

Background: DARPP-32 is implicated in l-DOPA-induced dyskinesia.
Results: PKA-dependent phosphorylation of DARPP-32 in a distinct subset of striatal neurons is required for l-DOPA-induced activation of ERK and mTORC1.
Conclusion: PKA-dependent phosphorylation of DARPP-32 plays a critical role in dyskinesia and associated signaling alterations.
Significance: The PKA/DARPP-32 cascade is a key target for the treatment of dyskinesia.
Keywords: Dopamine Receptors, ERK, mTOR Complex (mTORC), Parkinson Disease, Transgenic Mice
Abstract
Dyskinesia, a motor complication caused by prolonged administration of the antiparkinsonian drug l-3,4-dihydroxyphenylalanine (l-DOPA), is accompanied by activation of cAMP signaling and hyperphosphorylation of the dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32). Here, we show that the abnormal phosphorylation of DARPP-32 occurs specifically in medium spiny neurons (MSNs) expressing dopamine D1 receptors (D1R). Using mice in which DARPP-32 is selectively deleted in D1R-expressing MSNs, we demonstrate that this protein is required for l-DOPA-induced activation of the extracellular signal-regulated protein kinases 1 and 2 and the mammalian target of rapamycin complex 1 (mTORC1) pathways, which are implicated in dyskinesia. We also show that mutation of the phosphorylation site for cAMP-dependent protein kinase on DARPP-32 attenuates l-DOPA-induced dyskinesia and reduces the concomitant activations of ERK and mTORC1 signaling. These studies demonstrate that, in D1R-expressing MSNs, l-DOPA-induced activation of ERK and mTORC1 requires DARPP-32 and indicates the importance of the cAMP/DARPP-32 signaling cascade in dyskinesia.
Introduction
Parkinson disease is a frequent neurodegenerative disorder characterized by the progressive death of the dopaminergic neurons of the substantia nigra pars compacta (1). In the dorsal striatum, the area preferentially innervated by the substantia nigra pars compacta, medium spiny neurons (MSNs)3 react to the loss of dopamine by increasing their sensitivity to dopaminergic agonists (2). This phenomenon is particularly evident at the level of the MSNs that directly innervate the output nuclei of the basal ganglia. These cells are generally referred to as striatonigral MSNs and are selectively enriched in dopamine D1 receptors (D1Rs) (3). The development of sensitized D1R-mediated transmission in striatonigral MSNs has been proposed to play a critical role in the generation of the motor side effects, or dyskinesia, produced by repeated administration of l-DOPA, the most common antiparkinsonian medication (4, 5).
In the dopamine-depleted striatum, but not in the normal striatum, l-DOPA promotes the phosphorylation of the dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32) (6, 7), a key component of the canonical cAMP/cAMP-dependent protein kinase (PKA) signaling cascade activated by D1Rs (8). DARPP-32 has been causally linked to the emergence of l-DOPA-induced dyskinesia (LID) (7). In agreement with this idea, it has been recently shown that LID is decreased by selective inactivation of DARPP-32 in striatonigral MSNs (9).
In addition to the cAMP/PKA/DARPP-32 pathway, l-DOPA-mediated stimulation of D1Rs activates the extracellular signal-regulated kinases 1 and 2 (ERK) (7, 10–12) and the mammalian target of rapamycin complex 1 (mTORC1) (13), which are critical regulators of transcription and translation (14, 15). Abnormal activation of ERK and mTORC1 has been implicated in the development of l-DOPA-induced dyskinesia (LID) (7, 13, 16, 17).
It has been proposed that DARPP-32 is involved in the increase of ERK phosphorylation observed in the striatum following stimulation of D1Rs (7, 18). Previous work also showed that, in mice lesioned with 6-hydroxydopamine (6-OHDA; a toxin used to model Parkinson disease in rodents), inhibition of ERK prevents the ability of a D1R agonist to activate mTORC1 (13). Taken together, these findings suggest that l-DOPA may promote mTORC1 signaling via sequential and coordinated activation of DARPP-32 and ERK. However, the existence of a link between DARPP-32 and ERK signaling in the dorsal striatum has been challenged (10), prompting a more detailed analysis of the involvement of DARPP-32 in the regulation of ERK and mTORC1. In this study, we provide evidence demonstrating that l-DOPA increases PKA/DARPP-32 signaling selectively in striatonigral MSNs and that PKA-dependent phosphorylation of DARPP-32 is implicated in LID and in the activation of the ERK and mTORC1 cascades associated with this condition.
EXPERIMENTAL PROCEDURES
Animals
In this study, we used the following: 1) mice expressing FLAG-tagged DARPP-32 selectively in D1R-expressing striatonigral neurons and Myc-tagged DARPP-32 selectively in dopamine D2 receptor (D2R)-expressing striatopallidal neurons (D1-DARPP-32-FLAG/D2-DARPP-32-Myc transgenic mice) (19); 2) mice in which DARPP-32 is conditionally deleted in D1R- or D2R-expressing MSNs by means of the loxP/Cre recombinase system (D32F/FD1R Cre+ and D32F/FD2R Cre+ conditional knock-out mice) (9); 3) knock-in mice expressing a mutated form of DARPP-32, in which the phosphorylation site for PKA (Thr-34) is substituted with an Ala (A34T mutant mice) (20).
Drugs
l-DOPA (Sigma) was injected at a dose of 10 or 20 mg/kg in combination with the peripheral DOPA decarboxylase inhibitor, benserazide hydrochloride (Sigma) (7.5 or 12 mg/kg). Both drugs were dissolved in physiological saline (0.9% NaCl) and injected intraperitoneally in a total volume of 10 ml/kg body weight. When mice were not treated with l-DOPA, they received an equivalent volume of vehicle.
6-OHDA Lesion
Mice were anesthetized with a mixture of fentanyl citrate (0.315 mg/ml), fluanisone (10 mg/ml) (VetaPharma, Leeds, UK), midazolam (5 mg/ml) (Hameln Pharmaceuticals, Gloucester, UK), and water (1:1:2 in a volume of 10 ml/kg) and mounted in a stereotaxic frame (David Kopf Instruments, Tujunga, CA) equipped with a mouse adaptor. 6-OHDA-HCl (Sigma) was dissolved in 0.02% ascorbic acid in saline at a concentration of 3 μg of freebase 6-OHDA/μl. Each mouse received two unilateral injections of 6-OHDA (2 μl/injection) into the right dorsal striatum as described previously (7), according to the following coordinates (in mm) (21): anterior-posterior +1, medial-lateral −2.1, dorsal-ventral −3.2 and anterior-posterior +0.3, medial-lateral −2.3, and dorsal-ventral −3.2. Animals were allowed to recover for 3 weeks before behavioral evaluation and drug treatment. This procedure leads to a decrease in striatal tyrosine hydroxylase immunoreactivity ≥80% and to a marked akinesia affecting the side of the body contralateral to the lesioned striatum (7, 13).
Abnormal Involuntary Movements (AIMs)
Mice were treated for 10 days with 1 injection per day of l-DOPA (20 or 10 mg/kg) plus benserazide (12 or 7.5 mg/kg). AIMs were assessed after the last injection (day 10) using a previously established scale (22). Twenty minutes after l-DOPA administration, mice were placed in separate cages, and individual dyskinetic behaviors (i.e. AIMs) were assessed for 1 min every 20 min over a period of 120 min. AIMs were classified into four subtypes as follows: locomotive AIMs (contralateral turns), axial AIMs (dystonic posturing of the upper part of the body toward the side contralateral to the lesion), limb AIMs (abnormal movements of the forelimb contralateral to the lesion), and orolingual AIMs (vacuous jaw movements and tongue protrusion). Each subtype was scored on a severity scale from 0 to 4 as follows: 0, absent; 1, occasional; 2, frequent; 3, continuous; 4, continuous and not interruptible by outer stimuli.
FLAG- and Myc-tagged DARPP-32 Immunoprecipitations
Tagged DARPP-32 was immunoprecipitated from acutely dissected striata as described previously (18). Briefly, mice were sacrificed using focused microwave irradiation, and bilateral striata from each mouse were rapidly dissected and frozen on liquid nitrogen. Striata were then sonicated in lysis buffer with protease and phosphatase inhibitors, and homogenates were incubated simultaneously with EZView Red anti-FLAG M2 affinity gel (Sigma) and Myc antibody-coupled (Novus) magnetic beads (Invitrogen) overnight at 4 °C. Anti-FLAG beads were separated from the anti-Myc beads using a magnetic particle concentrator (Invitrogen). Anti-FLAG and anti-Myc beads were separately washed, and bound proteins were eluted by boiling in sample buffer. The unbound homogenate was retained for the total striatum sample.
Western Blotting
For the studies with D32F/FD1RCre+ and D32F/FD2RCre+, conditional knock-out mice and A34T mutant mice, the animals were treated with l-DOPA plus benserazide and killed by decapitation 30 min later. The heads of the animals were cooled in liquid nitrogen for 6 s, and the brains were removed. The dorsal striata were dissected out on an ice-cold surface, sonicated in 750 μl of 1% SDS, and boiled for 10 min. Aliquots (5 μl) of the homogenate were used for protein determination using a BCA (bicinchoninic acid) assay kit (Pierce). Equal amounts of protein (30 μg) for each sample were loaded onto 10% polyacrylamide gels. Proteins were separated by SDS-PAGE and transferred overnight to PVDF membranes (Amersham Biosciences) (23). The membranes were immunoblotted using antibodies against phospho-Thr-34-DARPP-32 (1:750) (24), phospho-Ser-10-acetyl-Lys-14-histone H3 (1:50000) (Millipore AB, Solna, Sweden), phospho-Thr-202/Tyr-204-ERK (1:2000), phospho-Ser-235/236-ribosomal protein S6 (rpS6) (1:1000), phospho-Ser-240/244-ribosomal protein S6 (1:1000) (Cell Signaling Technology, Beverly, MA), and phospho-Ser-845-Glu-1A (1:1000) (PhosphoSolutions, Aurora, CO). Antibodies against histone H3 (1:50,000, Abcam), ERK (1:1000), rpS6 (1:1000), Glu-1A (1:1000) (Cell Signaling Technology, Beverly, MA), and DARPP-32 (1:750) (25) that are not phosphorylation state-specific were used to estimate the total amount of protein. Detection was based on fluorescent secondary antibody binding and quantified using a Li-Cor Odyssey infrared fluorescent detection system (Li-Cor, Lincoln, NE). The levels of each phosphoprotein were normalized for the amount of the corresponding total protein detected in the sample.
Statistical Analysis
Data were analyzed using two-way ANOVA, in which treatment and cell type or treatment and genotype were the independent variables, followed by Bonferroni-Dunn post hoc test for specific comparisons.
RESULTS
l-DOPA Increases DARPP-32 Phosphorylation in Striatonigral MSNs
Previous work carried out in 6-OHDA-lesioned mice showed that systemic administration of l-DOPA activates ERK and mTORC1 specifically in the D1R-expressing MSNs of the striatonigral pathway (13, 26, 27). Therefore, we started by examining whether the increase in DARPP-32 phosphorylation produced by l-DOPA and implicated in dyskinesia (6, 7) occurred in the same population of striatal neurons. To address this question, D1-DARPP-32-FLAG/D2-DARPP-32-Myc transgenic mice were lesioned unilaterally with 6-OHDA and injected for 10 days with 20 mg/kg l-DOPA, a procedure that induces dyskinesia (7). LID was evaluated by scoring four types of AIMs immediately after the last injection of l-DOPA. At each time point, the scores for all types of AIMs were totaled, and the average score was 28.9 ± 2.3, with a median value of 31. This value was used to divide the mice into moderately dyskinetic (total AIMs score below the median value of 31) and severely dyskinetic (total AIMs score ranging above the median value of 31). The following day, the animals were injected with l-DOPA to induce DARPP-32 phosphorylation and killed 30 min later. It should be noted that chronic l-DOPA by itself does not affect protein phosphorylation in 6-OHDA-lesioned mice. Thus, when mice were killed 24 h following the last of 10 daily injections of l-DOPA, we did not find any alterations in the levels of Thr(P)-34 DARPP-32 (65 ± 10% versus 100 ± 47% in control unlesioned mice treated with saline), Ser(P)-845-Glu-1A (61 ± 9% versus 100 ± 26% in control mice), Thr(P)-202/Tyr-204-ERK2 (106 ± 6% versus 100 ± 3% in control mice), or Ser(P)-235/236-rpS6 (104 ± 4% versus 100 ± 3% in control mice).
Lesion with 6-OHDA per se did not affect phosphorylation of DARPP-32 at Thr-34 (Fig. 1, A and B). Similarly, 6-OHDA did not produce any significant change in the phosphorylation of DARPP-32 at Thr-75, a site implicated in the modulation of PKA activity (Fig. 1, C and D). When l-DOPA was administered to 6-OHDA-lesioned D1-DARPP-32-FLAG/D2-DARPP-32-Myc mice, a large increase in the phosphorylation of DARPP-32 was detected in DARPP-32 immunoprecipitated from the D1R-expressing striatonigral MSNs (p < 0.001 versus unlesioned control mice; two-way ANOVA, followed by Bonferroni-Dunn test) (Fig. 1B). In contrast, no change in Thr-34 phosphorylation was observed in DARPP-32 immunoprecipitated from D2R-expressing MSNs (Fig. 1B). We did not find any change in Thr-75 phosphorylation in response to l-DOPA (Fig. 1, A and B). Notably, the levels of phospho-Thr-34 DARPP-32 immunoprecipitated from the D1R-expressing MSNs of mice with severe AIMs were significantly higher than those detected in mice with moderate AIMs (Fig. 1, A and B). Thus, cell type-specific analysis of DARPP-32 phosphorylation demonstrates that LID is accompanied by increased phosphorylation of DARPP-32 at Thr-34 and that this change occurs specifically in the striatonigral MSNs of the direct pathway.
FIGURE 1.
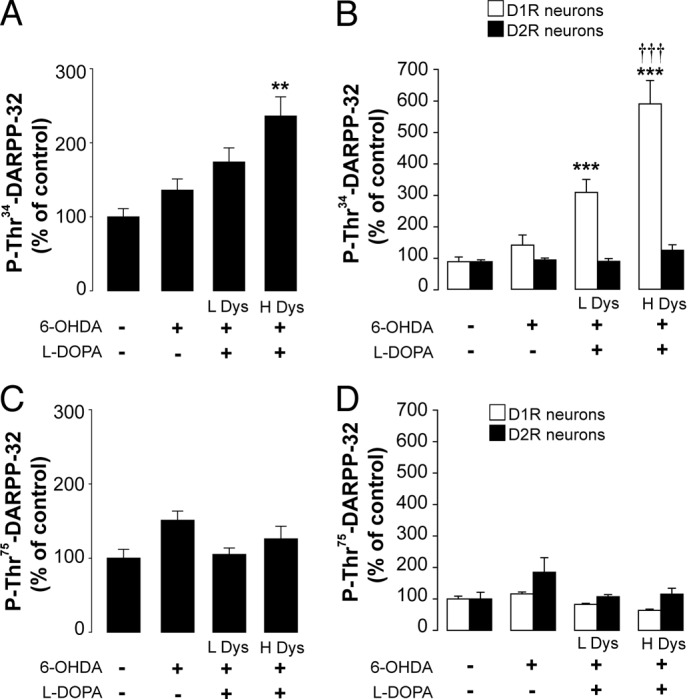
LID is accompanied by increased levels of phospho-Thr-34-DARPP-32 in D1R-expressing MSNs. D1-DARPP-32-FLAG/D2-DARPP-32-Myc transgenic mice were lesioned unilaterally with 6-OHDA and treated for 10 days with 20 mg/kg of l-DOPA. AIMs were determined immediately after the last injection of l-DOPA. The following day, mice were treated with vehicle or l-DOPA and killed 30 min later. Levels of phospho-Thr-34-DARPP-32 (A) and phospho-Thr-75-DARPP-32 (C) were determined in the striata of control mice (unlesioned, treated with vehicle) and mice lesioned with 6-OHDA and treated chronically with vehicle or l-DOPA. This latter group was divided into low dyskinetic (L Dys) and high dyskinetic (H Dys) (cf. “Results”). Data are expressed as percent of control (n = 4–12). **, p < 0.01 versus control; one-way ANOVA followed by Bonferroni-Dunn test. DARPP-32 was immunoprecipitated from D1R- and D2R-expressing MSNs using anti-FLAG and anti-Myc antibodies, respectively, and the levels of phospho-Thr-34-DARPP-32 (B) and phospho-Thr-75-DARPP-32 (D) were determined by Western blotting. Data are expressed as percent of control (n = 4–12). ***, p < 0.001 versus control; †††, p < 0.001 versus low dyskinetic group; one-way ANOVA followed by Bonferroni-Dunn test.
Lack of DARPP-32 in Striatonigral, but Not in Striatopallidal, MSNs Inhibits l-DOPA-induced ERK and mTORC1 Signaling
The studies performed in D1-DARPP-32-FLAG/D2-DARPP-32-Myc transgenic mice revealed that the increase in DARPP-32 phosphorylation implicated in LID occurred in the same neurons (i.e. the striatonigral MSNs) where activation of ERK had been previously demonstrated (26, 27). To determine the involvement of DARPP-32 in l-DOPA-induced ERK phosphorylation, we employed D32F/FD1RCre+ mice and D32F/FD2RCre+ mice, in which DARPP-32 is conditionally deleted in striatonigral and striatopallidal MSNs, respectively (9). In agreement with previous work (9), Western blot analysis revealed a reduction of DARPP-32 by 73.5 ± 0.5% in D32F/FD1RCre+ mice versus D32F/FCre− mice and by 48.7 ± 3.5% in D32F/FD2RCre+ mice versus D32F/FD2RCre− mice, reflecting loss of DARPP-32 protein from a subpopulation of MSNs in each mouse line.
D32F/FD1RCre+ mice, D32F/FD2RCre+ mice, and D32F/FCre− littermates were lesioned unilaterally with 6-OHDA, treated for 10 days with 10 mg/kg of l-DOPA, and killed 30 min after the last injection. In a previously published study, we have shown that the resultant dyskinetic response is strongly attenuated in D32F/FD1RCre+ mice, but not in D32F/FD2RCre+ mice (9). Administration of l-DOPA did not affect ERK phosphorylation in the intact striata (data not shown). This result is in line with several previous studies showing that l-DOPA, or dopaminergic agonists, does not affect ERK phosphorylation in the normal unlesioned striatum (7, 16, 27, 28). In contrast, the following lesion with 6-OHDA, l-DOPA increased phospho-ERK in control D32F/FCre− mice (Fig. 2A). This effect was reduced in D32F/FD1RCre+ mice, which lack DARPP-32 specifically in striatonigral MSNs, but preserved in D32F/FD2RCre+ mice, which lack DARPP-32 in striatopallidal MSNs (Fig. 2A). We also examined the state of phosphorylation of the Lys-14-acetylated form of histone H3, a downstream target of ERK (7, 29). l-DOPA produced a large increase in phospho-Ser-10-acetyl-Lys-14-histone H3 in the striata of 6-OHDA-lesioned D32F/FD2RCre− mice (Fig. 2B). This effect was attenuated in D32F/FD1RCre+ mice, although it was maintained in D32F/FD2RCre+ mice (Fig. 2B).
FIGURE 2.
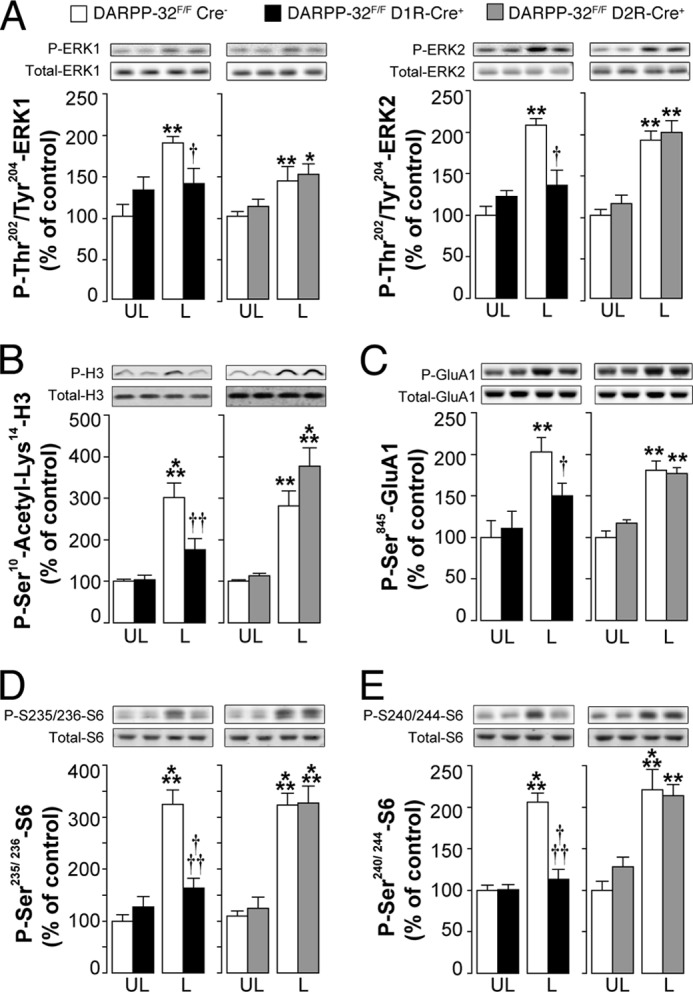
Selective deletion of DARPP-32 in striatonigral MSNs decreases l-DOPA-induced phosphorylation of ERK, histone H3, Glu-1A, and rpS6. DARPP-32F/FD1RCre+ mice, DARPP-32F/FD2RCre+ mice, and DARPP-32F/FCre− littermates were lesioned unilaterally with 6-OHDA, treated for 10 days with 10 mg/kg of l-DOPA, and killed 30 min after the last injection. Top rows show representative autoradiograms obtained using antibodies against total or phosphorylated ERK (A), histone H3 (B), Glu-1A (C), and rpS6 (D and E). Bottom rows are summary of results showing means ± S.E. (n = 6). *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus unlesioned (UL) DARPP-32F/FCre− and DARPP-32F/FD2RCre+ mice treated with l-DOPA; †, p < 0.05; ††, p < 0.01; †††, p < 0.001 versus 6-OHDA lesioned DARPP-32F/FCre− mice treated with l-DOPA (L); two-way ANOVA, followed by Bonferroni-Dunn test. A significant interaction between treatment and genotype was found in DARPP-32F/FD1RCre+ mice (F(1, 20) = 8.9, p < 0.01 for phospho-ERK1; F(1, 20) = 11.47, p < 0.01 for phospho-ERK2; F(1, 22) = 8.32, p < 0.01 for phospho-Ser10-acetyl-Lys-14-histone H3; F(1, 20) = 7.44, p < 0.05 for phospho-Ser-845-Glu-1A; F(1, 20) = 7.64, p < 0.05 for phospho-Ser-235/236-rpS6; F(1, 20) = 7.74, p < 0.05 for phospho-Ser-240/244-rpS6). A significant effect of the treatment was found in DARPP-32F/FD2RCre+ mice (F(1, 20) = 26.64, p < 0.001 for phospho-ERK1; F(1, 20) = 30.95, p < 0.001 for phospho-ERK2; F(1, 22) = 36.98, p < 0.001 for phospho-Ser-10-acetyl-Lys-14-histone H3; F(1, 20) = 35.44, p < 0.001 for phospho-Ser-845-Glu-1A; F(1, 20) = 80.3, p < 0.001 for phospho-Ser-235/236-rpS6; F(1, 20) = 44.41, p < 0.001 for phospho-Ser-240/244-rpS6).
Increased ERK signaling has been implicated in the activation of mTORC1 in striatonigral neurons (13). Therefore, we examined whether DARPP-32, by virtue of its ability to regulate ERK, was also implicated in the l-DOPA-induced phosphorylation of rpS6, a downstream target of mTORC1 (30). We found that l-DOPA induced phosphorylation of rpS6 at two sites, Ser-235/236 and Ser-240/244, and was reduced in the striata of 6-OHDA-lesioned D32F/FD1RCre+ mice, which lack DARPP-32 specifically in striatonigral MSNs (Fig. 2, D and E). In contrast, genetic inactivation of DARPP-32 in the D2R-expressing MSNs of the striatopallidal pathway did not alter the ability of l-DOPA to increase phosphorylation of S6 at Ser-235/236 and Ser-240/244 (Fig. 2, D and E).
In the same experiments, we also examined the ability of l-DOPA to induce PKA-mediated phosphorylation of the Glu-1A subunit of the glutamate AMPA receptor, which is increased in association with dyskinesia (7). We found that this effect of l-DOPA was also attenuated in D32F/FD1RCre+ but not in D32F/FD2RCre+ mice (Fig. 2C).
PKA-dependent Phosphorylation of DARPP-32 at Thr-34 Is Required for LID and for the Concomitant Increase in ERK and mTORC1 Signaling
The analysis of the changes of DARPP-32 phosphorylation in D1-DARPP-32-FLAG/D2-DARPP-32-Myc transgenic mice indicates that LID is accompanied by increased Thr-34 phosphorylation. To establish unequivocally the importance of this phosphorylation site in dyskinesia and to determine its involvement in the regulation of ERK and mTORC1 signaling, we made use of T34A DARPP-32 knock-in mice. T34A mutant mice and wild-type littermates were lesioned with 6-OHDA and injected for 10 days with l-DOPA (10 mg/kg). LID was determined on day 10 by assessing the severity of AIMs (cf. “Experimental Procedures”). We found that AIMs were significantly lower in the T34A mutant mice as compared with wild-type mice (Fig. 3A). The following day, the animals were treated with l-DOPA and killed 30 min later. In wild-type mice, administration of l-DOPA in combination with 6-OHDA lesion resulted in a large increase in the state of phosphorylation of ERK, Lys-14-acetylated histone H3, rpS6, and Glu-1A (Fig. 4). These responses were all reduced in the striata of T34A mutant mice (Fig. 4).
FIGURE 3.
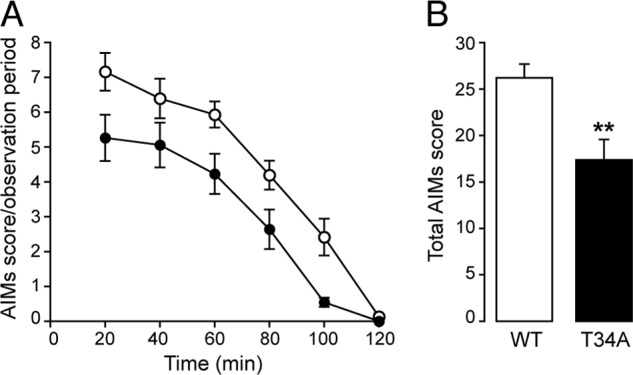
Mutation of Thr-34 on DARPP-32 decreases LID. Wild type (WT) and T34A mutant mice were lesioned unilaterally with 6-OHDA and treated for 10 days with 10 mg/kg of l-DOPA. AIMs were determined immediately after the last injection. A, time profile of the sum of locomotive, axial, limb, and orolingual AIMs scored every 20 min over a period of 120 min after the last drug administration. B, sum of total AIMs scored during all observation periods. *, p < 0.01 versus WT, two-way ANOVA followed by Bonferroni-Dunn test. A significant effect of the genotype was found (p < 0.05, F(1, 20) = 10.8).
FIGURE 4.
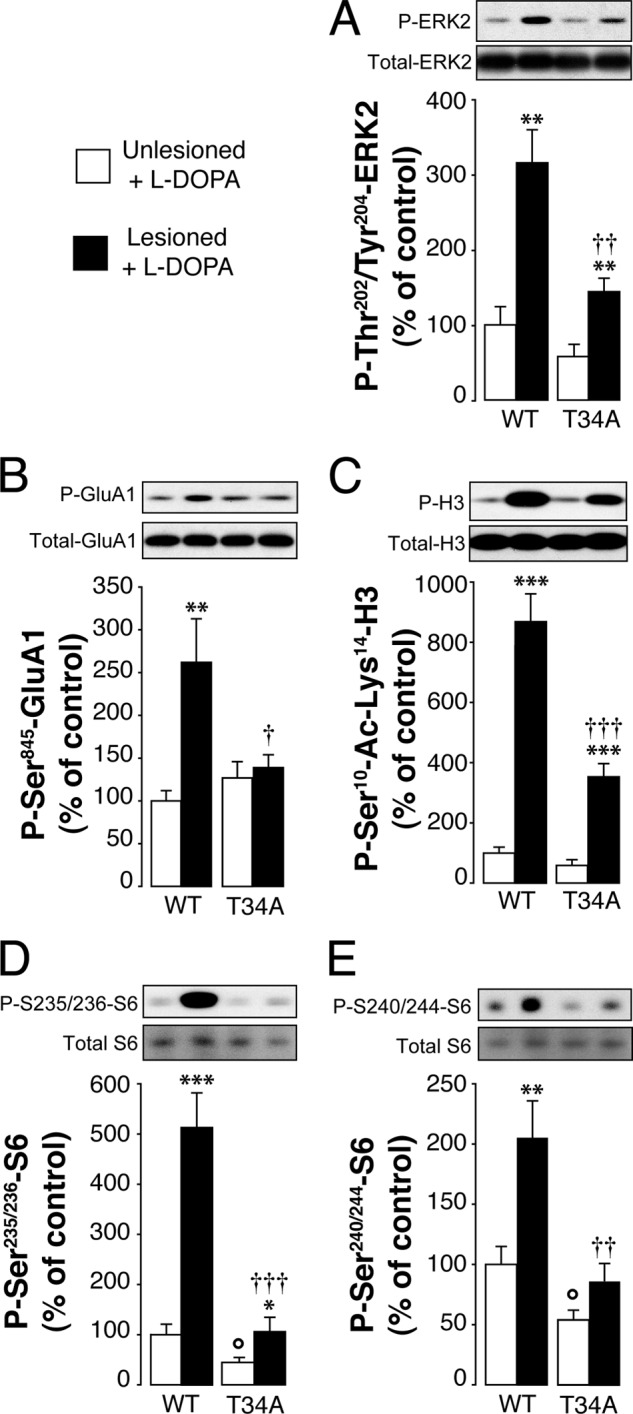
Mutation of Thr-34 on DARPP-32 decreases l-DOPA-induced phosphorylation of ERK, histone H3, Glu-1A, and rpS6. T34A mutant mice and wild-type littermates were lesioned with 6-OHDA, treated for 11 days with 10 mg/kg of l-DOPA, and killed 30 min after the last injection. Top rows show representative autoradiograms obtained using antibodies against total or phosphorylated ERK2 (A), Glu-1A (B), histone H3 (C), and rpS6 (D and E). Data are represented as means ± S.E. (n = 7–10). *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus unlesioned mice treated with l-DOPA (open bars); †, p < 0.05; ††, p < 0.01; †††, p < 0.001 versus 6-OHDA-lesioned mice treated with l-DOPA; °, p < 0.05 versus unlesioned mice treated with l-DOPA; two-way ANOVA, followed by Bonferroni-Dunn test. A significant interaction was found between treatment and genotype (F(1, 26) = 5.75, p < 0.05 for phospho-ERK2; F(1, 28) = 6.82, p < 0.05 for Glu-1A; F(1, 28) = 20.12, p < 0.001 for phospho-Ser-10-acetyl-Lys-14-histone H3; F(1, 28) = 19.74, p < 0.001 for phospho-Ser-235/236-rpS6; F(1, 28) = 4.26, p < 0.05 for phospho-Ser-240/244-rpS6).
DISCUSSION
In this study, we utilized various transgenic mouse lines to demonstrate the existence of a link between PKA-mediated phosphorylation of DARPP-32 and the concomitant activation of ERK and mTORC1 signaling produced in the dorsal striatum by l-DOPA. We also demonstrate that this regulation occurs exclusively in a well defined subgroup of MSNs, corresponding to the D1R-expressing striatonigral neurons of the direct pathway.
Previous work suggested that the ability of l-DOPA to activate PKA and DARPP-32 signaling in D1R-expressing MSNs was implicated in LID (7, 16). It was also found that, in addition to the activation of this canonical dopaminergic cascade, administration of l-DOPA increased the phosphorylation/activation of ERK (7, 10–12) and that this effect represented an additional mechanism contributing to the development of dyskinesia (7).
The present results demonstrate that activation of ERK/mTORC1 requires intact PKA/DARPP-32 signaling and that PKA-mediated phosphorylation of DARPP-32 is required for the full expression of LID. First, we show that the increase in phospho-Thr-34-DARPP-32 occurs in the same neuronal population, i.e. the striatonigral MSNs, in which abnormal ERK and mTORC1 regulation has been observed (13, 26, 27). Second, we show that cell type-specific inactivation of DARPP-32 in striatonigral MSNs strongly reduces the ability of l-DOPA to increase the phosphorylation of ERK and its downstream target, histone H3. Third, we provide evidence that a similar reduction in l-DOPA-mediated ERK and histone H3 phosphorylation occurs following a mutation of DARPP-32 that prevents its activation by PKA. Finally, we demonstrate that abolishment of PKA-mediated phosphorylation of DARPP-32 reduces dyskinetic behavior.
The involvement of DARPP-32 in the regulation of ERK exerted by l-DOPA is in line with the observation that the increase in ERK phosphorylation produced by dopaminergic drugs, such as cocaine and amphetamine, is lost in DARPP-32-deficient mice and in T34A mutant mice (31). PKA-mediated phosphorylation at Thr-34 converts DARPP-32 into an inhibitor of protein phosphatase-1 (32). Inhibition of protein phosphatase-1 has been proposed to modify the state of phosphorylation of protein kinases and protein phosphatases that control the state of phosphorylation of ERK, such as the mitogen-activated protein kinase/ERK kinase and the striatal-enriched protein phosphatase (31). The present data, indicating that mutation of Thr-34 on DARPP-32 decreases the ability of l-DOPA to promote ERK signaling, are compatible with the existence of a similar mechanism of regulation in the dorsal striatum. Thus, sensitized D1R transmission, developed in response to dopamine depletion, would potentiate the effect of l-DOPA on PKA and DARPP-32, leading to the concomitant phosphorylation/activation of ERK. This idea is supported by evidence indicating that inhibition of PKA prevents the ability of l-DOPA to increase ERK phosphorylation in a rat model of Parkinson disease (33). However, using a similar experimental model, it has been recently reported that a large reduction in l-DOPA-induced activation of PKA and DARPP-32 does not affect ERK phosphorylation (34). These results suggest that the ability of l-DOPA to promote ERK signaling depends on basal, rather than stimulated, PKA activity and DARPP-32 phosphorylation. Further studies will be necessary to clarify this issue.
The involvement of DARPP-32 in the development of LID has been previously shown using DARPP-32 knock-out mice (7), as well as D32F/FD1RCre+ and D32F/FD2RCre+ mice (16). In these studies it was found that dyskinesia was significantly reduced following systemic or cell-specific inactivation of DARPP-32. The present results, showing that LID is reduced in T34A mutant mice, support this idea and indicate that the involvement of DARPP-32 in dyskinesia depends on PKA-mediated phosphorylation at Thr-34.
One important finding of this study is that phosphorylation of DARPP-32 is required for the l-DOPA-induced activation of mTORC1 recently described in the striatonigral MSNs of dopamine-depleted mice (13). Enhanced mTORC1 signaling, which leads to the phosphorylation of rpS6 at multiple sites (30) and to increased 5′-cap-dependent initiation of mRNA translation (14), has been implicated in dyskinesia (13). The involvement of phospho-Thr-34-DARPP-32 in the control of striatal mTORC1 may be a direct consequence of the ability of PKA/DARPP-32 to promote ERK signaling. Indeed, activation of ERK is thought to promote the stimulation of the small heterotrimeric GTP-binding protein Rheb, which activates mTORC1 (35, 36). In support of this view, it has been shown that pharmacological inhibition of ERK prevents the ability of the D1R agonist SKF81297 to increase mTORC1-dependent phosphorylation of rpS6 in the dopamine-depleted striatum (13).
In conclusion, we show that the activation of three distinct signaling pathways, previously implicated in the development of LID, occurs along the same intracellular cascade. This cascade is induced specifically at the level of the striatonigral MSNs, which express D1Rs, and includes the activation of PKA/DARPP-32, ERK, and mTORC1. The finding that DARPP-32 is involved not only in the control of ERK, but also of mTORC1, highlights the key role played by this phosphoprotein in striatal signaling and provides information for the development of efficacious and selective interventions to be used in the treatment of the motor side effects produced by antiparkinsonian drugs.
Acknowledgment
We thank Simone Bido for excellent technical support.
This work was supported, in whole or in part, by National Institutes of Health Grants MH 090963 and DA 10044 (to P. G.). This work was also supported by Swedish Research Council Grants 20715 and 13482 (to G. F.), Karolinska Institutet/National Institute of Health Graduate Partnership Program (to M. F.), Italian Ministry of the University Grant FIRB Internazionalizzazione RBIN047W33 (to G. F.), The Michael Stern Parkinson Research Foundation (to P. G.), and Department of Defense/United States Army Medical Research Acquisition Activity Grants W81XWH-09-1-0402 and W81XWH-10-1-0640 (to P. G.).
- MSN
- medium spiny neuron
- D1R
- dopamine D1 receptor
- D2R
- dopamine D2 receptor
- DARPP-32
- dopamine- and cAMP-regulated phosphoprotein of 32 kDa
- mTORC1
- mammalian target of rapamycin complex 1
- LID
- l-DOPA-induced dyskinesia
- 6-OHDA
- 6-hydroxydopamine
- AIM
- abnormal involuntary movements
- rpS6
- ribosomal protein S6
- ANOVA
- analysis of variance
- l-DOPA
- l-3,4-dihydroxyphenylalanine.
REFERENCES
- 1. Kish S. J., Shannak K., Hornykiewicz O. (1988) Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson disease. Pathophysiologic and clinical implications. N. Engl. J. Med. 318, 876–880 [DOI] [PubMed] [Google Scholar]
- 2. Marshall J. F., Ungerstedt U. (1977) Supersensitivity to apomorphine following destruction of the ascending dopamine neurons. Quantification using the rotational model. Eur. J. Pharmacol. 41, 361–367 [DOI] [PubMed] [Google Scholar]
- 3. Gerfen C. R., Engber T. M., Mahan L. C., Susel Z., Chase T. N., Monsma F. J., Jr., Sibley D. R. (1990) D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250, 1429–1432 [DOI] [PubMed] [Google Scholar]
- 4. Guigoni C., Aubert I., Li Q., Gurevich V. V., Benovic J. L., Ferry S., Mach U., Stark H., Leriche L., Håkansson K., Bioulac B. H., Gross C. E., Sokoloff P., Fisone G., Gurevich E. V., Bloch B., Bezard E. (2005) Pathogenesis of levodopa-induced dyskinesia. Focus on D1 and D3 dopamine receptors. Parkinsonism Relat. Disord. 11, S25–S29 [DOI] [PubMed] [Google Scholar]
- 5. Santini E., Valjent E., Fisone G. (2008) Parkinson disease. Levodopa-induced dyskinesia and signal transduction. FEBS J. 275, 1392–1399 [DOI] [PubMed] [Google Scholar]
- 6. Picconi B., Centonze D., Håkansson K., Bernardi G., Greengard P., Fisone G., Cenci M. A., Calabresi P. (2003) Loss of bidirectional striatal synaptic plasticity in l-DOPA-induced dyskinesia. Nat. Neurosci. 6, 501–506 [DOI] [PubMed] [Google Scholar]
- 7. Santini E., Valjent E., Usiello A., Carta M., Borgkvist A., Girault J. A., Hervé D., Greengard P., Fisone G. (2007) Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in l-DOPA-induced dyskinesia. J. Neurosci. 27, 6995–7005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Greengard P. (2001) The neurobiology of slow synaptic transmission. Science 294, 1024–1030 [DOI] [PubMed] [Google Scholar]
- 9. Bateup H. S., Santini E., Shen W., Birnbaum S., Valjent E., Surmeier D. J., Fisone G., Nestler E. J., Greengard P. (2010) Distinct subclasses of medium spiny neurons differentially regulate striatal motor behaviors. Proc. Natl. Acad. Sci. U.S.A. 107, 14845–14850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gerfen C. R., Paletzki R., Worley P. (2008) Differences between dorsal and ventral striatum in Drd1a dopamine receptor coupling of dopamine- and cAMP-regulated phosphoprotein-32 to activation of extracellular signal-regulated kinase. J. Neurosci. 28, 7113–7120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pavón N., Martín A. B., Mendialdua A., Moratalla R. (2006) ERK phosphorylation and FosB expression are associated with l-DOPA-induced dyskinesia in hemiparkinsonian mice. Biol. Psychiatry 59, 64–74 [DOI] [PubMed] [Google Scholar]
- 12. Westin J. E., Vercammen L., Strome E. M., Konradi C., Cenci M. A. (2007) Spatiotemporal pattern of striatal ERK1/2 phosphorylation in a rat model of l-DOPA-induced dyskinesia and the role of dopamine D1 receptors. Biol. Psychiatry 62, 800–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Santini E., Heiman M., Greengard P., Valjent E., Fisone G. (2009) Inhibition of mTOR signaling in Parkinson disease prevents l-DOPA-induced dyskinesia. Sci. Signal. 2, ra36. [DOI] [PubMed] [Google Scholar]
- 14. Costa-Mattioli M., Sossin W. S., Klann E., Sonenberg N. (2009) Translational control of long lasting synaptic plasticity and memory. Neuron 61, 10–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thomas G. M., Huganir R. L. (2004) MAPK cascade signaling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173–183 [DOI] [PubMed] [Google Scholar]
- 16. Santini E., Sgambato-Faure V., Li Q., Savasta M., Dovero S., Fisone G., Bezard E. (2010) Distinct changes in cAMP and extracellular signal-regulated protein kinase signalling in l-DOPA-induced dyskinesia. PLoS One 5, e12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schuster S., Nadjar A., Guo J. T., Li Q., Ittrich C., Hengerer B., Bezard E. (2008) The 3-hydroxy-3-methylglutaryl-CoA reductase inhibitor lovastatin reduces severity of l-DOPA-induced abnormal involuntary movements in experimental Parkinson disease. J. Neurosci. 28, 4311–4316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Valjent E., Corvol J. C., Pages C., Besson M. J., Maldonado R., Caboche J. (2000) Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J. Neurosci. 20, 8701–8709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bateup H. S., Svenningsson P., Kuroiwa M., Gong S., Nishi A., Heintz N., Greengard P. (2008) Cell type-specific regulation of DARPP-32 phosphorylation by psychostimulant and antipsychotic drugs. Nat. Neurosci. 11, 932–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Svenningsson P., Tzavara E. T., Carruthers R., Rachleff I., Wattler S., Nehls M., McKinzie D. L., Fienberg A. A., Nomikos G. G., Greengard P. (2003) Diverse psychotomimetics act through a common signaling pathway. Science 302, 1412–1415 [DOI] [PubMed] [Google Scholar]
- 21. Franklin K. B. J., Paxinos G. (2008) The Mouse Brain in Stereotaxic Coordinates, 3rd Ed., Academic Press, San Diego [Google Scholar]
- 22. Lundblad M., Picconi B., Lindgren H., Cenci M. A. (2004) A model of l-DOPA-induced dyskinesia in 6-hydroxydopamine lesioned mice. Relation to motor and cellular parameters of nigrostriatal function. Neurobiol. Dis. 16, 110–123 [DOI] [PubMed] [Google Scholar]
- 23. Towbin H., Staehelin T., Gordon J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets. Procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 76, 4350–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Snyder G. L., Girault J. A., Chen J. Y., Czernik A. J., Kebabian J. W., Nathanson J. A., Greengard P. (1992) Phosphorylation of DARPP-32 and protein phosphatase inhibitor-1 in rat choroid plexus. Regulation by factors other than dopamine. J. Neurosci. 12, 3071–3083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hemmings H. C., Jr., Greengard P. (1986) DARPP-32, a dopamine- and adenosine 3′:5′-monophosphate-regulated phosphoprotein. Regional, tissue, and phylogenetic distribution. J. Neurosci. 6, 1469–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Darmopil S., Martín A. B., De Diego I. R., Ares S., Moratalla R. (2009) Genetic inactivation of dopamine D1 but not D2 receptors inhibits l-DOPA-induced dyskinesia and histone activation. Biol. Psychiatry 66, 603–613 [DOI] [PubMed] [Google Scholar]
- 27. Santini E., Alcacer C., Cacciatore S., Heiman M., Hervé D., Greengard P., Girault J. A., Valjent E., Fisone G. (2009) l-DOPA activates ERK signaling and phosphorylates histone H3 in the striatonigral medium spiny neurons of hemiparkinsonian mice. J. Neurochem. 108, 621–633 [DOI] [PubMed] [Google Scholar]
- 28. Gerfen C. R., Miyachi S., Paletzki R., Brown P. (2002) D1 dopamine receptor supersensitivity in the dopamine-depleted striatum results from a switch in the regulation of ERK1/2/MAP kinase. J. Neurosci. 22, 5042–5054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brami-Cherrier K., Valjent E., Hervé D., Darragh J., Corvol J. C., Pages C., Arthur S. J., Simon A. J., Girault J. A., Caboche J. (2005) Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J. Neurosci. 25, 11444–11454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ruvinsky I., Meyuhas O. (2006) Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem. Sci. 31, 342–348 [DOI] [PubMed] [Google Scholar]
- 31. Valjent E., Pascoli V., Svenningsson P., Paul S., Enslen H., Corvol J. C., Stipanovich A., Caboche J., Lombroso P. J., Nairn A. C., Greengard P., Hervé D., Girault J. A. (2005) Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc. Natl. Acad. Sci. U.S.A. 102, 491–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hemmings H. C., Jr., Greengard P., Tung H. Y., Cohen P. (1984) DARPP-32, a dopamine-regulated neuronal phosphoprotein, is a potent inhibitor of protein phosphatase-1. Nature 310, 503–505 [DOI] [PubMed] [Google Scholar]
- 33. Lebel M., Chagniel L., Bureau G., Cyr M. (2010) Striatal inhibition of PKA prevents levodopa-induced behavioral and molecular changes in the hemiparkinsonian rat. Neurobiol. Dis. 38, 59–67 [DOI] [PubMed] [Google Scholar]
- 34. Alcacer C., Santini E., Valjent E., Gaven F., Girault J. A., Hervé D. (2012) Gα(olf) mutation allows parsing the role of cAMP-dependent and extracellular signal-regulated kinase-dependent signaling in l-3,4-dihydroxyphenylalanine-induced dyskinesia. J. Neurosci. 32, 5900–5910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ma L., Chen Z., Erdjument-Bromage H., Tempst P., Pandolfi P. P. (2005) Phosphorylation and functional inactivation of TSC2 by ERK implications for tuberous sclerosis and cancer pathogenesis. Cell 121, 179–193 [DOI] [PubMed] [Google Scholar]
- 36. Roux P. P., Ballif B. A., Anjum R., Gygi S. P., Blenis J. (2004) Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. U.S.A. 101, 13489–13494 [DOI] [PMC free article] [PubMed] [Google Scholar]