

Abstract
BACKGROUND AND PURPOSE
Rimonabant (SR141716) and the structurally related AM251 are widely used in pharmacological experiments as selective cannabinoid receptor CB1 antagonists / inverse agonists. Concentrations of 0.5–10 µM are usually applied in in vitro experiments. We intended to show that these drugs did not act at GABAA receptors but found a significant positive allosteric modulation instead.
EXPERIMENTAL APPROACH
Recombinant GABAA receptors were expressed in Xenopus oocytes. Receptors were exposed to AM251 or rimonabant in the absence and presence of GABA. Standard electrophysiological techniques were used to monitor the elicited ionic currents.
KEY RESULTS
AM251 dose-dependently potentiated responses to 0.5 µM GABA at the recombinant α1β2γ2 GABAA receptor with an EC50 below 1 µM and a maximal potentiation of about eightfold. The Hill coefficient indicated that more than one binding site for AM251 was located in this receptor. Rimonabant had a lower affinity, but a fourfold higher efficacy. AM251 potentiated also currents mediated by α1β2, αxβ2γ2 (x = 2,3,5,6), α1β3γ2 and α4β2δ GABAA receptors, but not those mediated by α1β1γ2. Interestingly, the CB1 receptor antagonists LY320135 and O-2050 did not significantly affect α1β2γ2 GABAA receptor-mediated currents at concentrations of 1 µM.
CONCLUSIONS AND IMPLICATIONS
This study identified rimonabant and AM251 as positive allosteric modulators of GABAA receptors. Thus, potential GABAergic effects of commonly used concentrations of these compounds should be considered in in vitro experiments, especially at extrasynaptic sites where GABA concentrations are low.
LINKED ARTICLES
This article is part of a themed section on Cannabinoids in Biology and Medicine. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2012.165.issue-8. To view Part I of Cannabinoids in Biology and Medicine visit http://dx.doi.org/10.1111/bph.2011.163.issue-7
Keywords: GABA, GABAA receptors, AM251, rimonabant (SR141716), CB1 receptors, positive allosteric modulation
Introduction
In the CNS, endocannabinoids are synthesized post-synaptically in neurons, reach the pre-synaptic terminal in a yet poorly understood manner and act at G-protein-coupled cannabinoid CB1 receptors (nomenclature follows Alexander et al., 2011) to inhibit the release of excitatory and inhibitory neurotransmitters (see Hashimotodani et al., 2007; Katona and Freund, 2008). The central action of endocannabinoids may be modulated by inhibition of biosynthesis or degradation (Petrosino and Di Marzo, 2010; Piomelli, 2005) or by specifically blocking their action at CB1 receptors.
Antagonism of the CB1 receptor has a broad spectrum of pharmacological effects, including body weight reduction and improved metabolic profile (Kunos, 2007; Di Marzo, 2008; Kunos et al., 2008; Kunos et al., 2009), modulation of peripheral inflammatory processes (Karsak et al., 2007; Leonti et al., 2010), modulation of emotion (Lutz, 2009) and anti-craving effects (Solinas et al., 2008). The CB1 receptor inverse agonist rimonabant from Sanofi-Aventis was the first cannabinoid anorectic anti-obesity drug (Fernandez and Allison, 2004), but its approval in Europe was officially withdrawn by the European Medicines Agency in January 2009 because of central side effects. The iodinated rimonabant analogue AM251 was synthesized by the group of Makriyannis (Gatley et al., 1997). The CB1 receptor inverse agonist LY320135, which was developed by Eli Lilly (Felder et al., 1998), and the neutral CB1 receptor antagonist O-2050 (Gardner and Mallet, 2006), have also been reported to have selectivity towards CB1 receptors over CB2 receptors (see Pertwee et al., 2010).
To date, rimonabant and AM251 are regarded as selective and specific CB1 receptors inverse agonists characterized by Ki values <10 nM (reviewed by Pertwee et al., 2010). Both compounds are frequently used in in vitro experiments. A PubMed search with the term ‘AM251’ shows that this compound was mentioned in 427 publications either in the title or in the abstract. In brain slice experiments, the compounds are typically applied in a concentration range of 0.5–10 µM. As rimonabant was the first clinical CB1 receptor antagonist developed, its chemical scaffold was extensively profiled for off-target effects (Fong et al., 2009). Because such profiling is usually carried out with radioligand assays, any significant functional effect at GABAA receptors may have been overlooked.
The GABAA receptors are the major inhibitory neurotransmitter receptors in the CNS and have been estimated to be expressed at about 30% of all synapses. Each receptor is composed of five subunits that are arranged pseudosymmetrically around the central chloride ion selective channel (Macdonald and Olsen, 1994; Rabow et al., 1995). The subunits are selected from a total of 19 different but homologous subunits, indicating an as yet poorly understood diversity of GABAA receptors. The pharmacological properties of a GABAA receptor depend on subunit composition (Sieghart and Sperk, 2002) and arrangement (Minier and Sigel, 2004).
Here we report the unexpected direct effects of low concentrations of rimonabant and AM251 on different recombinant GABAA receptors, indicating that these compounds do not act solely at CB receptors. This observation may lead to reinterpretation of several scientific results.
Methods
AM251 and rimonabant
AM251 was obtained from Tocris and rimonabant (SR141716) from Fluka (Buchs, Switzerland). The drugs were dissolved at a concentration of 2.5 mM in DMSO and stored at −20°C. Immediately before the experiment the stock solutions were diluted in the experimental medium. The maximal final DMSO concentration was 0.4% (v/v). In control experiments, DMSO up to a concentration of 0.6% (v/v) did not affect the amplitude of the current response to GABA. Figure 1 compares the structures of the four CB antagonists used. While AM251 and rimonabant differ only by a halogen substituent, LY320135 and O-2050 belong to different structural classes.
Figure 1.
Chemical structure of the compounds studied.
Expression in Xenopus oocytes
Capped cRNAs were synthesized (Ambion, Austin, TX, USA) from the linearized vectors containing the cDNAs coding for the different rat subunits respectively. A poly-A tail of about 400 residues was added to each transcript using yeast poly-A polymerase (United States Biologicals, Cleveland, OH, USA). The concentration of the cRNA was quantified on a formaldehyde gel using Radiant Red stain (Bio-Rad) for visualization of the RNA. Known concentrations of RNA ladder (Invitrogen) were loaded as standard on the same gel. cRNAs were precipitated in ethanol/isoamylalcohol 19:1, the dried pellet dissolved in water and stored at −80°C. cRNA mixtures were prepared from these stock solutions and stored at −80°C.
Xenopus laevis oocytes were prepared, injected and defolliculated as described previously (Sigel, 1987; Sigel and Minier, 2005). They were injected with 50 nL of the cRNA solution containing rat α1, β2 and γ2 subunits at a concentration of 10 nM : 10 nM : 50 nM (Boileau et al., 2002), and then incubated in modified Barth's solution (10 mM HEPES, pH 7.5, 88 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, 0.82 mM MgSO4, 0.34 mM Ca(NO3)2, 0.41 mM CaCl2, 100 units·mL−1 penicillin, 100 µg·mL−1 streptomycin) at 18°C for at least 24 h before the measurement.
Functional characterization in Xenopus oocytes
Currents were measured using a specially built two-electrode voltage clamp amplifier in combination with a XY-recorder (90% response time 0.1 s) or digitized at 100 Hz using a MacLab/200 (AD Instruments). Tests with a model oocyte were performed to ensure linearity in the larger current range. The response was linear up to 20 μA. Electrophysiological experiments were performed at a holding potential of −80 mV. The perfusion medium contained 90 mM NaCl, 1 mM KCl, 1 mM MgCl2, 1 mM CaCl2 and 5 mM Na-HEPES (pH 7.4). The perfusion solution (6 mL·min−1) was applied through a glass capillary with an inner diameter of 1.35 mm, the mouth of which was placed about 0.4 mm from the surface of the oocyte (Baur and Sigel, 2007). The experiments were performed after the currents induced by GABA had become constant. Concentration response curves for GABA or AM251 were fitted with the equation I(c) = Imax/[1 + (EC50/c)n], where c is the concentration of GABA or AM251, EC50 the concentration of GABA eliciting half maximal current amplitude, Imax is the maximal current amplitude, I the current amplitude and n the Hill coefficient. Allosteric potentiation by the CB1 receptor antagonists was measured at a GABA concentration eliciting 2–5% of the maximal GABA current amplitude. GABA was applied for 20 s alone or in combination with allosteric compound. Relative current potentiation by the different compounds was determined as [I(compound + GABA)/IGABA− 1]× 100%. The perfusion system was cleaned between drug applications, by washing with 100% DMSO to avoid contamination. Care was taken to ensure full removal of DMSO after this cleaning procedure.
Results
Low concentrations of the cannabinoid receptor antagonist AM251 potentiate GABAA receptors
Recombinant α1β2γ2 GABAA receptors were expressed in Xenopus oocytes and currents induced by GABA measured. Figure 2A shows two applications of 0.5 µM GABA followed by combined application of the same concentration of GABA with 0.3 µM AM251. To our surprise, in the presence of such a small concentration of AM251 the current amplitude was enhanced more than threefold. Figure 2B shows averaged concentration response curves to AM251 and rimonabant. The curve for AM251 was characterized by an EC50 of 0.40 ± 0.13 µM and a maximal potentiation of 881 ± 167% (n= 4). The large Hill coefficient amounting to 1.5 ± 0.1 is indicative of the presence of more than one binding site in such a receptor. The curve for rimonabant was characterized by an EC50 of 7.3 ± 0.5 µM, a maximal potentiation of 3381 ± 165% (n= 4) and a Hill coefficient of 1.5 ± 0.1. In the absence of GABA, 3 µM AM251 or 3 µM rimonabant elicited small currents by itself, of about 1% and 2% of the maximal current amplitude elicited by GABA, respectively (not shown).
Figure 2.
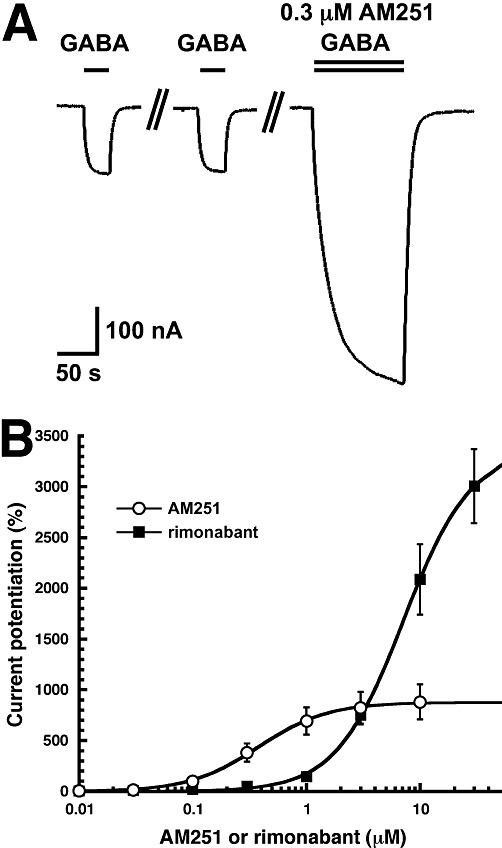
AM251 affects GABAA receptor currents. (A) Low concentrations of AM251 strongly potentiate currents mediated by recombinant α1β2γ2 GABAA receptors. Currents were measured in Xenopus oocytes expressing recombinant receptors. Two applications of 0.5 µM GABA were followed by a combined application of the same concentration of GABA with 0.3 µM AM251. (B) Concentration-response curves of the potentiation by AM251 and by rimonabant. Either no AM251 or increasing concentrations of AM251 or rimonabant were co-applied with 0.5 µM GABA. Individual curves were first normalized to the observed maximal current amplitude and subsequently averaged. Mean ± SEM of experiments carried out with four oocytes from two batches of oocytes are shown.
We then tested the effects of AM251 on the GABA concentration response curve of α1β2γ2 GABAA receptors. Increasing concentrations of GABA were applied to oocytes in the absence and presence of 1 µM AM251 (Figure 3). The curves were characterized by an EC50 of 15.4 ± 0.8 µM (n= 7) and 5.5 ± 0.4 µM (n= 4) and a Hill coefficient of 1.3 ± 0.1 and 1.0 ± 0.1 in the absence and presence of AM251 respectively. A GABA application without AM251 was carried out during the concentration response curves with AM251. This allowed an estimate of the relative maximal current amplitudes and indicated a 26 ± 15% (n= 4) increase by AM251.
Figure 3.
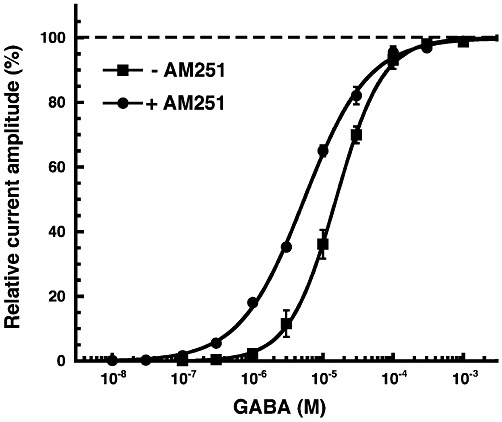
GABA concentration response curve in the absence and presence of 1 µM AM251. Increasing concentrations of GABA were applied in the absence and presence of 1 µM AM251. Individual curves obtained in the absence of AM251 were first normalized to the observed maximal current amplitude and subsequently averaged. Curves obtained in the presence of AM251 were treated in the same way. As such a procedure does not detect effects of AM251 on maximal current amplitudes, additional experiments were performed as described in the Results. Mean ± SEM of experiments carried out with at least four oocytes from two batches of oocytes are shown.
Site of action
Figure 4 shows that exclusion of γ2 subunits from GABAA receptors reduced the potentiation by AM251. Replacement of the α1 subunit in α1β2γ2 GABAA receptors by α2 did not significantly affect potentiation while replacement by α3, α5 or α6 led to a decrease in potentiation. Replacement of the β2 subunit in α1β2γ2 GABAA receptors by β1 lead to an almost complete loss of potentiation by AM251, while replacement by β3 tended to increase the potentiation.The α4β2δ GABAA receptors were also modulated.
Figure 4.
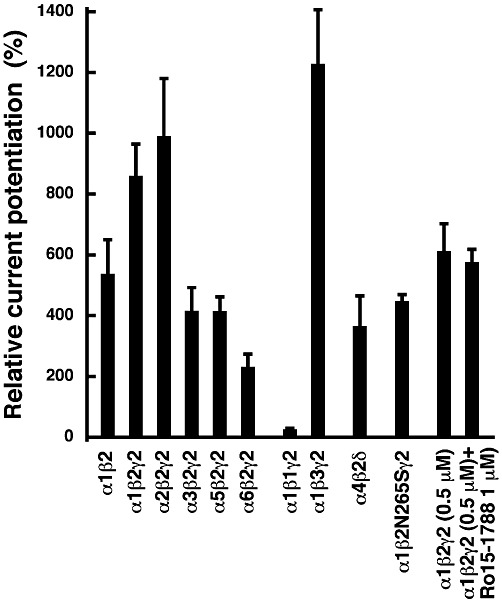
Subunit dependence and sensitivity to Ro15-1788 and to a point mutation of the potentiation by AM251. Currents were measured in Xenopus oocytes expressing recombinant α1β2, α1β2γ2, α2β2γ2, α3β2γ2, α5β2γ2, α6β2γ2, α1β1γ2, α1β3γ2 and α4β2δ GABAA receptors. Two applications of 0.5 µM GABA were followed by a combined application of the same concentration of GABA with 3 µM AM251. Mean ± SEM of experiments carried out with at least four oocytes are shown. Potentiation by 0.5 µM AM251 was tested as above and subsequently we tried to counteract potentiation by 1 µM Ro15-1788. Potentiation by 3 µM AM251 was also tested in point mutated α1β2N265Sγ2 receptors.
We were interested to see if AM251 acts at the same sites as the benzodiazepines or loreclezole. We found that 1 µM of the benzodiazepine antagonist Ro15-1788 did not counteract potentiation of the current by AM251 (Figure 4). In the point mutated receptor α1β2N265Sγ2 GABAA, where loreclezole has little effect, AM251 still potentiated the current response to GABA to about 50% of the wild-type receptor (Figure 4). These findings indicate that AM251 acts at neither of the mentioned sites.
Experiments with pentobarbital and the neurosteroid tetrahydrodeoxycorticosterone (THDOC) indicated that AM251 did not compete with these two ligands. Potentiation using the same oocytes for three subsequent measurements was 550 ± 77% (n= 4) for 5 µM pentobarbital, 908 ± 215% (n= 4) for 0.5 µM AM251 and 2003 ± 308% (n= 4) for the combined application of 5 µM pentobarbital and 0.5 µM AM251. Potentiation using the same oocytes for the three subsequent measurements was 483 ± 59% (n= 4) for 0.5 µM THDOC, 631 ± 80% (n= 4) for 0.5 µM AM251 and 1233 ± 125% (n= 4) for the combined application of 0.5 µM THDOC and 0.5 µM AM251.
The CB1 receptor antagonists LY320135 and O-2050
In AM251 the p-chloro group of rimonabant attached to a phenyl substituent is replaced with a p-iodo group. Both compounds robustly stimulated currents mediated by GABA (Figure 5). Two additional CB1 antagonists with an entirely different structure (Figure 1) were also examined. As shown in Figure 5 and 1 µM LY320135 and 1 µM O-2050 had no significant effect on the current elicited by GABA. A total of 10 µM LY320135 and 10 µM O-2050 potentiated these currents only by 19 ± 11% (n= 4) and 36 ± 11% (n= 4) respectively.
Figure 5.
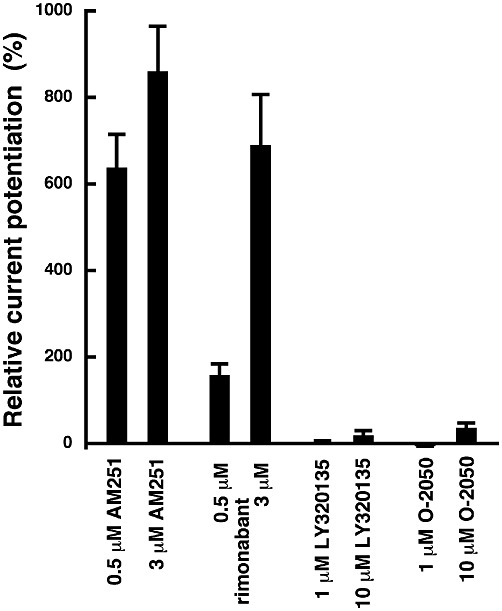
Effect of rimonabant, LY320135 and O-2050. The experiments were carried out as described under Figure 1A. Mean ± SEM of experiments carried out with 4–11 oocytes from at least two batches of oocytes are shown.
Discussion
AM251 is generally regarded as a selective and specific CB1 receptor ligand, while rimonabant has previously been shown to target and antagonize the transient receptor potential (TRP)V1 channels at µM concentrations (Gibson et al., 2008). Both compounds also bind at nM concentrations to the orphan GPCR GPR55 and act as agonists with yet unknown functional consequences (see Ross, 2009). Here we show that, in vitro, rimonabant and AM251 allosterically potentiate all but the β1 subunit containing GABAA receptors at nM concentrations. It is worth noting that also the putatively extrasynaptic α4β2δ GABAA receptors are subject to modulation. Interestingly, β3 subunit containing receptors, which are expressed in the spinal cord (Seo et al., 2010) and potentially associated with pain perception, are more strongly potentiated by rimonabant and AM251. The site of action is not identical to that of either the benzodiazepines, lorecezole, pentobarbital or neurosteroids. As the β1 subunit containing GABAA receptors were not modulated, the site is probably located in the β subunit.
Our experiments were performed in Xenopus oocytes. In brain slices, results are influenced by inhibition of GABA release caused by CB1 receptor activation by endocannabinoids produced in the post-synaptic cell upon depolarization. Indeed, AM251 and rimonabant lead to a CB1 receptor-mediated increase in GABA tone (Kim and Alger, 2010; Menzies et al., 2010). Chelation of Ca2+ in the post-synaptic cell to prevent production of endocannabinoids has been reported to suppress to a large extent this inhibition of GABA release as determined by the amplitude of inhibitory post-synaptic currents (IPSCs). In the presence of a Ca2+ chelator, rimonabant induced only a small increase in the amplitude of IPSCs (Kim and Alger, 2010). This observation does not contradict our results as they predict little direct stimulation of current mediated by GABAA receptors elicited by high concentrations of GABA, like those expected to be present in the synaptic cleft at the time of peak amplitudes of IPSCs. Our results would instead predict that the predominantly extrasynaptic GABAA receptors where concentrations of GABA should be low, would be most likely to be affected by AM251 or rimonabant. In support of our data, Mendiguren and Pineda (2009) have reported that AM251 and rimonabant decreased the firing rate of 5-HT dorsal raphe nucleus neurons in rat brain slices and that this effect was inhibited by the GABAA receptor antagonist picrotoxin.
We also studied CB1 receptor antagonists differing widely in structure (Figure 1). While LY320135 and low concentrations of O-2050 had no significant effect on the current elicited by GABA, rimonabant and AM251 showed strong potentiation of GABAA receptors. The fact that these two molecules, rimonabant and AM251, differ very little in structure (Figure 1), but differ about fourfold in the maximal current stimulation is surprising. The difference between these two compounds consists only of a halide substituent that is most probably located in a receptor region that upon interaction with the two ligands assumes different conformations, differing in their ability to stabilize the open channel state.
We are not aware of any report on sedative or anxiolytic properties of rimonabant or AM251 up to 10 mg·kg−1 i.p., which clearly suggests that either the drugs do not reach the brain in sufficient amounts, or that the effects on CB1 receptors dominate the in vivo pharmacology. There is very little information on levels of these antagonists in brain after peripheral administration. In one study, mice were injected i.p. with 0.3 mg·kg−1 rimonabant (Barna et al., 2009), corresponding to approximately the single oral dose of 20 mg recommended for human treatment. In this study, brain levels of about 0.25 nmol·g−1 tissue were reported. If a random distribution in the tissue is assumed, this corresponds to a concentration of 0.25 µM. At this concentration, we would predict some behavioural effects through interaction of rimonabant at GABAA receptors, unless the CB1 receptor-mediated pharmacology predominates. However, the above estimate of the free concentration may be too high as the hydrophobic drug binds to lipids.
We conclude that all the in vitro studies with brain slices or neuronal tissues that use either rimonabant or AM251 as selective inhibitors of CB1 receptors at concentrations higher than 0.03 µM AM251 or 0.1 µM rimonabant will need to take into account a potential direct modulation of GABAA receptors.
Acknowledgments
We thank Dr T.G. Smart for helpful discussions on possible influences of AM251 on GABAA receptors. This work was supported by the Swiss National Science Foundation grant 3100A0-105272/2.
Glossary
- CB1
cannabinoid receptor type 1
- GABAA receptor
γ-aminobutyric acid type A receptor
- IPSC
inhibitory post-synaptic current
Conflicts of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barna I, Till I, Haller J. Blood, adipose tissue and brain levels of the cannabinoid ligands WIN-55,212 and SR-141716A after their intraperitoneal injection in mice: compound-specific and area-specific distribution within the brain. Eur Neuropsychopharmacol. 2009;19:533–541. doi: 10.1016/j.euroneuro.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Baur R, Sigel E. Replacement of histidine in position 105 in the α5 subunit by cysteine stimulates zolpidem sensitivity of α5β2γ2 GABAA receptors. J Neurochem. 2007;103:2556–2564. doi: 10.1111/j.1471-4159.2007.04982.x. [DOI] [PubMed] [Google Scholar]
- Boileau AJ, Baur R, Sharkey LM, Sigel E, Czajkowski C. The relative amount of cRNA coding for γ2 subunits affects stimulation by benzodiazepines in GABAA receptors expressed in Xenopus oocytes. Neuropharmacology. 2002;43:695–700. doi: 10.1016/s0028-3908(02)00036-9. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. CB(1) receptor antagonism: biological basis for metabolic effects. Drug Discov Today. 2008;13:1026–1041. doi: 10.1016/j.drudis.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Felder CC, Joyce KE, Briley EM, Glass M, Mackie KP, Fahey KJ, et al. LY320135, a novel cannabinoid CB1 receptor antagonist, unmasks coupling of the CB1 receptor to stimulation of cAMP accumulation. J Pharmacol Exp Ther. 1998;284:291–297. [PubMed] [Google Scholar]
- Fernandez JR, Allison DB. Rimonabant Sanofi-Synthélabo. Curr Opin Investig Drugs. 2004;5:430–435. [PubMed] [Google Scholar]
- Fong TM, Shearman LP, Stribling DS, Shu J, Lao J, Huang CR-R, et al. Pharmacological efficacy and safety profile of taranabant in preclinical species. Drug Dev Res. 2009;70:349–362. [Google Scholar]
- Gardner A, Mallet PE. Suppression of feeding, drinking, and locomotion by a putative cannabinoid receptor silent antagonist. Eur J Pharmacol. 2006;530:103. doi: 10.1016/j.ejphar.2005.11.032. [DOI] [PubMed] [Google Scholar]
- Gatley SJ, Lan R, Pyatt B, Gifford AN, Volkow ND, Makriyannis A. Binding of the non-classical cannabinoid CP 55,940, and the diarylpyrazole AM251 to rodent brain cannabinoid receptors. Life Sci. 1997;61:191–197. doi: 10.1016/s0024-3205(97)00690-5. [DOI] [PubMed] [Google Scholar]
- Gibson HE, Edwards JG, Page RS, Van Hook MJ, Kauer JA. TRPV1 channels mediate long-term depression at synapses on hippocampal interneurons. Neuron. 2008;57:746–759. doi: 10.1016/j.neuron.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Kano M. Endocannabinoids and synaptic function in the CNS. Neuroscientist. 2007;13:127–137. doi: 10.1177/1073858406296716. [DOI] [PubMed] [Google Scholar]
- Karsak M, Gaffal E, Date R, Wang-Eckhardt L, Rehnelt J, Petrosino S, et al. Attenuation of allergic contact dermatitis through the endocannabinoid system. Science. 2007;316:1494–1497. doi: 10.1126/science.1142265. [DOI] [PubMed] [Google Scholar]
- Katona I, Freund TF. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat Med. 2008;14:923–930. doi: 10.1038/nm.f.1869. [DOI] [PubMed] [Google Scholar]
- Kim J, Alger BE. Reduction in endocannabinoid tone is a homeostatic mechanism for specific inhibitory synapses. Nat Neurosci. 2010;13:592–600. doi: 10.1038/nn.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunos G. Understanding metabolic homeostasis and imbalance: what is the role of the endocannabinoid system? Am J Med. 2007;120:S18–S24. doi: 10.1016/j.amjmed.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Kunos G, Osei-Hyiaman D, Liu J, Godlewski G, Bátkai S. Endocannabinoids and the control of energy homeostasis. J Biol Chem. 2008;283:33021–33305. doi: 10.1074/jbc.R800012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunos G, Osei-Hyiaman D, Bátkai S, Sharkey KA, Makriyannis A. Should peripheral CB(1) cannabinoid receptors be selectively targeted for therapeutic gain? Trends Pharmacol Sci. 2009;30:1–7. doi: 10.1016/j.tips.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonti M, Casu L, Raduner S, Cottiglia F, Floris C, Altmann KH, et al. Falcarinol is a covalent cannabinoid CB1 receptor antagonist and induces pro-allergic effects in skin. Biochem Pharmacol. 2010;79:1815–1826. doi: 10.1016/j.bcp.2010.02.015. [DOI] [PubMed] [Google Scholar]
- Lutz B. Endocannabinoid signals in the control of emotion. Curr Opin Pharmacol. 2009;9:46–52. doi: 10.1016/j.coph.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Olsen RW. GABAA receptor channels. Annu Rev Neurosci. 1994;17:569–602. doi: 10.1146/annurev.ne.17.030194.003033. [DOI] [PubMed] [Google Scholar]
- Mendiguren A, Pineda J. Effect of the CB(1) receptor antagonists rimonabant and AM251 on the firing rate of dorsal raphe nucleus neurons in rat brain slices. Br J Pharmacol. 2009;158:1579–1587. doi: 10.1111/j.1476-5381.2009.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies JR, Ludwig M, Leng G. Direct and indirect effects of cannabinoids on in vitro GABA release in the rat arcuate nucleus. J Neuroendocrinol. 2010;22:585–592. doi: 10.1111/j.1365-2826.2010.01990.x. [DOI] [PubMed] [Google Scholar]
- Minier F, Sigel E. Positioning of the alpha-subunit isoforms confers a functional signature to gamma-aminobutyric acid type A receptors. Proc Natl Acad Sci USA. 2004;101:7769–7774. doi: 10.1073/pnas.0400220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol Rev. 2010;62:588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosino S, Di Marzo V. FAAH and MAGL inhibitors: therapeutic opportunities from regulating endocannabinoid levels. Curr Opin Investig Drugs. 2010;11:51–62. [PubMed] [Google Scholar]
- Piomelli D. The endocannabinoid system: a drug discovery perspective. Curr Opin Investig Drugs. 2005;6:672–679. [PubMed] [Google Scholar]
- Rabow LE, Russek SJ, Farb DH. From ion currents to genomic analysis: recent advances in GABAA receptor research. Synapse. 1995;21:189–127. doi: 10.1002/syn.890210302. [DOI] [PubMed] [Google Scholar]
- Ross RA. The enigmatic pharmacology of GPR55. Trends Pharmacol Sci. 2009;30:156–163. doi: 10.1016/j.tips.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Seo K, Seino H, Yoshikawa H, Petrenko AB, Baba H, Fujiwara N, et al. Genetic reduction of GABA receptor gamma2 subunit expression potentiates the immobilizing action of isoflurane. Neurosci Lett. 2010;472:1–4. doi: 10.1016/j.neulet.2010.01.031. [DOI] [PubMed] [Google Scholar]
- Sieghart W, Sperk G. Subunit composition, distribution and function of GABAA receptor subtypes. Curr Top Med Chem. 2002;2:795–816. doi: 10.2174/1568026023393507. [DOI] [PubMed] [Google Scholar]
- Sigel E. Properties of single sodium channels translated by Xenopus oocytes after injection with messenger ribonucleic acid. J Physiol. 1987;386:73–90. doi: 10.1113/jphysiol.1987.sp016523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigel E, Minier F. Educational paper: the Xenopus oocyte: system for the study of functional expression and modulation of proteins. Mol Nutr Food Res. 2005;49:228–234. doi: 10.1002/mnfr.200400104. [DOI] [PubMed] [Google Scholar]
- Solinas M, Goldberg SR, Piomelli D. The endocannabinoid system in brain reward processes. Br J Pharmacol. 2008;154:369–383. doi: 10.1038/bjp.2008.130. [DOI] [PMC free article] [PubMed] [Google Scholar]