

Abstract
Crosstalk between the Notch and wingless-type MMTV integration site (WNT) signaling pathways has been investigated for many developmental processes. However, this negative correlation between Notch and WNT/β-catenin signaling activity has been studied primarily in normal developmental and physiological processes in which negative feedback loops for both signaling pathways are intact. We found that Notch1 signaling retained the capability of suppressing the expression of WNT target genes in colorectal cancers even when β-catenin destruction by the adenomatous polyposis coli (APC) complex was disabled. Activation of Notch1 converted high-grade adenoma into low-grade adenoma in an Apcmin mouse colon cancer model and suppressed the expression of WNT target genes in human colorectal cancer cells through epigenetic modification recruiting histone methyltransferase SET domain bifurcated 1 (SETDB1). Extensive microarray analysis of human colorectal cancers also showed a negative correlation between the Notch1 target gene, Notch-regulated ankyrin repeat protein 1 (NRARP), and WNT target genes. Notch is known to be a strong promoter of tumor initiation, but here we uncovered an unexpected suppressive role of Notch1 on WNT/β-catenin target genes involved in colorectal cancer.
Introduction
Activation of the adenomatous polyposis coli/β-catenin (APC/β-catenin) pathway is a crucial initiating event in human colorectal cancer (CRC) (1, 2). The multistage progression of CRC is then followed by sequential activation of oncogenes and inactivation of tumor-suppressor genes, including K-RAS, TGFβ/SMAD4, and TP53 (2, 3). Although the implications of these genetic alterations are well characterized in the adenoma-carcinoma sequence, limited studies have been conducted to examine the influence of other molecular signaling pathways on these tumors.
Signaling pathways, such as wingless-type MMTV integration site (WNT), BMP, Hedgehog, and Notch, are important not only in embryonic development, but also in adult intestinal homeostasis (4, 5). In the intestine, WNT signaling is crucial for the proliferation and maintenance of intestinal stem cells and progenitor cells (6). Notch signaling regulates cell fate decisions between secretory and absorptive cell lineages (7, 8). The Notch signaling pathway also plays a role in the maintenance of proliferating progenitors (9, 10). Under normal conditions, these 2 pathways are exclusively required for maintaining intestinal stem cells (7). Activation of the WNT/β-catenin signaling pathway is the main rate-limiting step of CRC initiation (1), and Notch signaling has been shown to promote CRC initiation in a mouse Apcmin tumor model (11, 12). Therefore, the Notch and WNT pathways may function cooperatively in intestinal epithelium and tumors.
γ-Secretase inhibitor has been utilized to prevent intestinal tumor growth in Apcmin mice (10), which showed reduced proliferation of tumor cells and increased goblet cell conversion following γ-secretase inhibitor treatment. In contrast, overexpression of the Notch intracellular domain facilitates tumor formation in Apcmin mice (11), suggesting an oncogenic role of Notch in intestinal tumor formation. However, other reports have examined the efficacy of γ-secretase inhibitors in blocking the growth of CRCLs and have obtained disappointing results (13). A genetic analysis involving Apc and Rbpjκ floxed mice also failed to demonstrate that Notch signaling is essential for tumorigenic transformation (14), and the Apc;Rbpjκ compound mice did not show a survival advantage. Thus, the precise role of Notch signaling in CRCs is controversial.
We investigated the role of Notch signaling in established CRC tumors. We did not examine this signaling during the initial phase of transformation because it has been clearly shown in many studies that Notch signaling promotes the onset of intestinal tumors (11). Thus, after tumor onset, we reexamined the characteristics of Notch-activated tumors in RosaN1+/RN1;Apcmin mice in which tumor initiation had progressed compared with Apcmin mice (11). Unexpectedly, these tumors showed low-grade adenoma features, including columnar epithelial morphology and a restoration of the adherens junction. Concomitant microarray analysis demonstrated Notch-mediated downregulation of Tcf4/β-catenin target genes, even in the absence of functional Apc. Microarray analysis of human CRC patient data further reinforced the negative correlation between Notch and WNT signaling activity in CRC. Our study examining the effect of Notch signaling in CRC cell lines (CRCLs) revealed that Notch signaling leads to a repressive histone status of WNT target gene promoters through the activities of Nemo-like kinase (NLK) and SET domain bifurcated 1 (SETDB1). While Notch signaling is a strong promoter in intestinal tumor initiation, it also has an unexpected suppressive role in the expression of WNT/β-catenin target genes in established tumors, even in the absence of functional Apc.
Results
Histological characterization of Notch-activated Apcmin tumor after its onset.
To examine how Notch activation affects intestinal tumors, we generated an intestinal gain-of-Notch model by crossing Apcmin background mice with Rosa-N1icd (RN1) mice, which have a transgene composed of a floxed Neo/STOP cassette followed by N1ICD without the PEST domain in the Rosa26 locus (15) and the Villin-Cre (Vil-Cre) transgene (16). These mice contain Notch-activated intestinal epithelium in which intestinal tumors occur upon the loss of heterogeneity of the Apcmin locus. A detailed description of these mice is provided in Supplemental Information and in Supplemental Figure 1 (supplemental material available online with this article; doi: 10.1172/JCI61216DS1). Consistent with the results of a previous study (11), 7-week-old Vil-Cre;RosaN1+/RN1;Apcmin mice developed several tumors in their small and large intestines, while tumors were not or were rarely detected in control Apcmin mice. Since the onset of tumor formation was clearly accelerated in Vil-Cre;RosaN1+/RN1;Apcmin mice compared with control Apcmin mice in our study as well as in a previous study (11), we collected tumor tissues from 7-week-old Vil-Cre;RosaN1+/RN1;Apcmin mice and 10- to 15-week-old Apcmin mice. For histological analysis, we used tumors of similar size from each genotype for comparison.
Interestingly, a detailed histological analysis of tumors in Vil-Cre;RosaN1+/RN1;Apcmin mice, particularly those in the colon, showed low-grade adenoma characteristics with enhanced epithelial cell morphology, while control tumors from Apcmin mice showed high-grade adenoma characteristics (Figure 1, A and B). Tumors from Vil-Cre;RosaN1+/RN1;Apcmin mice did not form tightly packed cell agglomerations as observed in Apcmin tumors. Instead, they formed an extended single layer of epithelial cells. Low-grade adenoma characteristics were also observed in older (10- to 15-week-old) moribund Vil-Cre;RosaN1+/RN1;Apcmin mice (not shown), suggesting that mortality of Vil-Cre;RosaN1+/RN1;Apcmin mice was not due to further progression of tumor severity.
Figure 1. Notch-activated Apcmin tumors show low-grade adenoma features.
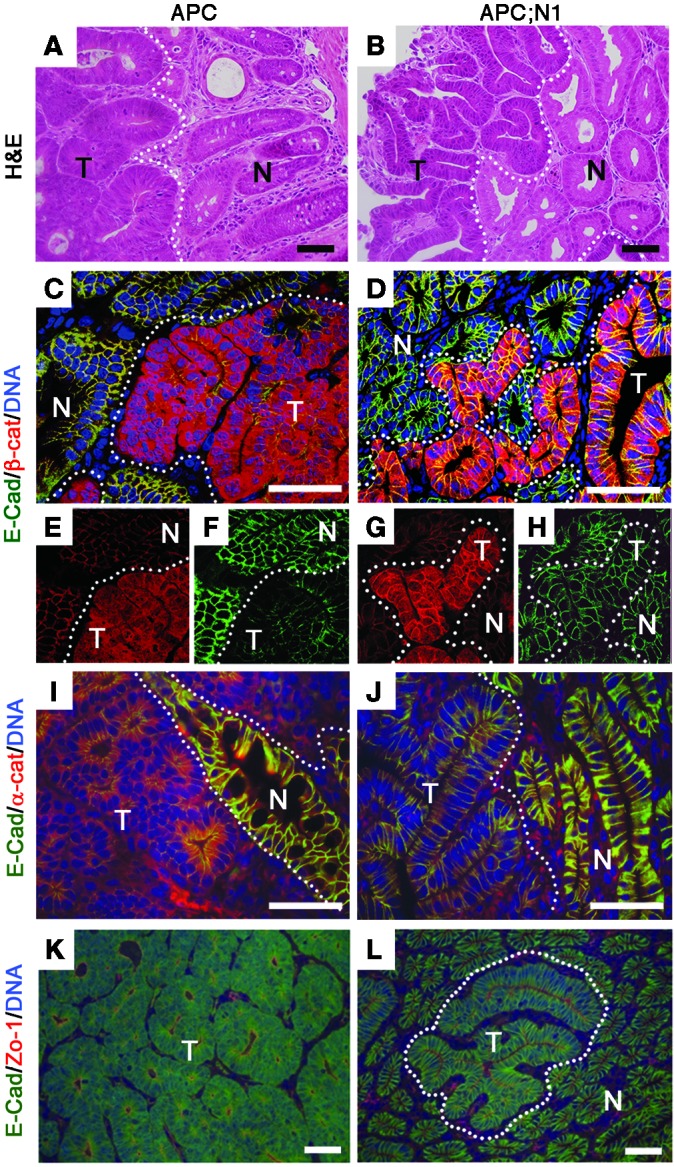
Colons from 10- to 15-week-old Apcmin (APC: A, C, E, F, I, and K) and 7-week-old Vil-Cre;RosaN1+/RN1;Apcmin (APC;N1: B, D, G, H, J, and L) mice were used for histological analysis. Note that APC;N1 tumors have well-branched tubular structures (B), and more membranous β-catenin (G) and E-cadherin (H) staining with extended apical membranes (L) compared with APC tumors. Staining: H&E (A and B); E-cadherin (E-cad) (green), and β-catenin (β-cat) (red) (C, D, E, F, G, and H); E-cadherin (green) and α-catenin (red) (I and J); and E-cadherin (green) and ZO-1 (red) (K and L). N, normal tissue; T, tumor. Dotted lines indicate tumor regions. Scale bars: 50 εm.
Next, we investigated the localization of β-catenin. Control Apcmin tumors showed prominent nuclear and cytoplasmic staining of β-catenin, a hallmark of active WNT signaling (Figure 1, C and D). Although nuclear β-catenin was still detectable in Notch-activated Apcmin tumors, most of the β-catenin was present in the plasma membrane (Figure 1, F and G). Importantly, the expression level of E-cadherin was restored in the plasma membrane of Notch-activated Apcmin tumors (Figure 1, E and H). Coimmunostaining with E-cadherin and α-catenin showed that, while adherens junctions were disrupted in Apcmin tumors (Figure 1I), E-cadherin and α-catenin were colocalized in the plasma membrane of Notch-activated Apcmin tumors (Figure 1J). Moreover, double staining of E-cadherin and ZO-1, a marker for tight junctions, clearly showed that lateral and apical membranes were well established in Notch-activated Apcmin tumors (Figure 1, K and L). These results show that Notch-activated Apcmin tumors display low-grade adenoma with restored epithelial characteristics.
Downregulated WNT target genes in Notch-activated Apcmin tumors.
Milder histological characteristics of Notch-activated Apcmin tumors compared with those of normal Apcmin tumors prompted us to further analyze molecular changes in these tumor models. Because tumor onset was accelerated in the Vil-Cre;RosaN1+/RN1;Apcmin mice as described above, we collected tumors of similar size from 7-week-old Vil-Cre;RosaN1+/RN1;Apcmin mice and 10-week-old Apcmin mice. First, we determined the changes in expression between tumors from these 2 genotypes and then compared the entire signature to changes between normal intestinal epithelium and Apcmin tumors (Figure 2A and Supplemental Table 1). Among 2,750 genes significantly altered during Apcmin tumorigenesis (normal tissue vs. tumor), activation of Notch signaling suppressed approximately 40% of tumor-associated genes toward normal levels (Figure 2A). In cluster A, depicted in the figure, we identified genes related to cell metabolism and differentiation using Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, confirming the histological observation that Notch signaling promotes epithelial differentiation of intestinal tumor cells (Supplemental Table 2).
Figure 2. Decreased levels of WNT target genes in Notch-activated Apcmin tumors.

Colons (normal) from 10-week-old WT and 7-week-old Vil-Cre;RosaN1+/RN1 (N1) mice and colonic tumor tissues (tumor) from 10-week-old Apcmin (APC) and 7-week-old Vil-Cre;RosaN1+/RN1;Apcmin mice were used for gene expression and histological analysis. (A) Heat map of gene expression levels. Clusters A and B indicate genes that reverted to levels in normal tissues upon Notch activation in the tumors. (B–E) Real-time qRT-PCR analysis (B–E). Note the reduced expression levels of WNT target genes (Axin2, Wif1, Nkd1, and Apcdd1) in APC;N1 tumors compared with those of APC tumors. Bars indicate mean + SD. *P < 0.05; **P < 0.01; ***P < 0.001. (F and G) In situ hybridization (F) and fluorescent immunostaining (G) of Apcmin (upper panels) and Vil-Cre;RosaN1+/RN1;Apcmin (lower panels) tumors. Note that the expression levels of Axin2, Wif1, Apcdd1, and PROX1 were reduced or absent in Vil-Cre;RosaN1+/RN1;Apcmin tumors. Dotted lines indicate tumor regions. Scale bars: 100 εm (F); 50 εm (G).
Strikingly, the KEGG pathway analysis of cluster B showed that WNT signaling pathway genes were downregulated in Notch-activated tumors (Supplemental Table 3; P = 1.3 × 10–4). Quantitative RT-PCR (qRT-PCR) analysis confirmed that the WNT target genes Axin2, Wif1, Nkd1, and Apcdd1 (17–20) were downregulated in Notch-activated Apcmin tumors compared with normal Apcmin tumors (Figure 2, B–E). Consistent with these results, in situ hybridization and immunohistological analysis clearly showed that expression levels of Axin2, Wif1, and Apcdd1 were reduced in Notch-activated Apcmin tumors (Figure 2, F and G). Importantly, we also observed that PROX1, a β-catenin target gene known to promote intestinal tumor progression (21), was nearly absent in Notch-activated Apcmin tumors (Figure 2G). These data show that Notch signaling specifically counteracts deregulation of approximately 40% of Apcmin tumor-associated genes by modulating factors downstream of the WNT signaling pathway.
Negative correlation between Notch and WNT target genes in colonic tumors in mice and humans.
Based on our observations using mouse tumor models, we explored gene expression profiles of human CRC patients to determine whether similar features, such as the negative correlation between Notch and WNT target genes, could also be observed in human CRC. To address this issue, we first sought to identify a reliable marker gene for Notch signaling activity. Although Hes1 is a well-known Notch target gene, a recent study reported that Hes1 is also directly transcribed by β-catenin signaling through conserved Tcf motifs as well as Notch (14). We also did not observe a clear correlation in Hes1 expression between control and Notch-activated Apcmin tumors (Figure 3A). Therefore, we sought to identify another known Notch target gene and identified Notch-regulated ankyrin repeat protein 1 (Nrarp) from our mouse models as reported (22–24). To confirm the correlation between Nrarp expression and Notch signaling activity, we used transgenic Notch reporter (TNR) mice, which have a Notch-responsive GFP reporter that faithfully recapitulates endogenous Notch activity (25). In Apcmin;TNR mice, expression levels of GFP and Nrarp were reduced to approximately half in the tumor compared with nontumor intestinal epithelial tissues, while the expression level of Hes1 slightly increased (Figure 3A). Consistently, in situ hybridization and real-time qRT-PCR analysis showed that the Nrarp expression level was increased in Notch-activated Apcmin tumors compared with normal Apcmin tumors (Figure 3, B and C). These data show that Nrarp reports Notch signaling activity more accurately than Hes1 in intestinal tumors.
Figure 3. Nrarp represents Notch signaling activity in intestinal tumors.

(A) Real-time qRT-PCR analysis of Notch target genes (GFP of TNR, Nrarp, and Hes1) and WNT target gene (Axin2) in normal and tumor tissues from 15-week-old Apc and Apcmin;TNR (Apc;TNR) mice. Note that Notch activity (TNR) was well correlated with the expression level of Nrarp, but not Hes1. Bars indicate mean + SD. *P < 0.05. (B and C) In situ hybridization (B) and real-time qRT-PCR (C) analysis of Nrarp in the colonic tumor tissues from Apc and Vil-Cre;RosaN1+/RN1;Apcmin mice. Dotted lines indicate tumor regions (B). Scale bars: 100 εm. Bars indicate mean + SD. ***P < 0.001.
Using Nrarp as an indicator of Notch signaling activity in CRC, we used both Pearson’s and Spearman’s correlation analysis (P ≤ 0.05; see Methods) to identify genes correlated with Nrarp in 2 independent human microarray data sets (GEO GSE5206 and GSE2109). These data sets contained a sufficient amount of human patient data to perform our statistical analysis. Coexpression analysis provided a set of genes whose expression was positively or negatively correlated with Nrarp (human Nrarp CO-DEG, where DEG indicates differentially expressed gene) expression. If the Nrarp expression level accurately predicts Notch activity in human CRC, genes that are filtered using this coexpression analysis would be similar to genes identified by comparing control and Notch-activated Apcmin tumors. We analyzed genes that commonly occur in mouse Notch DEG sets between control and Notch-activated Apcmin tumors (Supplemental Table 1; P < 0.05, 2-fold) and human Nrarp CO-DEG (genes that correlate with Nrarp; P ≤ 0.05) (Supplemental Table 4; 239 genes coregulated in mouse Notch-DEG and human Nrarp CO-DEG). The heat map of these genes, sorted based on the expression level of Nrarp, showed an 85.8% identical expression change between mouse and human (Figure 4A), suggesting that Nrarp is a reliable marker for Notch signaling activity in human CRC. Genes associated with the differentiation of intestinal epithelium, CDX1 and CDX2, showed higher expression levels in Nrarphi human colonic tumors (Figure 4B and ref. 26). VIMENTIN (27) and PROX1, which are associated with tumorigenic activities such as EMT and tumor progression, exhibited lower levels of expression in Nrarphi human colonic tumors (Figure 4B). In particular, a negative correlation between Nrarp and PROX1, a known WNT target gene important in CRC progression, further supports our previous observation that Notch signaling can attenuate WNT/β-catenin activity under destruction complex–deregulated conditions.
Figure 4. Conserved Notch signatures in both mouse and human intestinal tumors.

(A) Heat map comparison between Notch-responsive genes (clusters A and B in Figure 3A) from mouse models and microarray data sets from human CRC patients (GEO GSE5206 and GSE2109). Note that genes upregulated upon Notch activation in Apcmin tumors have higher expression levels in Nrarphi tumors compared with those of Nrarplo tumors and vice versa. Asterisk indicates the group of genes that do not correlate between mouse and human data. APC, Vil-Cre;Apcmin. (B) Positive correlation of CDX1 and CDX2 (transcription factor for epithelial differentiation) and negative correlation of VIMENTIN (epithelial-mesenchymal transition marker) and PROX1 (tumor progression related gene) with Nrarp, respectively. ρ, correlation coefficient. Statistics are by Pearson’s correlation. (C) Expression levels of Nrarp in the 3 independent microarray data sets (GEO GSE5206, GSE4107, and GSE 8671). AD, adenoma; AC, adenocarcinoma; WD, well differentiated; MD, moderately differentiated; PD, poorly differentiated; C, cancer. Note the reduced Nrarp expression in CRCs compared with normal tissue. ***P < 0.001; **P < 0.005. Statistics for far-left graph are by ANOVA. (D) Positive correlation between Nrarp expression and patient survival from microarray data set (GEO GSE17538). The Kaplan-Meier method was used to estimate survival of the 2 groups; Nrarphi (solid line) and Nrarpmid/lo (dotted line).
Next, to investigate Notch signaling activity during CRC development in humans, we compared Nrarp expression levels among 3 human microarray data sets. Nrarp expression was significantly decreased along with human CRC progression (Figure 4C; GEO GSE5206, normal to adenoma, adenocarcinoma). Although the stepwise differences were minor, the overall pattern showed a significant decrease in expression (Figure 4C; P = 0.0002 using 1-way ANOVA). Data sets (GEO GSE4107 and GSE8671) were used to confirm the reduction of Nrarp expression in carcinomas compared with that of normal intestinal epithelium (Figure 4C; P = 0.0012 and P < 0.0001, respectively). Recently, Smith et al. reported microarray data coupled with patient survival information (GEO GSE17538) (28). We divided this patient data into 2 groups, Nrarphi (upper 25%) and Nrarpmid/lo, and constructed a Kaplan–Meier plot (Figure 4D). Interestingly, significantly longer and higher survival was observed for patients with high Nrarp expression, suggesting that Nrarp expression is a strong predictor of patient survival (P = 0.022). Based on Nrarp expression as a surrogate marker, we show that Notch activity in CRCs decreases during CRC progression.
Notch signaling suppresses the expression of WNT target genes in human CRC cells.
To further investigate the crosstalk between Notch and WNT signaling, we sorted Nrarp in human CRCLs based on Nrarp expression levels. Interestingly, Nrarphi CRCLs highly expressed CDX1 and CDX2, but did not express or only minimally expressed VIMENTIN and PROX1 (Figure 5A). In contrast, Nrarplo/– CRCLs highly expressed VIMENTIN and PROX1, but did not express or only minimally expressed CDX1 and CDX2 (Figure 5A). In order to determine whether Notch signaling is really active in the Nrarphi CRCLs, but not in Nrarplo/– CRCLs, we examined the expression of cleaved Notch1. As expected, cleaved Notch1 was readily detected in the Nrarphi CRCLs, such as SNU61 and LOVO, but not in Nrarplo/– CRCLs, such as SW620 and COLO205 (Figure 5B). In order to investigate whether the NRARP expression is directly induced by Notch signaling, we tested the effect of cycloheximide, a new protein synthesis inhibitor, on Notch signaling-induced NRARP expression in SNU61 and LOVO cells. The decrease of NRARP expression by γ-secretase inhibitor (DAPT) treatment in SNU61 and LOVO cells was reverted by washout of DAPT in the absence or presence of cycloheximide (Figure 5C), indicating that the induction of NRARP by Notch activation in the Nrarphi CRCLs does not require de novo protein synthesis. In accordance with the previous reports (22–24), our data show that NRARP is a direct target of Notch signaling in the Nrarphi CRCLs.
Figure 5. Notch signaling promotes differentiation of CRCLs.

(A) Real-time qRT-PCR analysis of Nrarp, CDX1, CDX2, VIMENTIN, and PROX1 in various human CRCLs. Note that Nrarphi CRCLs showed high expression levels of CDX1 and CDX2, while Nrarplo/– CRCLs highly expressed VIMENTIN and PROX1. (B) Western blot analysis of NICD and NRARP in CRCLs. Note that the expression of cleaved Notch 1 was well correlated with the expression levels of NRARP in SNU61, LOVO, SW620, and COLO205 cells. (C) Real-time qRT-PCR analysis of Nrarp expression in cycloheximide-treated CRCLs. LOVO and SNU61 cells were treated with DAPT (1 εM) for 48 hours to block Notch-signaling activity. Cells were then washed, and medium containing DAPT (mock washout) or medium lacking DAPT (washout) with or without 20 μM cycloheximide was added. After 12 hours of additional culture, cells were harvested, and Nrarp RNA levels were determined. (D) Real-time qRT-PCR analysis of CDX1, CDX2, and VIMENTIN in CRCLs. SNU1040, LOVO and SNU61 cells were treated daily with vehicle or 1 εM DAPT. SW620 and COLO205 cells were transfected with 2 εg of mock vector or ΔEN1, a constitutively active form of Notch1. Levels of CDX1, CDX2, and VIMENTIN transcripts were measured after 48 hours. Bars indicate mean + SD. *P < 0.05; **P < 0.001.
To determine whether the characteristics of CRCLs could be affected by Notch activity, Nrarphi CRCLs were treated with DAPT. Surprisingly, CDX1 and CDX2 expression levels in Nrarphi CRCLs, such as LOVO and SNU61, decreased following treatment with DAPT, whereas VIMENTIN expression increased (Figure 5D). In contrast, when the Nrarplo/– CRCLs SW620 and COLO205 were transfected with constitutively active Notch1 (ΔEN1), expression levels of CDX1 and CDX2 increased, while VIMENTIN expression decreased (Figure 5D). Taken together, these results suggest that Notch activity affects the characteristics of CRCLs.
Since Notch signaling downregulates the WNT target genes Axin2, Wif1, Nkd1, and Apcdd1 in our mouse models (Figure 2, B–G), we further investigated whether Notch signaling also affects the expression of WNT target genes in human CRCLs. We determined whether inhibition of Notch signaling results in the expression of WNT target genes in Nrarphi CRCLs. Interestingly, PROX1 expression was significantly increased by DAPT treatment in the Nrarphi CRCLs SNU1040, SNU61, SNU283, LOVO, and SW480 (Figure 6A). Similar results were also obtained when dominant-negative mastermind-like (DN-MAML) gene, which blocks the transcriptional activity of Notch, was transfected into these CRCLs (Figure 6A). In addition to PROX1, other β-catenin–responsive genes, such as c-MYC and Axin2, were increased following DAPT treatment in LOVO and SNU61 cells, while expression of an unrelated gene, MMP-2, did not change (Figure 6B). We also determined whether activation of Notch signaling in Nrarplo/– CRCLs leads to inhibition of WNT target gene expression. Expression levels of WNT/β-catenin target genes, including PROX1, AXIN2, c-MYC, and APCDD1, were decreased by ΔEN1 in SW620 and COLO205 CRCLs (Figure 7, A and B), while MMP-2 expression did not change, suggesting that Notch signaling suppresses WNT target genes.
Figure 6. Inhibition of Notch signaling increases expression of WNT target genes in CRCLs.

(A) Real-time qRT-PCR analysis of WNT target genes PROX1 in Nrarphi CRCLs. SNU1040, SNU61, SNU283, LOVO, and SW480 cells were treated daily with vehicle or 1 εM DAPT (upper graphs). Each cell was transfected with 2 εg of mock vector or DN-MAML (dominant-negative form of MAML) (lower graphs). Levels of PROX1 transcripts were measured at the indicated times (72 and 96 hours). Bars indicate mean + SD. (B) Real-time qRT-PCR analysis of WNT target genes in DAPT-treated CRCLs. LOVO (upper graphs) and SNU61 (lower graphs) cells were treated daily with vehicle or 1 εM DATP. Levels of Axin2 and c-MYC transcripts were measured at the indicated times (72 and 96 hours). MMP2 and Nrarp were used for the control and Notch target genes, respectively. β-Actin was used for normalization. Bars indicate mean + SD. *P < 0.05; **P < 0.001.
Figure 7. Activation of Notch signaling downregulated expression of WNT target genes in CRCLs.

(A and B) Real-time qRT-PCR analysis of WNT target genes (PROX1, Axin2, c-MYC, and ApcDd1) in Nrarplo/– CRCLs. SW620 and COLO205 cells were transfected with 2 εg of mock vector or ΔEN1. Levels of PROX1, Axin2, c-MYC, and ApcDd1 transcripts were measured after 48 hours. MMP2 and Nrarp were used as control and Notch target genes, respectively. β-Actin was used for normalization. Bars indicate mean + SD. *P < 0.05.
We next investigated whether Notch signaling influences tumor growth. As expected, the treatment of DAPT in the Nrarphi SNU61 and LOVO CLCLs increased the proliferation of these cells, although the increase was not dramatic (Supplemental Figure 2A). Consistently, when ΔEN1 was expressed on the NRARPlo/– COLO205 and SW620 CRCLs, it significantly inhibited the proliferation of these CLCs (Supplemental Figure 2B). Taking these data together, we concluded that Notch signaling suppresses the expression of WNT target genes in human CRC cells.
Notch signaling suppresses the expression of WNT target genes by modifying epigenetic status.
Since Notch signaling suppresses WNT/β-catenin activity under destruction complex–deregulated conditions, we speculated that Notch signaling could modulate the transcriptional activity of WNT target genes. To examine this possibility, we tested the binding activity of β-catenin/TCF4 to WNT/β-catenin target promoter regions. Interestingly, both DAPT treatment and DN-MAML overexpression in LOVO cells resulted in increased binding activity of β-catenin/TCF4 to the PROX1, Axin2, and c-MYC promoter regions (Figure 8A and Supplemental Figure 3), suggesting that Notch activation affects the binding affinity of β-catenin/TCF4 to target gene promoters.
Figure 8. Notch signaling regulated expression of WNT target genes through epigenetic modification in CRCLs.

(A and B) ChIP and PCR analysis of promoter regions of WNT target genes in Nrarphi LOVO cells and Nrarplo/– SW620 cells, and LOVO cells transfected with 2 εg of mock and DN-MAML (right panels) were treated for 96 hours. SW620 cells were transfected with 2 εg of mock vector or ΔEN1 for 48 hours. Soluble chromatin prepared from each cultured cells was immunoprecipitated with indicated Abs. The final DNA extracts were amplified using pairs of primers that cover the WNT/β-catenin–binding sites in promoter regions of WNT target genes. The exon 6 of GAPDH served as negative controls. (C and D) Real-time qRT-PCR analysis of WNT target genes (PROX1, c-MYC, and Axin2) in Nrarplo/– SW620 cells transfected with control siRNA, NLK siRNA, SETDB1 siRNA, mock vector (white bars), and ΔEN1 (black bars), as indicated. Levels of NLK, SETDB1, PROX1, Axin2, and c-MYC transcripts were measured after 48 hours. (E) ChIP analysis of WNT target genes (PROX1, c-MYC, and Axin2) in Nrarplo/– SW620 cells. SW620 cells were transfected with 2 εg of mock vector or ΔEN1 for 48 hours. Soluble chromatin prepared from each cultured cells was immunoprecipitated with indicated Abs. The final DNA extracts were amplified using pairs of primers that cover the WNT/β-catenin binding sites in promoter regions of WNT target genes. The exon 6 of GAPDH served as a negative control.
We next determined whether the binding affinity of β-catenin/TCF4 to the promoter is affected by Notch signaling through epigenetic modification. Deacetylation and methylation of histones play an important role in transcription regulation. Specifically, trimethylation of the histone H3 tail at lysine-9 (H3K9me3) and lysine-27 (H3K27me3) is associated with gene silencing, while trimethylation of the histone H3 tail at lysine-4 (H3K4me3) is associated with gene activation (29–31). The levels of H3K9me3 and H3K27me3 apparently decreased following DAPT treatment and overexpression of DN-MAML in LOVO cells, while H3K4me3, a marker of active gene transcription, increased (Figure 8A and Supplemental Figure 3). In contrast, the levels of H3K9me3 and H3K27me3 in Nrarplo/– SW620 CRCLs apparently increased by ΔEN1 in WNT/β-catenin target promoter regions (Figure 8B and Supplemental Figure 4), while H3K4me3 and H3K4ac, markers of active genes, did not change (data not shown). These results show that Notch signaling can inhibit the expression of WNT/β-catenin target genes through epigenetic modification in CRC.
We further investigated how Notch signaling negatively regulates WNT target gene expression through histone modification. NLK, known to antagonize WNT signaling, acts downstream of the Notch pathway to inhibit TCF/β-catenin signaling during mesoderm induction in sea urchin embryos (32). NLK also phosphorylates SETDB1, a histone methyltransferase, leading to the formation of a corepressor complex that inactivates the activity of the transcriptional factor PPARγ through histone 3-K9 methylation (33). This suggests that NLK and SETDB1 affect Notch-mediated inhibition of WNT target gene expression by modifying histone status. To examine this possibility, we transfected Nrarplo/– SW620 cells with NLK siRNA and SETDB1 siRNA (Supplemental Figure 5). These cells were then retransfected with control and ΔEN1. Intriguingly, downregulation of the WNT target genes PROX1, c-MYC, and Axin2 by Notch signaling was significantly abolished by both NLK siRNA (Figure 8C) and SETDB1 siRNA (Figure 8D) treatment, suggesting that NLK and SETDB1 are required for downregulation of WNT target genes by Notch signaling.
To investigate whether Notch signaling affects the recruitment of NLK and SETDB1 in WNT/β-catenin target promoter regions, we performed a ChIP assay using Nrarplo/– SW620 cells transfected with ΔEN1. As shown in Figure 8E, NLK and SETDB1 were readily recruited to WNT/β-catenin target promoter regions by ΔEN1. These results demonstrate that NLK and SETDB1 are involved in the epigenetic regulation of gene expression by Notch signaling.
Finally, we investigated the effects of DAPT in NRARPlo/– CRCLs. As expected, we could not detect any significant differences in NRARPlo/– CRCLs after DAPT treatment. Thus, we concluded that the DAPT treatment in the NRARPlo/– cell lines did not further affect the epigenetic status and characteristics of these cells. A detailed Description is provided in Supplemental Information and Supplemental Figure 6.
Discussion
In the intestine, Notch signaling is a crucial signaling component for the maintenance of intestinal progenitors and stem cells and for the regulation of binary cell fate decisions (7, 8). Notch activation also significantly accelerates tumor formation in terms of number and onset timing (11). Since this signaling pathway has been shown to positively regulate the proliferation of intestinal progenitors and the initiation of tumor formation, this pathway is defined as oncogenic. Generally, understanding the oncogenic role of specific signaling components is important for identifying therapeutic targets of cancers. Consequently, γ-secretase inhibitors, which were originally developed to treat Alzheimer patients, are also therapeutic candidates for the treatment of CRCs. However, our study shows that the role of Notch signaling in CRCs is complex. An incomplete understanding of the role of this pathway in CRC will prevent the successful development of treatment methods. Here, we discuss a new role for Notch in CRCs and the significance of this finding.
Dual roles of Notch signaling in intestinal tumorigenesis.
Notch signaling is generally known to be oncogenic in various tissues, although the loss of this signaling can also result in tumor formation (34, 35). In the intestine, Notch signaling was thought to exhibit oncogenic potential by regulating the proliferation of intestinal progenitors (9, 36). In accordance with these reports, activation of Notch signaling strongly prompts tumorigenic activity in Apcmin backgrounds (11, 12). We also observed that Vil-Cre;RosaN1+/RN1;Apcmin mice developed numerous tumors in the small and large intestines at an early age, and the onset of tumor formation was accelerated compared with that of control Apcmin mice. Our results are consistent with a recent report stating that Notch and WNT signals cooperate to trigger intestinal tumorigenesis (11). Since a loss of heterozygosity (LOH) of the Apc locus is dependent upon centromeric somatic recombination (37, 38), a higher level of proliferation will increase the probability of acquiring this somatic mutation. Therefore, the increased tumor formation in Notch-activated epithelium may be caused by increased proliferation of intestinal progenitors (9).
Recently, Sonoshita et al. reported that disruption of Aes, a negative regulator of Notch signaling, in Apcmin mice resulted in marked tumor invasion and intravasation, suggesting that Notch signaling may be critical for tumor metastasis when tumor cells interact with their surrounding endothelial cells and smooth muscle cells (39). However, tumor initiation and the number of tumor cells were not enhanced by genetic depletion of Aes in Apcmin mice compared with control mice. Additionally, overexpression or knockdown of Aes did not affect the proliferation of colon cancer cells in culture or in grafting experiments. These results are inconsistent with those of previous studies, demonstrating that activated Notch signaling facilitates tumor formation in Apcmin mice through increased proliferation of tumor cells (10, 11). Other studies have also reported that Aes can modulate other signaling pathways, such as WNT, TGF-β, and Hedgehog, which have critical roles in CRCs (40–42). Therefore, further characterization to determine whether Aes affects CRCs through Notch or other signaling pathways is required. Previously, Babaei-Jadidi et al. reported that the loss of Fbxw7 in Apcmin mice resulted in early tumor initiation compared with Apcmin mice (43). However, genetic deletion of Fbxw7 in the intestine resulted in multiple deregulations of other molecules, such as Jun and DEK, as well as in Notch signaling. Fbxw7 mutant tumors showed accumulation of the DEK protooncogene. Thus, it is not clear whether accelerated tumor initiation in Fbxw7 in Apcmin mice is due to activated Notch1.
In the present study, we directly activated Notch signaling in Apcmin mice using the RosaN1+/RN1 knockin mouse, with which we could activate components downstream of Notch and rescue and/or reverse the loss-of-Notch mutant phenotype (44). Histological analysis of Vil-Cre;RosaN1+/RN1;Apcmin mice revealed that Notch-activated tumors in these mice exhibit low-grade adenoma characteristics despite an initial increase in tumor formation. Additionally, Notch activation can revert approximately 40% of gene expression changes associated with Apcmin tumors. The most striking finding was that WNT/β-catenin target gene expression was downregulated upon activation of Notch in Apcmin tumors. This was also demonstrated in human CRCs and CRCLs. Furthermore, we found that Notch signaling regulates the expression of WNT/β-catenin target genes through epigenetic modification in CRC cells. Based on this observation, our present study demonstrates that Notch signaling acts negatively on CRC progression through downregulation of WNT/β-catenin target genes, which explains how Notch-activated Apcmin tumor remains in a low-grade state.
Crosstalk between Notch and WNT signaling in intestinal tumorigenesis and Nrarp, a reliable marker for Notch signaling activity.
Several genetic observations suggest that functional crosstalk exists between WNT and Notch signaling (45). Initial insights on how Notch signaling represses WNT signaling arose from studies conducted using Drosophila, in which a direct physical interaction between NICD and Dishevelled has been suggested as mediating the mutual inhibition of these 2 signaling pathways (46). In the skin, Notch1 activation inhibits the WNT signaling pathway by downregulating WNT ligand gene expression, which is mediated by p21 transcription (47). Additionally, GSK3β has been suggested to be a crucial link between these 2 signaling pathways (44, 48, 49). However, downregulation of WNT/β-catenin target genes in Apcmin tumors by active Notch signaling is not easily explained by such mechanisms because they are upstream regulators of the APC/β-catenin complex. Here, we observed molecular crosstalk between WNT and Notch signaling, which takes place downstream of the APC/β-catenin complex; Notch signaling can suppress the transcriptional activity of WNT target genes by modulating their histone status. Additionally, the negative correlation between Notch and WNT activity was observed in our animal model as well as in human CRC patient arrays and human CRCLs.
Recently, Alves-Guerra et al. reported that MAML1 is a coactivator of β-catenin activity and that the C-terminal region of MAML1 (aa 640–840) is critical to β-catenin activity (50). In our study, we used DN-MAML (aa 13–74) to block Notch activity by binding to NICD. When DN-MAML (aa 13–74) was transiently expressed in various NRARPhi colon cancer cells, it did not affect β-catenin activity, while Notch activity was effectively blocked. Our data show that the N-terminal domain of MAML1 is not required for β-catenin activity. Alves-Guerra et al. also demonstrated that the increase in transcriptional activity of the β-catenin pathway is due to MAML in addition to crosstalk with the Notch signaling pathway. However, we observed that γ-secretase inhibitor (DAPT) treatment of NRARPhi colon cancer cells inhibited Notch activity similarly to DN-MAML (aa 13–74), suggesting that the expression of WNT/β-catenin target genes is regulated in a Notch signaling–dependent manner in Nrarphi CRCLs.
The best characterized targets of Notch-mediated activation include members of the hairy and enhancer of split (HES) and HES-related repressor protein (HERP) families of basic helix-loop-helix (bHLH) transcriptional repressors (51). Although Hes1 is a well-known Notch target gene, it is also regulated by Notch-independent signaling pathways, such as the WNT pathway. Recently, Peignon et al. reported that Hes1 was induced directly by β-catenin signaling via the conserved Tcf-binding site of the Hes1 promoter region (14). Indeed, Hes1 expression is elevated in Apcmin tumors and human CRCs (10). Moreover, Hes1 expression is not decreased in Apc and Rbp double-knockout mice compared with Apc-null mice (14). In this study, we identified Nrarp as a reliable Notch target gene for representing the endogenous level of Notch signaling activity in intestinal tumors. Nrarp is a Notch target gene for feedback inhibition of its transcriptional activity. Notch signaling activity influences the expression of Nrarp as it does other feedback inhibitors such as Axin2 (WNT/β-catenin signaling) and Socs3 (Jak/Stat3 signaling) (17). The expression level of Nrarp is significantly reduced in Notch1–/– embryos (24). Moreover, the expression of Nrarp, but not Hes1, was well correlated with the expressional changes of a TNR (TNR-GFP) (Figure 3A). These data show that Nrarp is a reliable sensitivity and dependency marker for Notch signaling activity.
In conclusion, we demonstrate the dual roles of Notch signaling in intestinal tumor progression; Notch both suppresses tumor progression and enhances tumor initiation. In particular, Notch signaling has a negative effect on CRC progression by downregulating WNT/β-catenin target genes. Thus, the results of our study will help further the understanding of the dual roles of Notch signaling in intestinal tumorigenesis and provide critical clues for improving treatments for colonic neoplasia and cancers. We are only beginning to understand the complex interplay between these signaling pathways, and further studies will reveal more detailed mechanisms by which crosstalk contributes to intestinal tumor progression. This will help us to unveil a promising tool for restoring the deregulated WNT pathway in CRCs.
Methods
Mice.
The Apcmin mouse line and Villin-Cre (Vil-Cre) transgenic mouse line were purchased from The Jackson Laboratories and maintained in our animal colony under our institutional guidelines. Rosa-Notch1 mice (a gift from D. Melton, Harvard Stem Cell Institute, Harvard University, Boston, Massachusetts, USA) harbor the intracellular domain of mouse Notch1 in the ubiquitously expressed Rosa26 locus, the expression of which is blocked in the absence of Cre. Rosa-Notch1 mice were crossed with the Vil-Cre mice and Apcmin mice to generate Vil-Cre;RosaN1+/RN1;Apcmin. The TNR mice were a gift from N. Gaiano (Johns Hopkins University, Institute for Cell Engineering, School of Medicine, Baltimore, Maryland, USA).
Cell culture.
Human CRC cells were obtained from ATCC and the Korean Cell Line Bank (52). Cells were maintained in RPMI 1640 medium (100 U/ml penicillin; 100 εg/ml streptomycin) supplemented with 10% FBS (HyClone Labs) in an atmosphere of 95% air and 5% CO2 at 37°C. All experiments were performed with cells at 50%–70% confluence.
Tissue preparation, immunohistochemistry, and in situ hybridization.
The intestinal tract was flushed gently with cold PBS, followed by a flush with 4% paraformaldehyde in PBS. For histological analysis, tissues were fixed in 4% paraformaldehyde overnight at 4°C and embedded in paraffin wax for sectioning. Sections (3–4 εm) were stained using H&E. For immunohistochemistry, paraffin-embedded sections were rehydrated, and antigenic epitopes were exposed using boiling citrate buffer or Tris/EDTA buffer. Sections were incubated in blocking solution (3% BSA, 5% goat serum or horse serum, and 0.5% Tween 20 in PBS) at room temperature (RT) for 2 hours, followed by an additional incubation with Abs to β-catenin (1:200, BD Biosciences; 1:200, Santa Cruz Biotechnology Inc.), E-cadherin (1:200, BD Biosciences), α-catenin (1:50, Santa Cruz Biotechnology Inc.), ZO-1 (1:200, Zymed), Prox1 (1:100, Millipore), Axin2 (1:200, Abcam), and Apcdd1 (1:200, Abcam). Specific binding was detected using an Envision kit (DAKO) or Alexa Fluor 488–labeled (green) and/or Alexa Fluor 594–labeled (red) Abs (Molecular Probes). For in situ hybridization, tissues were fixed in 10% formalin overnight at RT and embedded in paraffin wax for sectioning. Dig-labeled antisense RNA transcripts were used for hybridization and visualized using anti-Dig-alkaline phosphatase Ab (BM) and BM purple reagent (BM).
Microarray analysis and statistics.
For mouse tissue mRNA analysis, total RNA was isolated using Trizol (Invitrogen) and purified using RNeasy columns (QIAGEN). The RNA was reverse transcribed, amplified, and hybridized onto Sentrix Mouse-6 v1 BeadChips (Illumina) according to the manufacturer’s instructions. Probe intensity was normalized using the quantile method (53) at the log2 scale. To identify tumor DEGs (T-DEG), fold changes between WT normal tissues and Apcmin tumors were computed, and P values were calculated against null fold changes generated by 100 permutations of sample labels. T-DEG was defined as a set of genes having fold changes greater than plus or minus 2 and P values of less than 0.05. Notch-DEG was identified using the same method. To identify genes whose expression levels correlated with that of Nrarp in 2 microarray data sets, we first calculated Pearson’s and Spearman’s correlations between Nrarp and other genes in each data set (GEO GSE2109 and GSE5206). We then calculated the P values for each type of correlation using null correlations between Nrarp and randomly permuted genes. Random permutation experiments were repeated 100 times. For each gene in the data set, 2 P values from Pearson’s and Spearman’s correlations were integrated into a combined P value by using the Liptak-Stouffer Z-method (54). Finally, 2 combined P values from 2 data sets were recombined to generate an overall P value, which indicates the significance of correlation of a gene with Nrarp in both data sets. Human NRARP CO-DEG was then identified as a gene with an overall P value of less than 0.05. KEGG pathways significantly associated with a set of genes were identified using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) (55).
Retroviral expression vectors and siRNAs.
The ΔEN1 cDNA was cloned into the HpaI site of pMSCV. ΔEN1 is a constitutively active form of Notch1 that lacks the extracellular domain. This form of Notch1 can bypass S2 cleavage and is readily processed by γ-secretase in a ligand-independent manner, but can still be blocked by a γ-secretase inhibitor. The MigR1-DN-MAML (dominant-negative form of MAML1) was a gift of J. Aster (Brigham and Women’s Hospital, Boston, Massachusetts, USA). siRNA to NLK and SETDB1 was designed and synthesized by Dharmacon (Thermo Scientific).
Western blot and RT-PCR analyses.
For Western blot analysis, equal amounts of whole-cell extracts or tissue extracts were separated using SDS-PAGE and transferred to PVDF membranes. Membranes were incubated with Abs to Mib1 (gift from P. Gallagher, Department of Cellular and Integrative Physiology, Indiana University School of Medicine, Indianapolis, Indiana, USA), cleaved Notch1 (Abcam), Hes1 (gift from T. Sudo, Pharmaceutical Research Laboratories, Toray Industries Inc., Tebiro, Kamakura, Japan), Math1 (Abcam), NRARP (BD Biosciences — Clontech), Prox1 (Chemicon), Axin2 (Abcam), C-myc (Santa Cruz Biotechnology Inc.), APCDD1 (Abcam), MMP2 (Cell Signaling Technology), and β-actin (Sigma-Aldrich). Protein bands were detected by enhanced chemiluminescence (Amersham Biosciences). For RT-PCR and real-time qRT-PCR analysis, total RNA was isolated from freshly dissected intestines using Trizol reagent (Life Technologies), and complementary DNA synthesis was performed according to the manufacturer’s instructions (Omniscript Kit; QIAGEN). PCR quantification was conducted using the SYBR green method. Primer information is available in Supplemental Data.
ChIP analysis.
ChIP analyses were performed using the EZ-Chip kit according to the manufacturer’s protocol (Upstate Biotechnology). Specific PCR primers were designed to contain putative β-catenin/TCF4–binding sites as determined by TFSEARCH, version. 1.3, and MatInspector, version 3.0, Genomatix Software. Immunoprecipitation was performed using Abs to β-catenin (Santa Cruz Biotechnology Inc.), TCF-4 (Santa Cruz Biotechnology Inc.), H3K27me3 (Abcam), H3K9me3 (Abcam), H3K4me3 (Abcam), NLK (Abcam), and SETDB1 (Abcam). Immunoprecipitated complexes were isolated and 1 μl of immunoprecipitated purified DNA was amplified using pairs of primers that cover the WNT/β-catenin–binding sites in promoter regions of WNT target genes. Exon 6 of GAPDH served as a negative control. Primer information is available in the Supplemental Data.
Statistics.
All values are given as mean ± SD. Statistical comparisons were made by 2-tailed Student’s t test. A P value of less than 0.05 was considered to be statistically significant.
Study approval.
All mouse lines were maintained in specific pathogen–free conditions at the Institute of Laboratory Animal Resources, Seoul National University. All animal experiments were approved by the ethical committees at Seoul National University (permit number SNU-081001-9).
Supplementary Material
Acknowledgments
This work was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (2012-0000121), a Global Frontier Project grant (NRF-M1AXA002-2011-0028413) of the National Research Foundation (NRF) funded by the Ministry of Education, Science and Technology of Korea, the Bio and Medical Technology Development Program of the NRF funded by the Korean government (MEST) (2011-0019269), and the National R&D Program for Cancer Control, Ministry of Health and Welfare, the Republic of Korea (0920310).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(9):3248–3259. doi:10.1172/JCI61216.
References
- 1.Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001;1(1):55–67. doi: 10.1038/35094067. [DOI] [PubMed] [Google Scholar]
- 2.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87(2):159–170. doi: 10.1016/S0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 3.Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135(4):1079–1099. doi: 10.1053/j.gastro.2008.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sancho E, Batlle E, Clevers H. Signaling pathways in intestinal development and cancer. Annu Rev Cell Dev Biol. 2004;20:695–723. doi: 10.1146/annurev.cellbio.20.010403.092805. [DOI] [PubMed] [Google Scholar]
- 5.Katoh M. Networking of WNT, FGF, Notch, BMP, and Hedgehog signaling pathways during carcinogenesis. Stem Cell Rev. 2007;3(1):30–38. doi: 10.1007/s12015-007-0006-6. [DOI] [PubMed] [Google Scholar]
- 6.van de Wetering M, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111(2):241–250. doi: 10.1016/S0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 7.Nakamura T, Tsuchiya K, Watanabe M. Crosstalk between Wnt and Notch signaling in intestinal epithelial cell fate decision. J Gastroenterol. 2007;42(9):705–710. doi: 10.1007/s00535-007-2087-z. [DOI] [PubMed] [Google Scholar]
- 8.Vooijs M, Liu Z, Kopan R. Notch: architect, landscaper, and guardian of the intestine. Gastroenterology. 2011;141(2):448–459. doi: 10.1053/j.gastro.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fre S, Huyghe M, Mourikis P, Robine S, Louvard D, Artavanis-Tsakonas S. Notch signals control the fate of immature progenitor cells in the intestine. Nature. 2005;435(7044):964–968. doi: 10.1038/nature03589. [DOI] [PubMed] [Google Scholar]
- 10.van Es JH, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435(7044):959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 11.Fre S, et al. Notch and Wnt signals cooperatively control cell proliferation and tumorigenesis in the intestine. Proc Natl Acad Sci U S A. 2009;106(15):6309–6314. doi: 10.1073/pnas.0900427106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodilla V, et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc Natl Acad Sci U S A. 2009;106(15):6315–6320. doi: 10.1073/pnas.0813221106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akiyoshi T, et al. Gamma-secretase inhibitors enhance taxane-induced mitotic arrest and apoptosis in colon cancer cells. Gastroenterology. 2008;134(1):131–144. doi: 10.1053/j.gastro.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 14.Peignon G, et al. Complex interplay between beta-catenin signalling and Notch effectors in intestinal tumorigenesis. Gut. 2011;60(2):166–176. doi: 10.1136/gut.2009.204719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A. 2003;100(25):14920–14925. doi: 10.1073/pnas.2436557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, Gumucio DL. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem. 2002;277(36):33275–33283. doi: 10.1074/jbc.M204935200. [DOI] [PubMed] [Google Scholar]
- 17.Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22(4):1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi M, et al. Isolation of a novel human gene, APCDD1, as a direct target of the beta-Catenin/T-cell factor 4 complex with probable involvement in colorectal carcinogenesis. Cancer Res. 2002;62(20):5651–5656. [PubMed] [Google Scholar]
- 19.Yan D, et al. Elevated expression of axin2 and hnkd mRNA provides evidence that Wnt/beta -catenin signaling is activated in human colon tumors. Proc Natl Acad Sci U S A. 2001;98(26):14973–14978. doi: 10.1073/pnas.261574498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zirn B, et al. Target genes of the WNT/beta-catenin pathway in Wilms tumors. Genes Chromosomes Cancer. 2006;45(6):565–574. doi: 10.1002/gcc.20319. [DOI] [PubMed] [Google Scholar]
- 21.Petrova TV, et al. Transcription factor PROX1 induces colon cancer progression by promoting the transition from benign to highly dysplastic phenotype. Cancer Cell. 2008;13(5):407–419. doi: 10.1016/j.ccr.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 22.Pirot P, van Grunsven LA, Marine JC, Huylebroeck D, Bellefroid EJ. Direct regulation of the Nrarp gene promoter by the Notch signaling pathway. Biochem Biophys Res Commun. 2004;322(2):526–534. doi: 10.1016/j.bbrc.2004.07.157. [DOI] [PubMed] [Google Scholar]
- 23.Lamar E, et al. Nrarp is a novel intracellular component of the Notch signaling pathway. Genes Dev. 2001;15(15):1885–1899. doi: 10.1101/gad.908101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krebs LT, Deftos ML, Bevan MJ, Gridley T. The Nrarp gene encodes an ankyrin-repeat protein that is transcriptionally regulated by the notch signaling pathway. Dev Biol. 2001;238(1):110–119. doi: 10.1006/dbio.2001.0408. [DOI] [PubMed] [Google Scholar]
- 25.Duncan AW, et al. Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat Immunol. 2005;6(3):314–322. doi: 10.1038/ni1164. [DOI] [PubMed] [Google Scholar]
- 26.Soubeyran P, et al. Cdx1 promotes differentiation in a rat intestinal epithelial cell line. Gastroenterology. 1999;117(6):1326–1338. doi: 10.1016/S0016-5085(99)70283-0. [DOI] [PubMed] [Google Scholar]
- 27.Chen X, Halberg RB, Burch RP, Dove WF. Intestinal adenomagenesis involves core molecular signatures of the epithelial-mesenchymal transition. J Mol Histol. 2008;39(3):283–294. doi: 10.1007/s10735-008-9164-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith JJ, et al. Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer. Gastroenterology. 2010;138(3):958–968. doi: 10.1053/j.gastro.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410(6824):116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 30.Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001;292(5514):110–113. doi: 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- 31.Schotta G, et al. Central role of Drosophila SU(VAR)3-9 in histone H3-K9 methylation and heterochromatic gene silencing. EMBO J. 2002;21(5):1121–1131. doi: 10.1093/emboj/21.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rottinger E, Croce J, Lhomond G, Besnardeau L, Gache C, Lepage T. Nemo-like kinase (NLK) acts downstream of Notch/Delta signalling to downregulate TCF during mesoderm induction in the sea urchin embryo. Development. 2006;133(21):4341–4353. doi: 10.1242/dev.02603. [DOI] [PubMed] [Google Scholar]
- 33.Takada I, et al. A histone lysine methyltransferase activated by non-canonical Wnt signalling suppresses PPAR-gamma transactivation. Nat Cell Biol. 2007;9(11):1273–1285. doi: 10.1038/ncb1647. [DOI] [PubMed] [Google Scholar]
- 34.Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11(5):338–351. doi: 10.1038/nrc3035. [DOI] [PubMed] [Google Scholar]
- 35.Radtke F, Raj K. The role of Notch in tumorigenesis: oncogene or tumour suppressor? Nat Rev Cancer. 2003;3(10):756–767. doi: 10.1038/nrc1186. [DOI] [PubMed] [Google Scholar]
- 36.Riccio O, et al. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. EMBO Rep. 2008;9(4):377–383. doi: 10.1038/embor.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luongo C, Moser AR, Gledhill S, Dove WF. Loss of Apc+ in intestinal adenomas from Min mice. Cancer Res. 1994;54(22):5947–5952. [PubMed] [Google Scholar]
- 38.Shoemaker AR, Luongo C, Moser AR, Marton LJ, Dove WF. Somatic mutational mechanisms involved in intestinal tumor formation in Min mice. Cancer Res. 1997;57(10):1999–2006. [PubMed] [Google Scholar]
- 39.Sonoshita M, et al. Suppression of colon cancer metastasis by Aes through inhibition of Notch signaling. Cancer Cell. 2011;19(1):125–137. doi: 10.1016/j.ccr.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 40.Chen G, Courey AJ. Groucho/TLE family proteins and transcriptional repression. Gene. 2000;249(1–2):1–16. doi: 10.1016/S0378-1119(00)00161-X. [DOI] [PubMed] [Google Scholar]
- 41.Wang WF, Wang YG, Reginato AM, Plotkina S, Gridley T, Olsen BR. Growth defect in Grg5 null mice is associated with reduced Ihh signaling in growth plates. Dev Dyn. 2002;224(1):79–89. doi: 10.1002/dvdy.10089. [DOI] [PubMed] [Google Scholar]
- 42.Roose J, et al. The Xenopus Wnt effector XTcf-3 interacts with Groucho-related transcriptional repressors. Nature. 1998;395(6702):608–612. doi: 10.1038/26989. [DOI] [PubMed] [Google Scholar]
- 43.Babaei-Jadidi R, et al. FBXW7 influences murine intestinal homeostasis and cancer, targeting Notch, Jun, and DEK for degradation. J Exp Med. 2011;208(2):295–312. doi: 10.1084/jem.20100830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koo BK, et al. Notch signaling promotes the generation of EphrinB1-positive intestinal epithelial cells. Gastroenterology. 2009;137(1):145–155. doi: 10.1053/j.gastro.2009.03.046. [DOI] [PubMed] [Google Scholar]
- 45.Hayward P, Kalmar T, Arias AM. Wnt/Notch signalling and information processing during development. Development. 2008;135(3):411–424. doi: 10.1242/dev.000505. [DOI] [PubMed] [Google Scholar]
- 46.Axelrod JD, Matsuno K, Artavanis-Tsakonas S, Perrimon N. Interaction between Wingless and Notch signaling pathways mediated by dishevelled. Science. 1996;271(5257):1826–1832. doi: 10.1126/science.271.5257.1826. [DOI] [PubMed] [Google Scholar]
- 47.Devgan V, Mammucari C, Millar SE, Brisken C, Dotto GP. p21WAF1/Cip1 is a negative transcriptional regulator of Wnt4 expression downstream of Notch1 activation. Genes Dev. 2005;19(12):1485–1495. doi: 10.1101/gad.341405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Foltz DR, Santiago MC, Berechid BE, Nye JS. Glycogen synthase kinase-3beta modulates notch signaling and stability. Curr Biol. 2002;12(12):1006–1011. doi: 10.1016/S0960-9822(02)00888-6. [DOI] [PubMed] [Google Scholar]
- 49.Brack AS, Conboy IM, Conboy MJ, Shen J, Rando TA. A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell. 2008;2(1):50–59. doi: 10.1016/j.stem.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 50.Alves-Guerra MC, Ronchini C, Capobianco AJ. Mastermind-like 1 Is a specific coactivator of beta-catenin transcription activation and is essential for colon carcinoma cell survival. Cancer Res. 2007;67(18):8690–8698. doi: 10.1158/0008-5472.CAN-07-1720. [DOI] [PubMed] [Google Scholar]
- 51.Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol. 2003;194(3):237–255. doi: 10.1002/jcp.10208. [DOI] [PubMed] [Google Scholar]
- 52.Ku JL, Park JG. Biology of SNU cell lines. Cancer Res Treat. 2005;37(1):1–19. doi: 10.4143/crt.2005.37.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 54.Hwang D, et al. A data integration methodology for systems biology. Proc Natl Acad Sci U S A. 2005;102(48):17296–17301. doi: 10.1073/pnas.0508647102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.