly, the T582P mutation, that had no effect on localization of BRCA2 to the centrosome and midbody, but was associated with reduced binding to CEP55, Alix and Tsg101 and a significantly increased frequency of cytokinetic defects, displayed wildtype BRCA2 HDR activity. Thus, these missense mutations effectively distinguish between the contributions of BRCA2 to HR DNA repair and cytokinesis and suggest a direct role for BRCA2 at the midbody in regulation of cytokinesis. Whether the specific defects in cytokinesis influence cancer risk and/or promote tumor progression remains to be determined.
In conclusion, we show that BRCA2 plays an important role in regulation of cytokinesis by mediating the assembly of factors at the midbody. Thus, the delays or failure of cytokinesis associated with BRCA2 mutation may account in part for the numerical chromosomal defects in BRCA2 mutant cells and may contribute to the development of cancer in BRCA2 mutation carriers.
Experimental Procedures
Detailed descriptions of all experimental methods are provided in the supplemental experimental procedures. Briefly:
Plasmid construction
FLAG-tagged full-length BRCA2 (WT-BRCA2) and fragments of BRCA2 were cloned into the pcDNA3.1 vector. Point mutants of BRCA2 were generated using the Quikchange site-directed mutagenesis kit (Stratagene). EGFP-BRCA2 was cloned in pEGFP-C2 vector (Clonetech).
Live cell imaging
Live cell imaging was performed using brca2+/+ and brca2-/- mouse tumor cells and HeLa cells transiently transfected with BRCA2 siRNA (Dharmacon and Qiagen), CEP55 siRNAs (IDT), or BRCA2 siRNAs and the GFP-tagged BRCA2 mini-gene. BRCA2 localization studies were also conducted by live cell imaging of 293T cells transfected with mCherry tagged α-tubulin and GFP-tagged BRCA2 using a Zeiss LSM510 confocal microscope.
Homologous recombination repair assay
The homologous recombination repair assay was carried out as described previously (Wu et al., 2005).
Quantitation and statistical analysis of immunofluorescence and live cell imaging data
To compare the relative counts of the numbers of cells displaying categorized phenotypes (normal, delayed cytokinesis, or failed cytokinesis) (Figure 1), (Lap2β positive versus negative) (Figure S2), (MKLP1, MKLP2 and PRC1 localized or mislocalized) (Fig 2), (present or absent of Endobrevin) or (prominent, weak, absence of Alix and Tsg101) (Figure 2) between different cell types in each experiment we applied a Poisson regression approach (McCullagh and Nelder, 1989) implemented in SAS (SAS Institute, Inc., Cary, NC). A separate Poisson regression model was fit to the observed data for each specific comparison.
Supplementary Material
Acknowledgments
We thank Jennifer Scott and James Tarara for technical support and Thomas P. Stossel and Fumihika Nakamura, Brigham and Women’s Hospital for FLNA deficient (M2) and FLNA reconstituted (A7) melanoma cell lines. This research was supported by the National Institutes of Health Grant CA116167, a Specialized Program of Research Excellence (SPORE) in Breast Cancer CA116201, and a Specialized Program of Research Excellence (SPORE) in Pancreatic Cancer CA102701. G. Mondal was supported by a grant from the Komen Foundation for the Cure.
Footnotes
Supplemental information includes seven supplemental figures, supplemental experimental procedures, supplemental movie legends and six movies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Carlton JG, Martin-Serrano J. Parallels between cytokinesis and retroviral budding: a role for the ESCRT machinery. Science. 2007;316:1908–1912. doi: 10.1126/science.1143422. [DOI] [PubMed] [Google Scholar]
- Cunningham CC, Gorlin JB, Kwiatkowski DJ, Hartwig JH, Janmey PA, Byers HR, Stossel TP. Actin-binding protein requirement for cortical stability and efficient locomotion. Science. 1992;255:325–327. doi: 10.1126/science.1549777. [DOI] [PubMed] [Google Scholar]
- Daniels MJ, Wang Y, Lee M, Venkitaraman AR. Abnormal cytokinesis in cells deficient in the breast cancer susceptibility protein BRCA2. Science. 2004;306:876–879. doi: 10.1126/science.1102574. [DOI] [PubMed] [Google Scholar]
- Fabbro M, Zhou BB, Takahashi M, Sarcevic B, Lal P, Graham ME, Gabrielli BG, Robinson PJ, Nigg EA, Ono Y, et al. Cdk1/Erk2- and Plk1-dependent phosphorylation of a centrosome protein, Cep55, is required for its recruitment to midbody and cytokinesis. Dev Cell. 2005;9:477–488. doi: 10.1016/j.devcel.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Farrugia DJ, Agarwal MK, Pankratz VS, Deffenbaugh AM, Pruss D, Frye C, Wadum L, Johnson K, Mentlick J, Tavtigian SV, et al. Functional assays for classification of BRCA2 variants of uncertain significance. Cancer Res. 2008;68:3523–3531. doi: 10.1158/0008-5472.CAN-07-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Walsh CA. The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat Cell Biol. 2004;6:1034–1038. doi: 10.1038/ncb1104-1034. [DOI] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. doi: 10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromley A, Yeaman C, Rosa J, Redick S, Chen CT, Mirabelle S, Guha M, Sillibourne J, Doxsey SJ. Centriolin anchoring of exocyst and SNARE complexes at the midbody is required for secretory-vesicle-mediated abscission. Cell. 2005;123:75–87. doi: 10.1016/j.cell.2005.07.027. [DOI] [PubMed] [Google Scholar]
- Iwamori T, Iwamori N, Ma L, Edson MA, Greenbaum MP, Matzuk MM. TEX14 interacts with CEP55 to block cell abscission. Mol Cell Biol. 30:2280–2292. doi: 10.1128/MCB.01392-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsdottir AB, Vreeswijk MP, Wolterbeek R, Devilee P, Tanke HJ, Eyfjord JE, Szuhai K. BRCA2 heterozygosity delays cytokinesis in primary human fibroblasts. Cell Oncol. 2009;31:191–201. doi: 10.3233/CLO-2009-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurasawa Y, Earnshaw WC, Mochizuki Y, Dohmae N, Todokoro K. Essential roles of KIF4 and its binding partner PRC1 in organized central spindle midzone formation. Embo J. 2004;23:3237–3248. doi: 10.1038/sj.emboj.7600347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Elia N, Ghirlando R, Lippincott-Schwartz J, Hurley JH. Midbody targeting of the ESCRT machinery by a noncanonical coiled coil in CEP55. Science. 2008;322:576–580. doi: 10.1126/science.1162042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Daniels MJ, Garnett MJ, Venkitaraman AR. A mitotic function for the high-mobility group protein HMG20b regulated by its interaction with the BRC repeats of the BRCA2 tumor suppressor. Oncogene. 2011;30:3360–3369. doi: 10.1038/onc.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekomtsev S, Guizetti J, Pozniakovsky A, Gerlich DW, Petronczki M. Evidence that the tumor-suppressor protein BRCA2 does not regulate cytokinesis in human cells. J Cell Sci. 2010:1395–1400. doi: 10.1242/jcs.068015. [DOI] [PubMed] [Google Scholar]
- Liu J, Yuan Y, Huan J, Shen Z. Inhibition of breast and brain cancer cell growth by BCCIPalpha, an evolutionarily conserved nuclear protein that interacts with BRCA2. Oncogene. 2001;20:336–345. doi: 10.1038/sj.onc.1204098. [DOI] [PubMed] [Google Scholar]
- Matuliene J, Kuriyama R. Kinesin-like protein CHO1 is required for the formation of midbody matrix and the completion of cytokinesis in mammalian cells. Mol Biol Cell. 2002;13:1832–1845. doi: 10.1091/mbc.01-10-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullagh P, Nelder JA. Generalized Linear Models. Second Edition. Chapter 10. Chapman and Hall; London: 1989. [Google Scholar]
- Mollinari C, Kleman JP, Saoudi Y, Jablonski SA, Perard J, Yen TJ, Margolis RL. Ablation of PRC1 by small interfering RNA demonstrates that cytokinetic abscission requires a central spindle bundle in mammalian cells, whereas completion of furrowing does not. Mol Biol Cell. 2005;16:1043–1055. doi: 10.1091/mbc.E04-04-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Sandrin V, Chung HY, Morham SG, Gygi SP, Rodesch CK, Sundquist WI. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. Embo J. 2007;26:4215–4227. doi: 10.1038/sj.emboj.7601850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnally MH, D’Angelo JM, Craig SW. Filamin concentration in cleavage furrow and midbody region: frequency of occurrence compared with that of alpha-actinin and myosin. J Cell Biol. 1980;87:219–226. doi: 10.1083/jcb.87.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Playford MP, Lyons PD, Sastry SK, Schaller MD. Identification of a filamin docking site on PTP-PEST. J Biol Chem. 2006;281:34104–34112. doi: 10.1074/jbc.M606277200. [DOI] [PubMed] [Google Scholar]
- Prekeris R, Gould GW. Breaking up is hard to do - membrane traffic in cytokinesis. J Cell Sci. 2008;121:1569–1576. doi: 10.1242/jcs.018770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryser S, Dizin E, Jefford CE, Delaval B, Gagos S, Christodoulidou A, Krause KH, Birnbaum D, Irminger-Finger I. Distinct roles of BARD1 isoforms in mitosis: full-length BARD1 mediates Aurora B degradation, cancer-associated BARD1beta scaffolds Aurora B and BRCA2. Cancer Res. 2009;69:1125–1134. doi: 10.1158/0008-5472.CAN-08-2134. [DOI] [PubMed] [Google Scholar]
- Sakai W, Swisher EM, Jacquemont C, Chandramohan KV, Couch FJ, Langdon SP, Wurz K, Higgins J, Villegas E, Taniguchi T. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009;69:6381–6386. doi: 10.1158/0008-5472.CAN-09-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar S, Gerlich DW. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell. 2009;136:473–484. doi: 10.1016/j.cell.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Vinciguerra P, Godinho SA, Parmar K, Pellman D, D’Andrea AD. Cytokinesis failure occurs in Fanconi anemia pathway-deficient murine and human bone marrow hematopoietic cells. J Clin Invest. 2010;120:3834–3842. doi: 10.1172/JCI43391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HF, Takenaka K, Nakanishi A, Miki Y. BRCA2 and nucleophosmin co-regulate centrosome amplification and form a complex with Rho effector kinase ROCK2. Cancer Res. 2011;71:68–77. doi: 10.1158/0008-5472.CAN-10-0030. [DOI] [PubMed] [Google Scholar]
- Wu K, Hinson SR, Ohashi A, Farrugia D, Wendt P, Tavtigian SV, Deffenbaugh A, Goldgar D, Couch FJ. Functional evaluation and cancer risk assessment of BRCA2 unclassified variants. Cancer Res. 2005;65:417–426. [PubMed] [Google Scholar]
- Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, Zheng N, Chen PL, Lee WH, Pavletich NP. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297:1837–1848. doi: 10.1126/science.297.5588.1837. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Shen Z. Interaction with BRCA2 suggests a role for filamin-1 (hsFLNa) in DNA damage response. J Biol Chem. 2001;276:48318–48324. doi: 10.1074/jbc.M102557200. [DOI] [PubMed] [Google Scholar]
- Zhao WM, Seki A, Fang G. Cep55, a microtubule-bundling protein, associates with centralspindlin to control the midbody integrity and cell abscission during cytokinesis. Mol Biol Cell. 2006;17:3881–3896. doi: 10.1091/mbc.E06-01-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.