

Abstract
Stavudine (d4T) is used extensively as part of HAART in resource poor settings, despite its toxicities. The revised WHO guidelines specify replacement of d4T with less toxic but more expensive drugs when feasible, and that d4T doses be standardized to 30 mg twice daily (bid) (irrespective of body-weight), from the approved 40 mg bid in adults (body-weight ≥ 60 kg). Therefore, an in silico population pharmacokinetic and biochemical model was utilized to compare relative efficacies of the two doses in humans. Assessment of predicted quartile ranges of simulated concentrations of the triphosphate of d4T suggested sufficient trough concentrations to inhibit wild type HIV-1 reverse transcriptase at the reduced dose, lending support to the revised WHO recommendations.
Keywords: antiretroviral, nucleosides, NONMEM, simulation, HIV, d4T
Nucleoside reverse transcriptase inhibitors (NRTIs) remain the backbone of highly active antiretroviral therapy (HAART), in combination with protease (PI), non-nucleoside reverse transcriptase (NNRTI), integrase, or entry/fusion inhibitors. NRTIs are essentially prodrugs which undergo intracellular phosphorylation to their active triphosphate forms (NRTI-TP), which exert antiviral activity primarily via competitive inhibition of HIV-1 reverse transcriptase (HIV-1 RT) (Schinazi et al., 2006). Most HIV-1-infected persons live in poor and middle-income countries, and rely on the availability of inexpensive generic fixed-dose combinations of HAART (McCutchan, 2009; van Griensven et al., 2009). The low cost of stavudine (d4T) has enabled the scale up of HAART in these settings. According to the WHO, 56 % of adults on HAART in low- and middle-income settings received d4T as part of their HIV regimens (WHO 2010, Casiro, A.D., 2011). This figure is even higher in Sub-Saharan Africa where 80 % of infected individuals received d4T between 2005 and 2006, compared with 66% in Asia and 15% in South America (WHO 2009; Keiser et al., 2008). Several fixed dose combinations are currently marketed in Africa, usually in combination with 150 mg lamivudine and 200 mg nevirapine. The original formulations contained 40 mg d4T (e.g., GPO-VIR S 40™, Thai Government Pharmaceutical Organization; Triomune-40™, Cipla Ltd; Virolans™ capsules, d4T 40 mg version, Ranbaxy Laboratories Ltd). However, d4T is associated with debilitating long term side effects, associated with mitochondrial dysfunction, which include hyperlactemia, lipodystrophy, neuropathies, and lactic acidosis, some of which are irreversible (McComsey and Lonergan, 2004; van Griensven et al., 2009). Randomized studies on first-line HAART in first-world and in resource limited settings, indicated that zidovudine (ZDV) and tenofovir had similar efficacy and produced fewer metabolic side effects compared with d4T (Bygrave et al., 2011; Dube et al., 2007; Dube et al., 2005; Gallant et al., 2004; Mercier et al., 2009; Pujari et al., 2005). These toxicities prompted the World Health Organization (WHO) in 2009, to recommend that countries phase out d4T (WHO 2009; Wainberg, 2009). Although d4T (and ZDV) is off patent and is manufactured as a low cost generic medication in combination for use in 80 poor countries (ViiV Healthcare Web site), and tenofovir disoproxil fumarate is being offered by Gilead, its manufacturer, at a tiered discounted price in low- and middle-income countries (Gilead Web site), prices still remain beyond the reach of the poorest countries. Given the magnitude of the infected population, together with economic and other constraints, the WHO revised its recommendation to specify that “safer but currently more expensive first-line ARTs should be progressively introduced as currently they may not be feasible or affordable in many high-burden settings with low coverage, less developed health systems, limited laboratory capacity, finite budgets and competing health priorities” The recommendations also specify that a reduced dosage of 30 mg twice daily (bid) should be administered to all individuals on d4T in an attempt to reduce side effects (WHO 2010).
Pilot and retrospective studies suggested that reduction in d4T dosage from 40 to 30 mg twice daily (bid), in individuals weighing less than 60 kg and possibly heavier individuals as well, may decrease the incidence of side effects, without compromising antiviral efficacy (Ait-Mohand et al., 2008; Hoffmann et al., 2009; Karara et al., 2010; Pedrol et al., 2007; Sanchez-Conde et al., 2005). However, d4T dose reduction to control toxicity remains controversial. Domingo et al., 2010, reported a statistically significant (p = 0.013) higher median stavudine triphosphate (d4T-TP) content in the lymphocyte peripheral blood mononuclear (PBM) cells of 17 individuals with HAART associated lipodystrophy syndrome (HALS) (20.6 femtomoles/106 cells in individuals with HALS, interquartile range (IQR) = 14.90 – 26.92), compared to 16 individuals without HALS. (13.85 fmol/106 cells, IQR = 8.65 – 20.15) (Domingo et al., 2010). However, it is possible that the differences in d4T-TP levels between the two groups would have been even greater had d4T-TP measurements been normalized with the fraction of dividing cells (d4T is primarily phosphorylated in dividing lymphocytes), and had blood sampling times been standardized according to the time of the last d4T administration. Another recent study reported a lower risk of developing lipoatrophy in subjects who received 30 mg d4T exclusively (regardless of body weight, as suggested by WHO 2007 guidelines) since the start of treatment (n = 69), than in individuals who received weight adjusted d4T doses (30 mg if ≤ 60 kg, and 40 mg otherwise, patients treated before WHO 2007 guidelines) (n = 64) since treatment initiation. Furthermore, the odds ratio for the 30 mg bid only cohort was similar to a control group treated with AZT (n = 110), a drug which is not associated with lipoatrophy (Cournil et al., 2010). In contrast, a study involving 47 HIV-infected individuals did not find a correlation between lipodystrophy and d4T plasma exposures (AUC), with some individuals demonstrating this side effect even at a 20 mg bid dose (Sinxadi et al., 2010). The revised WHO guideline (WHO, 2010) now recommended that where d4T use is continued, it be administered at 30 mg BID for all individuals, irrespective of body weight. Based on the expectation of reduced toxicity with maintenance of adequate viral inhibition, fixed dose combination formulations containing d4T at the reduced 30 mg dosage with the standard lamivudine and nevirapine dosages are presently in use (e.g., Stavex-30 LN).
Population pharmacokinetic and pharmacodynamic simulations are useful for consolidating all available drug information into a usable form for the design of clinical trials of antiretroviral agents. These simulations may allow detailed analysis of of dosage regimens before actual studies are conducted. For example, based on predicted antiviral response, Rosario et al utilized clinical trial simulations to streamline the phase 2 development of the CCR5 receptor blocking agent maraviroc (Rosario et al., 2005). Furthermore, our group developed simulation models for NRTI to predict the antiviral response of lamivudine, and to suggest a reduced dosage regimen for zidovudine (Hurwitz et al., 2008; Hurwitz et al., 2007). The pharmacological and biochemical parameters of d4T necessary for modeling, including population pharmacokinetics, cellular phosphorylation in activated PBM cells (the major target for HIV-1 infection and replication), and the potency of inhibition of inhibition of the HIV-1 RT reverse transcription enzyme by d4T-TP have been studied (Becher et al., 2004; Panhard et al., 2007; Ueno and Mitsuya, 1997). Therefore, it was considered feasible to merge these data in silico using mechanistic data, and to conduct a virtual dosing study to assess whether d4T is expected to maintain efficacy at the 30 mg bid dose, as suggested by the clinical pilot studies.
Becher, et al., reported a linear correlation (r2 = 0.45), between the accumulation of d4T-TP in the PBM cells of subjects and d4T plasma concentrations of individuals taking d4T in a HAART cocktail. However, d4T is a thymidine analog and is phosphorylated primarily in activated (CD4+) lymphocytes (Jacobsson et al., 1995). The proportion of activated PBM cells varies between infected individuals and is not usually reported in clinical studies, making it difficult to assess the linearity of d4T-TP versus extracellular d4T using pooled in vivo data from individuals, each with differing activated lymphocyte fractions. Therefore the linearity between d4T and cellular accumulation of d4T-TP was tested in phytohaemagglutinin (PHA) stimulated PBM cells in cell culture, in which the degree of activation is more uniform (Hernandez-Santiago et al., 2005; Hernandez-Santiago et al., 2007a; Hernandez-Santiago et al., 2007b). The concentrations measured in vitro were then compared to average concentrations in humans after normalizing using 8% as an average estimate of dividing lymphocytes measured in infected individuals (Mohri et al., 2001). Since maximal d4T plasma concentrations (Cmax) in individuals receiving 40 mg bid rarely exceed 7 μM, are reached within 0.5-1 h (Tmax), and subsequently decay with a plasma half-life (t1/2) of 1 to 1.5 h (Horton et al., 1995), cells were incubated with 0.5 to 30 μM d4T in triplicate for 4 h before rinsing in ice cold phosphate buffered saline and extracted with ice cold 60% methanol in water and drying under a gently filtered air flow. The extracted d4T-TP was redissolved in mobile phase and assayed by LC-MS/MS (Fromentin et al., 2009; Hernandez-Santiago et al., 2007a). The cellular accumulation of d4T-TP increased linearly with d4T concentration at clinically relevant concentrations (r2 between 0.5 and 10 μM = 0.99), but appeared to plateau at > 10 μM (Fig. 1). d4T-TP concentrations in the linear range of the curve (0.05 – 3.4 μM) included maximal cellular concentrations observed in the clinic after adjusting for an average of 8% activated lymphocytes in HIV-infected humans (Fig. 2) (Becher et al., 2004). Therefore, saturation of phosphorylation is not a concern at clinically relevant doses. This supported our model assumption that the accumulation rate of d4T-TP in dividing PBM cells in humans was proportional to extracellular d4T concentrations at clinically relevant doses (below).
Fig. 1.
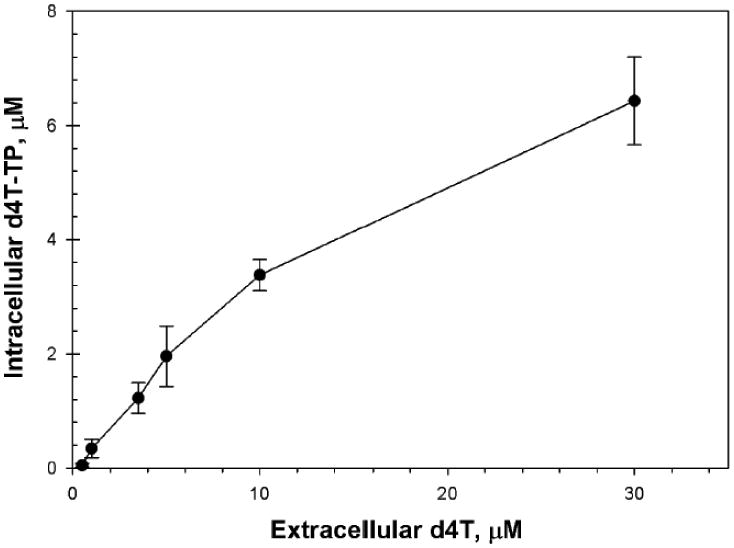
Concentrations of d4T-TP (μM) in activated human PBM cells following a 4 h exposure to varying concentrations of d4T (•). Data shown are mean +/- SD. Plasma concentrations in the clinic are usually in the range of 0.5 to 10 μM. There was good linearity up to the highest clinical concentration range of 7 μM (r2 > 0.99).
Fig. 2.
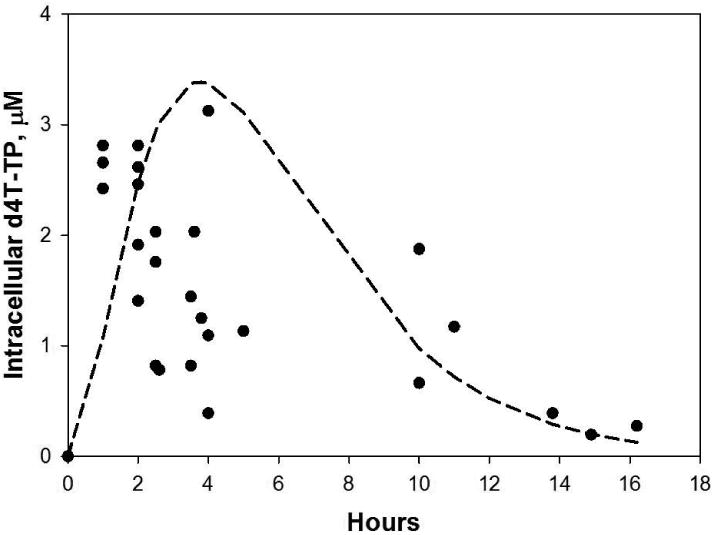
Concentrations of d4T-TP (μM) in activated PBM cells versus time after a single orally administered d4T dose (•). These concentrations were calculated from in vivo d4T-TP measurements reported in lymphocytes (stimulated and non-stimulated) (Becher et al., 2004). Calculations assumed that d4T is primarily phosphorylated in dividing lymphocytes, and an average 8% dividing fraction of lymphocytes (Mohri et al., 2001). The proportion of dividing lymphocytes was not reported in the study of Becher et al., (2004). Also shown is the predicted curve derived from co-fitting the median plasma pharmacokinetic model (without inter-individual variations) (Panhard et al., 2007) with the intracellular data of Becher et al. (2004).
An in vivo population pharmacokinetic and cellular biochemical model was used to simulate plasma d4T and cellular d4T-TP concentrations versus time and dose for HIV-1 infected individuals administered d4T, using the ADVAN9 differential equation routine of NONMEM (6.2, ICON Development Solutions, Ellicott City, MD), together with PLTtools (3.0, PLTsoft, San Francisco, CA). The model assumed that the nucleoside analog d4T achieves rapid equilibration between extra- and intra- cellular concentrations. This may be justified, since nucleoside analog are substrates for rapidly functioning equilibrative transporters which are present on the cell membranes of lymphocytes and have ∼ millisecond equilibrative half-lives (Balimane and Sinko, 1999; Plagemann et al., 1988). Thymidine analogs are primarily phosphorylated in activated (dividing) lymphocytes. Therefore, the cellular accumulation of d4T-TP in vitro and in vivo could be compared by normalizing the respective quantities of d4T-TP by the respective ratios of dividing cells. It follows that since d4T-TP accumulation in vitro was proportional to extracellular d4T, d4T-TP would also remain proportional in vivo. Average and between subject variations in plasma concentrations of d4T were assumed to follow the model proposed by Panhard et al. (Panhard et al., 2007) which was fitted using data from 39 subjects taking d4T at dosages of 30 mg bid (if < 60 kg, n = 8), or 40 mg bid (if > 60 kg, n = 31), either with indinavir (n= 9), with indinavir + ritonavir (n = 2), or with nelfinavir (n = 15). d4T was assumed to be unbound to plasma proteins (Schinazi et al., 2006).
In vivo measurements of d4T-TP in PBM cells were obtained from a study of d4T-TP in 25 of 26 compliant HIV-1 infected subjects administered 30 (if < 60 kg, n = 13), or 40 mg bid (if ≥ 60 kg, n = 12) of d4T with ddI and efavirenz (Becher et al., 2004). The pooled d4T-TP concentrations versus time were co-fitted with the median plasma concentration versus time profile predicted by the population pharmacokinetic model of Panhard for individuals not taking indinavir (i.e., inter-subject variations were not included) (Table 1) (Panhard et al., 2007). Dividing cell concentrations of d4T-TP were calculated assuming an 8% activated lymphocyte fraction (assumed equal to dividing cells), and a cell volume of 0.32 pL (Table 1). Since d4T-TP measurements were not stratified by dose, and the number of individuals receiving either dosage were evenly matched (30 mg bid, n = 13 versus 40 mg bid, n =12), the average dosage of 35 mg bid was used to fit the model. Considering the relative total volumes of plasma and PBM cells, phosphorylation was assumed to have a negligible effect on plasma d4T concentrations (CP). First-order accumulation (KPP) and decay (KDP) rate constants of d4T-TP accumulation were fitted according to:
Table 1. Parameters used to simulate CP/d4T-TP concentrations in activated PBM cells.
| Population pharmacokinetics (Panhard et al., 2007) |
Estimate (%CV) | 95% CI |
|---|---|---|
| N | 39 subjects | |
| V/F (L) | 23.9 | 13.4-42.6 |
| Cl/F/ (L/h) | 15.9 | 10.4-24.4 |
| IND | 1.53 | 0.91-2.57 |
| ka (h-1) | 0.452 | 0.311-0.656 |
| ω2CL/F | (74.0) | 57.1-96.0 |
| ω2V/F | (80.6) | 50.5-128.7 |
| ω2Ka | 0 | 1 (fixed) |
| σ (%) | 37.7 | 32.7-43.4 |
| a (ng/ml) | 110 | |
| Cellular parameters | Reference | |
| Cell volume (pL) | 0.32 | (Diamond et al., 2004) |
| Cells stimulated | ||
| PHA stimulated | 40 % | (Jacobsson et al., 1995) |
| in humans | 8 % | (Mohri et al., 2001) |
| Phosphorylation in vivo | 95% CI | |
| KPP (h-1) | 1.45 | 1 (fixed) |
| KDP (h-1) | 0.526 | 1 (fixed) |
| σ2 (%) | 58 | |
| Mol. weight of d4T | 242.2 |
Model fitting and simulations were performed using the ADVAN9 differential equation routine of NONMEM (6.2, ICON Development Solutions, Ellicott City, MD). CL and V are systemic clearance and distribution volumes, respectively and are divided by F, the unknown fraction of drug which is orally absorbed. Ka is the first-order oral absorption rate constant. IND is the potentiating factor of CL/F. Thus, when d4T is coadministered with indinavir, CL/F was boosted by a factor of 53% (Panhard, et al., 2007). KPP and KDP are first-order rate constants which describe d4T-TP accumulation and decay, respectively, and were obtained by co-fitting the pooled d4T-TP concentration to the median plasma concentration versus time curve. Pharmacokinetic parameters were assumed to be log-normally distributed with variances ω2. %CV of a log-normally distributed parameter = √(e(ω2)-1) × 100. The residual variance σ2 in plasma concentrations of d4T was modeled using a combined proportional and additive error structure, commonly used in population pharmacokinetics. According to this error structure residual error becomes proportional to concentration and tends to “a” at lower concentrations (Panhard et al., 2007). The residual variance of the d4T-TP values ( ) was modeled using a proportional error structure. Thus, observed [d4T-TP] = “true” [d4T-TP] × (1 + ε), where ε is normally distributed with variance . The model fitted for the in vivo cellular pharmacology of d4T-TP converged to > 3 decimal places, and produced a minimum value of the NONMEM objective function (-2 × log-likelihood, -2LL) = 278, using the FOCE with interaction option.
These parameters were considered invariant in the model.
| Eq.1 |
Becher et al., reported a large inter-subject variation in the accumulation of d4T-TP in PBM cells versus time. However, that study had a paucity of d4T-TP measurements within 2 hr after d4T administration (Fig. 2). Therefore, some of the variation in the earlier time points may have resulted from a variable lag time in the onset of d4T absorption following oral administration of d4T. Becher, et al., postulated that some of the substantial variability of d4T-TP concentrations could have resulted from variation in the percentage of activated PBM cells between subjects, a parameter not measured in their study. d4T is phosphorylated primarily in dividing lymphocytes. The proportion of dividing lymphocytes may be elevated in uncontrolled HIV infections, and may decrease towards baseline during adequate HAART treatment (Mohri et al., 2001). d4T-TP concentrations were less variable in measurements taken at periods > 10 h following drug administration. Not surprisingly, the model shown in Fig. 2 fit the later time points better than the earlier time points, while adequately estimating the maximal d4T-TP concentrations.
Plasma d4T concentrations (Cp) were simulated for cohorts of 2,000 individuals (sufficient to ensure reproducible median concentrations versus time) at 30 at 40 mg bid d4T, using the population pharmacokinetic model of Panhard, et al. The Cp versus time profiles were then used to generate corresponding d4T-TP concentration versus time profiles for each simulated individual using Eq. 1 (details and parameters in Table 1). Since the model of Panhard et al. described a 53% increase in the oral clearance (CL/F, where CL = clearance and F is the unknown fraction of oral drug absorbed), when co-administered with indinavir, this analysis was repeated for individuals receiving 30 or 40 mg d4T bid without and with indinavir.The CP and d4T-TP concentrations versus time profiles from all simulated individuals in each cohort were then used to calculate median, 5th, 25th 75th and 95th percentile ranges. The d4T and d4T-TP percentile ranges of individuals taking 30 and 40 mg of d4T bid without indinavir are plotted in Fig 3A and B, respectively, while the corresponding curves for a d4T dosage regimen with indinavir are shown in Fig. 3C and D, respectively. The median inhibitory constant (Ki) of d4T-TP versus wild type (wt) HIV-1 (0.28 μM), measured using a steady-state kinetic assay in the presence of physiological concentrations of competing thymidine triphosphate (TTP) was also plotted for comparison (Ueno and Mitsuya, 1997).
Fig. 3.
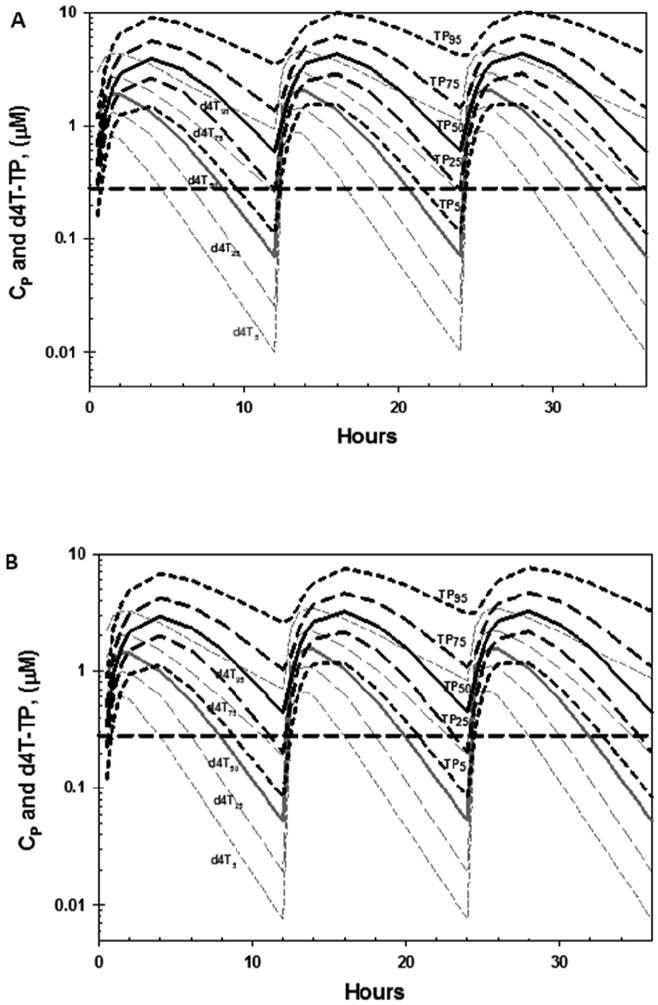
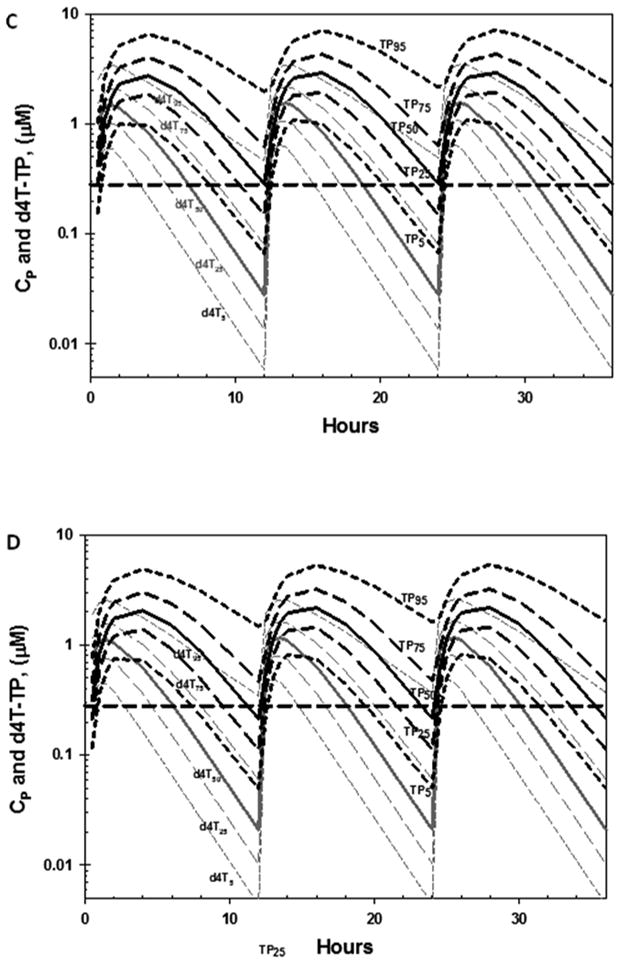
Predicted plasma d4T (grey) and cellular d4T-TP (TP, black) concentrations are shown for individuals taking d4T (without indinavir) at dosages of: 40 mg twice a day (bid) (A) or 30 mg bid (B). Analogous predicted concentration are also shown for individuals taking d4T (with indinavir) at 40 mg bid (C), and 30 mg bid (D). Medians (solid curves), 5th 25th and 75th and 95th percentiles (dashed curves), were calculated using data from 2,000 simulated individuals from each cohort, and are plotted together with the Ki of d4T-TP versus wild type HIV-1 RT measured in vitro in the presence of competing thymidine-5′-triphosphate (TTP).
Concentrations versus time profiles became reproducible after 2 doses for both dosage simulations (Fig. 3A - D). When d4T was administered at a 40 mg bid dosage in the absence of indinavir (Fig. 3A), post 2 day trough concentrations exceeded the Ki of d4T-TP versus wild-type HIV-1 (0.28 μM) in ∼ 75% of individuals, as shown by the intersection of the 75th percentile d4T-TP curve (P25) with the Ki (Fig. 3A and 3B). Reducing the d4T dosage to 30 mg bid did not have a major predicted effect, since the 75th percentile curve of d4T-TP was below the Ki for only 1 to 2 out of 12 hr (Fig. 3B). Co-administration of 40 mg bid of d4T with indinavir was predicted to produce post- 2 day trough concentrations > Ki in only ∼ 50% of individuals (Fig. 3C), due to the boosted clearance in d4T CL/F predicted by the population pharmacokinetic model. Decreasing the d4T dosage to 30 mg bid during co-administration with indinavir was not predicted to have a major effect on trough d4T-TP concentrations relative to 40 mg d4T bid coadministered with indinavir (Fig. 3D). None of the simulations produced trough concentrations > Ki of clinically resistant HIV-1 isolates (2.5 μM) (Ueno and Mitsuya, 1997).
The net terminal half-life (t1/2) in cellular d4T-TP in humans reported by Becher et al. (2004), based on the terminal half-life using the data summarized in Fig. 2 was 6.6 h, while that calculated based on the cellular dephosphorylation rate constant (KDP) derived by co-fitting the in vivo cellular d4T-TP concentrations with median plasma concentration versus time profile of d4T (t1/2 = 0.693/KDP) was 1.3 h. This is consistent with some replacement of cellular d4T-TP in vivo due to phosphorylation of d4T entering lymphocytes from the plasma (d4T-TP itself does not penetrate the cell membrane). Interestingly, an extracellular t½ of 3.5 h was previously reported in PHA stimulated PBM cells (Zhu et al., 1990).
The model had certain limitations since for example, calculations assumed an 8% dividing lymphocyte fraction. However, in practice this fraction may vary between 3 and 16% depending on disease state (Hellerstein et al., 1999; Mohri et al., 2001). The d4T-TP cell contents reported by Becher et al. (2004) were measured in fmol per 106 cells, rather than as fmol per volume of cells, and cell volume varies with the fraction of activated lymphocytes (not measured in the study). In the modeling the phosphorylation rates and cell sizes were assumed constant. The population pharmacokinetic parameters used were obtained from a relatively small study (n = 39 subjects) (Panhard et al., 2007). In that study body-weight and creatinine clearance were not demonstrated to be significant covariates of CL/F and orally determined distribution volume (V/F, where V is volume and F is the unknown fraction of dose absorbed) of d4T. However, it is possible that these covariates may have significant impact if larger and more diverse populations are modeled. Concentrations of d4T-TP in activated lymphocytes were assessed relative to the Ki of HIV-1 measured in vitro and used as an indicator of antiviral efficacy. However, Ki measurements are sensitive to a variety of factors, including the use of homopolymer/heteropolymer templates, pH variations, competing levels of natural dNTP's including ATP, and possibly other experimental conditions. Levels of HIV-1 RT heterodimer p66/p51 and homodimer form p66/p66 can also affect the Ki values (Shao, 2004). Therefore, further analysis may be warranted once additional data become available.
This study in association with early clinical studies provides support that a d4T dose reduction from 40 to 30 mg bid to reduce toxicity, would not substantially compromise antiviral efficacy against wild type HIV-1. However, a substantial decrease in efficacy was predicted for both d4T doses when co-administered with indinvavir which was reported to increase the CL/F of d4T (Panhard, et al., 2007). Therefore, the possibility of other drug interactions should also be considered.
Lipodystrophy is associated with depletion of mitochondrial DNA in adipocytes, which could result from certain NRTI-TP acting as inhibitors of mitochondrial DNA polymerase-γ (pol–γ), and as chain terminators, if replicating mitochondrial DNA strands incorporate NRTI-TP (Igoudjil et al., 2008; Wendelsdorf et al., 2009). However, an enzymatic study using cultured mouse 3T3-L1 adipocytes revealed NRTI phosphorylation was much lower in adipocytes than in lymphocytes and concluded that toxicity may result at least in part from the depletion of natural dNTP in adipocytes (Rylova et al., 2005). To date no pharmacokinetic studies have been performed to quantify the accumulation and in vivo stability of d4T-TP in adipocytes during d4T administration. Therefore, although a similar simulation study could in principle be conducted by linking the pharmacokinetics of d4T with the kinetics of d4T-TP accumulation in the adipocytes of infected individuals, the data required to derive the required model parameters are not readily available.
The majority of people in Africa and other third world countries still rely on cocktails containing d4T and Nevirapine as first line therapy thanks to the generic drugs imported primarily from Brazil, China and India, which are manufactured in most cases under license from US or European companies. These cocktails of fixed dose combinations are not suboptimal with regard to efficacy, and d4T may have advantages in relation to resistance development, specifically the counter-selection of K65R due to the bidirectional antagonism of thymidine analog mutations and K65R pathways in optimized regimens (Parikh et al., 2006). Therefore, it is likely that the use of d4T containing regimens will continue in developing countries for the immediate future until a safer effective alternative becomes globally available, including in resource limited settings (Walensky et al., 2010). d4T usage worldwide has declined significantly, however, the WHO estimates that 56% of adults receiving antiretroviral therapy in low-and middle-income countries were receiving d4T compared with 8% receiving tenofovir-containing therapy (World Health Organization. Towards universal access: scaling up priority HIV/AIDS interventions in the health sector: progress report 2009). The modeling and pilot clinical trial findings suggest that dose reduction from 40 to 30 mg d4T in existing HAART regimens could maintain efficacy. Whether a concurrent decrease in HIV associated lipodystrophy syndrome (HALS) occurs at the reduced dosage can only be assessed by well-controlled clinical trials.
Highlights.
Stavudine (d4T) is widely used in resource poor settings despite toxicity.
WHO recommended reducing d4T dose from 40 to 30 mg bid when replacement is not feasible.
A population PK/biochemical model was used to simulate cellular d4T-TP versus dose.
d4T-TP concentrations for 30 mg bid exceeded wt HIV-1 EC50 in most virtual subjects,
This suggests that this low dose could maintain antiviral efficacy
Acknowledgments
This work was supported in part by NIH CFAR grant 2P30-AI-050409 and the Department of Veterans Affairs (to RFS). We thank Dr. E. Fromentin for her excellent technical support for the cellular biochemical studies.
Footnotes
Part of this work was presented at HIV-DART (Dec 7-10, 2010), Los Cabos, Mexico.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ait-Mohand H, Bonmarchand M, Guiguet M, Slama L, Marguet F, Behin A, Amellal B, Bennai Y, Peytavin G, Calvez V, Pialoux G, Murphy R, Katlama C. Viral efficacy maintained and safety parameters improved with a reduced dose of stavudine: a pilot study. HIV Med. 2008;9:738–746. doi: 10.1111/j.1468-1293.2008.00616.x. [DOI] [PubMed] [Google Scholar]
- Balimane PV, Sinko PJ. Involvement of multiple transporters in the oral absorption of nucleoside analogues. Adv Drug Deliv Rev. 1999;39:183–209. doi: 10.1016/s0169-409x(99)00026-5. [DOI] [PubMed] [Google Scholar]
- Becher F, Landman R, Mboup S, Kane CNT, Canestri A, Liegeois F, Vray M, Prevot MH, Leleu G, Benech H. Monitoring of didanosine and stavudine intracellular trisphosphorylated anabolite concentrations in HIV-infected patients. Aids. 2004;18:181–187. doi: 10.1097/00002030-200401230-00006. [DOI] [PubMed] [Google Scholar]
- Bygrave H, Ford N, van Cutsem G, Hilderbrand K, Jouquet G, Goemaere E, Vlahakis N, Trivino L, Makakole L, Kranzer K. Implementing a tenofovir-based first-line regimen in rural Lesotho: clinical outcomes and toxicities after two years. J Acquir Immune Defic Syndr. 2011;56:e75–78. doi: 10.1097/QAI.0b013e3182097505. [DOI] [PubMed] [Google Scholar]
- Casiro' AD. The Evolving Role of Stavudine as Part of Initial Antiretroviral Therapy in Resource-Constrained Settings. [accessed 06-03-2011];Clinical Care Options 04 2011. http://www.clinicaloptions.com/HIV/Resources/News%20and%20Comment/Expert%20Viewpoints/April%202011.aspx.
- Cournil A, Coudray M, Kouanfack C, Essomba CN, Tonfack CA, Biwole-Sida M, Delaporte E, Bork K, Laurent C. Reduced dose of stavudine and lipoatrophy in HIV-infected patients in Cameroon. Antivir Ther. 2010;15:1039–1043. doi: 10.3851/IMP1664. [DOI] [PubMed] [Google Scholar]
- Diamond TL, Roshal M, Jamburuthugoda VK, Reynolds HM, Merriam AR, Lee KY, Balakrishnan M, Bambara RA, Planelles V, Dewhurst S, Kim B. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J Biol Chem. 2004;279:51545–51553. doi: 10.1074/jbc.M408573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo P, Cabeza MC, Pruvost A, Salazar J, Gutierrez Mdel M, Mateo MG, Domingo JC, Fernandez I, Villarroya F, Munoz J, Vidal F, Baiget M. Relationship between HIV/Highly active antiretroviral therapy (HAART)-associated lipodystrophy syndrome and stavudine-triphosphate intracellular levels in patients with stavudine-based antiretroviral regimens. Clin Infect Dis. 2010;50:1033–1040. doi: 10.1086/651117. [DOI] [PubMed] [Google Scholar]
- Dube MP, Komarow L, Mulligan K, Grinspoon SK, Parker RA, Robbins GK, Roubenoff R, Tebas P. Long-term body fat outcomes in antiretroviral-naive participants randomized to nelfinavir or efavirenz or both plus dual nucleosides. Dual X-ray absorptiometry results from A5005s, a substudy of Adult Clinical Trials Group 384. J Acquir Immune Defic Syndr. 2007;45:508–514. doi: 10.1097/QAI.0b013e3181142d26. [DOI] [PubMed] [Google Scholar]
- Dube MP, Parker RA, Tebas P, Grinspoon SK, Zackin RA, Robbins GK, Roubenoff R, Shafer RW, Wininger DA, Meyer WA, 3rd, Snyder SW, Mulligan K. Glucose metabolism, lipid, and body fat changes in antiretroviral-naive subjects randomized to nelfinavir or efavirenz plus dual nucleosides. AIDS. 2005;19:1807–1818. doi: 10.1097/01.aids.0000183629.20041.bb. [DOI] [PubMed] [Google Scholar]
- Fromentin E, Asif G, Obikhod A, Hurwitz SJ, Schinazi RF. Simultaneous quantification of 9-(beta-D-1,3-dioxolan-4-yl)guanine, Amdoxovir and Zidovudine in human plasma by liquid chromatography-tandem mass spectrometric assay. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3482–3488. doi: 10.1016/j.jchromb.2009.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallant JE, Staszewski S, Pozniak AL, DeJesus E, Suleiman JM, Miller MD, Coakley DF, Lu B, Toole JJ, Cheng AK. Efficacy and safety of tenofovir DF vs stavudine in combination therapy in antiretroviral-naive patients: a 3-year randomized trial. JAMA. 2004;292:191–201. doi: 10.1001/jama.292.2.191. [DOI] [PubMed] [Google Scholar]
- Gilead Web site. Enabling access to treatment. [accessed 07-12-2011]; Available at: http://www.gilead.com/enabling_accesshttp://www.gilead.com/pdf/access_fact_sheet.pdf.
- Hellerstein M, Hanley MB, Cesar D, Siler S, Papageorgopoulos C, Wieder E, Schmidt D, Hoh R, Neese R, Macallan D, Deeks S, McCune JM. Directly measured kinetics of circulating T lymphocytes in normal and HIV-1-infected humans. Nat Med. 1999;5:83–89. doi: 10.1038/4772. [DOI] [PubMed] [Google Scholar]
- Hernandez-Santiago BI, Chen H, Asif G, Beltran T, Mao S, Hurwitz SJ, Grier J, McClure HM, Chu CK, Liotta DC, Schinazi RF. Pharmacology and pharmacokinetics of the antiviral agent beta-D-2′,3′-dideoxy-3′-oxa-5-fluorocytidine in cells and rhesus monkeys. Antimicrob Agents Chemother. 2005;49:2589–2597. doi: 10.1128/AAC.49.7.2589-2597.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Santiago BI, Mathew JS, Rapp KL, Grier JP, Schinazi RF. Antiviral and cellular metabolism interactions between dexelvucitabine and lamivudine. Antimicrob Agents Chemother. 2007a;51:2130–2135. doi: 10.1128/AAC.01543-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Santiago BI, Obikhod A, Fromentin E, Hurwitz SJ, Schinazi RF. Short communication cellular pharmacology of 9-(beta-D-1,3-dioxolan-4-yl) guanine and its lack of drug interactions with zidovudine in primary human lymphocytes. Antivir Chem Chemother. 2007b;18:343–346. doi: 10.1177/095632020701800606. [DOI] [PubMed] [Google Scholar]
- Hoffmann CJ, Charalambous S, Fielding KL, Innes C, Chaisson RE, Grant AD, Churchyard GJ. HIV suppression with stavudine 30 mg versus 40 mg in adults over 60kg on antiretroviral therapy in South Africa. AIDS. 2009;23:1784–1786. doi: 10.1097/QAD.0b013e32832e0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton CM, Dudley MN, Kaul S, Mayer KH, Squires K, Dunkle L, Anderson R. Population pharmacokinetics of stavudine (d4T) in patients with AIDS or advanced AIDS-related complex. Antimicrob Agents Chemother. 1995;39:2309–2315. doi: 10.1128/aac.39.10.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz SJ, Asif G, Kivel NM, Schinazi RF. Development of an optimized dose for coformulation of zidovudine with drugs that select for the K65R mutation using a population pharmacokinetic and enzyme kinetic simulation model. Antimicrob Agents Chemother. 2008;52:4241–4250. doi: 10.1128/AAC.00054-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz SJ, Asif G, Schinazi RF. Development of a population simulation model for HIV monotherapy virological outcomes using lamivudine. Antivir Chem Chemother. 2007;18:329–341. doi: 10.1177/095632020701800605. [DOI] [PubMed] [Google Scholar]
- Igoudjil A, Massart J, Begriche K, Descatoire V, Robin MA, Fromenty B. High concentrations of stavudine impair fatty acid oxidation without depleting mitochondrial DNA in cultured rat hepatocytes. Toxicol In Vitro. 2008;22:887–898. doi: 10.1016/j.tiv.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Jacobsson B, Britton S, He Q, Karlsson A, Eriksson S. Decreased thymidine kinase levels in peripheral blood cells from HIV-seropositive individuals: implications for zidovudine metabolism. AIDS Res Hum Retroviruses. 1995;11:805–811. doi: 10.1089/aid.1995.11.805. [DOI] [PubMed] [Google Scholar]
- Karara MW, Okalebo FA, Oluka MN, Ombega J, Guantai AN, Osanjo GO. Comparative tolerability and efficacy of stavudine 30 mg versus stavudine 40 mg in patients on combination antiretroviral therapy in Kenya. Journal of AIDS and HIV Research. 2010;2:24–31. [Google Scholar]
- Keiser O, Anastos K, Schechter M, Balestre E, Myer L, Boulle A, Bangsberg D, Toure H, Braitstein P, Sprinz E, Nash D, Hosseinipour M, Dabis F, May M, Brinkhof MW, Egger M. Antiretroviral therapy in resource-limited settings 1996 to 2006: patient characteristics, treatment regimens and monitoring in sub-Saharan Africa, Asia and Latin America. Trop Med Int Health. 2008;13:870–879. doi: 10.1111/j.1365-3156.2008.02078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McComsey G, Lonergan JT. Mitochondrial dysfunction: patient monitoring and toxicity management. J Acquir Immune Defic Syndr. 2004;37(1):S30–35. doi: 10.1097/01.qai.0000137004.63376.27. [DOI] [PubMed] [Google Scholar]
- McCutchan JA. Management of HIV in resource limited settings. Curr Opin Infect Dis. 2009;22:464–470. doi: 10.1097/QCO.0b013e3283308fe2. [DOI] [PubMed] [Google Scholar]
- Mercier S, Gueye NF, Cournil A, Fontbonne A, Copin N, Ndiaye I, Dupuy AM, Cames C, Sow PS, Ndoye I, Delaporte E, Simondon KB. Lipodystrophy and metabolic disorders in HIV-1-infected adults on 4- to 9-year antiretroviral therapy in Senegal: a case-control study. J Acquir Immune Defic Syndr. 2009;51:224–230. doi: 10.1097/QAI.0b013e31819c16f4. [DOI] [PubMed] [Google Scholar]
- Mohri H, Perelson AS, Tung K, Ribeiro RM, Ramratnam B, Markowitz M, Kost R, Hurley A, Weinberger L, Cesar D, Hellerstein MK, Ho DD. Increased turnover of T lymphocytes in HIV-1 infection and its reduction by antiretroviral therapy. J Exp Med. 2001;194:1277–1287. doi: 10.1084/jem.194.9.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panhard X, Legrand M, Taburet AM, Diquet B, Goujard C, Mentre F. Population pharmacokinetic analysis of lamivudine, stavudine and zidovudine in controlled HIV-infected patients on HAART. Eur J Clin Pharmacol. 2007;63:1019–1029. doi: 10.1007/s00228-007-0337-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh UM, Bacheler L, Koontz D, Mellors JW. The K65R mutation in human immunodeficiency virus type 1 reverse transcriptase exhibits bidirectional phenotypic antagonism with thymidine analog mutations. J Virol. 2006;80:4971–4977. doi: 10.1128/JVI.80.10.4971-4977.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrol E, Martin T, del Pozo MA, Flores J, Sanz J, Carton JA, Jusdado JJ, Arazo P, Ribera E, Deig E. Efficacy and safety of a reduced-dose of stavudine in HIV-infected patients under immunological and virological stable conditions. Med Clin (Barc) 2007;129:361–365. doi: 10.1157/13110209. [DOI] [PubMed] [Google Scholar]
- Plagemann PG, Wohlhueter RM, Woffendin C. Nucleoside and nucleobase transport in animal cells. Biochim Biophys Acta. 1988;947:405–443. doi: 10.1016/0304-4157(88)90002-0. [DOI] [PubMed] [Google Scholar]
- Pujari SN, Dravid A, Naik E, Bhagat S, Tash K, Nadler JP, Sinnott JT. Lipodystrophy and dyslipidemia among patients taking first-line, World Health Organization-recommended highly active antiretroviral therapy regimens in Western India. J Acquir Immune Defic Syndr. 2005;39:199–202. [PubMed] [Google Scholar]
- Rosario MC, Jacqmin P, Dorr P, van der Ryst E, Hitchcock C. A pharmacokinetic-pharmacodynamic disease model to predict in vivo antiviral activity of maraviroc. Clin Pharmacol Ther. 2005;78:508–519. doi: 10.1016/j.clpt.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Rylova SN, Albertioni F, Flygh G, Eriksson S. Activity profiles of deoxynucleoside kinases and 5′-nucleotidases in cultured adipocytes and myoblastic cells: insights into mitochondrial toxicity of nucleoside analogs. Biochem Pharmacol. 2005;69:951–960. doi: 10.1016/j.bcp.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Sanchez-Conde M, de Mendoza C, Jimenez-Nacher I, Barreiro P, Gonzalez-Lahoz J, Soriano V. Reductions in stavudine dose might ameliorate mitochondrial-associated complications without compromising antiviral activity. HIV Clin Trials. 2005;6:197–202. doi: 10.1310/ed57-eu48-rk6a-e5u0. [DOI] [PubMed] [Google Scholar]
- Schinazi RF, Hernandez-Santiago BI, Hurwitz SJ. Pharmacology of current and promising nucleosides for the treatment of human immunodeficiency viruses. Antiviral Res. 2006;71:322–334. doi: 10.1016/j.antiviral.2006.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao X. Ph.D. Thesis. Karolinska Institute; Stockholm, Sweden: 2004. Reverse transcriptase assays of resistance to anti-HIV drugs and their Mechanism of action. [Google Scholar]
- Sinxadi PZ, van der Walt JS, McIlleron HM, Badri M, Smith PJ, Dave JA, Levitt NS, Maartens G. Lack of association between stavudine exposure and lipoatrophy, dysglycaemia, hyperlactataemia and hypertriglyceridaemia: a prospective cross sectional study. AIDS Res Ther. 2010;7:23. doi: 10.1186/1742-6405-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno T, Mitsuya H. Comparative enzymatic study of HIV-1 reverse transcriptase resistant to 2′,3′-dideoxynucleotide analogs using the single-nucleotide incorporation assay. Biochemistry. 1997;36:1092–1099. doi: 10.1021/bi962393d. [DOI] [PubMed] [Google Scholar]
- van Griensven J, Zachariah R, Rasschaert F, Mugabo J, Atte EF, Reid T. Stavudine- and nevirapine-related drug toxicity while on generic fixed-dose antiretroviral treatment: incidence, timing and risk factors in a three-year cohort in Kigali, Rwanda. Trans R Soc Trop Med Hyg. 2009 doi: 10.1016/j.trstmh.2009.07.009. [DOI] [PubMed] [Google Scholar]
- ViiV Healthcare Web site. Not-for-profit pricing. [accessed 07-12-2011]; Available at: http://www.viivhealthcare.com/
- Wainberg MA. Two standards of care for HIV: why are Africans being short-changed? Retrovirol. 2009;6:109. doi: 10.1186/1742-4690-6-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky RP, Wood R, Ciaranello AL, Paltiel AD, Lorenzana SB, Anglaret X, Stoler AW, Freedberg KA. Scaling Up the 2010 World Health Organization HIV Treatment Guidelines in Resource-Limited Settings: A Model-Based Analysis. PLoS Med. 2010;7:e1000382. doi: 10.1371/journal.pmed.1000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendelsdorf KV, Song Z, Cao Y, Samuels DC. An analysis of enzyme kinetics data for mitochondrial DNA strand termination by nucleoside reverse transcription inhibitors. PLoS Comput Biol. 2009;5:e1000261. doi: 10.1371/journal.pcbi.1000261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. Antiretroviral therapy for HIV infection in adults and adolescents: recommendations for a public health approach—2010 rev. [Accessed June 05, 2011]; Available at: http://whqlibdoc.who.int/publications/2010/9789241599764_eng.pdf. [PubMed]
- World Health Organization. Towards universal access: scaling up priority HIV/AIDS interventions in the health sector: progress report 2009 website. [Accessed July 2011]; Available at: http://www.who.int/hiv/pub/tuapr_2009_en.pdf.
- Zhu Z, Ho HT, Hitchcock MJ, Sommadossi JP. Cellular pharmacology of 2′,3′- didehydro-2′,3′-dideoxythymidine (D4T) in human peripheral blood mononuclear cells. Biochem Pharmacol. 1990;39:R15–19. doi: 10.1016/0006-2952(90)90418-k. [DOI] [PubMed] [Google Scholar]