

Abstract
Abdominal aortic aneurysms (AAAs) and heart failure are complex life-threatening diseases whose etiology is not completely understood. In this study, we investigated whether deficiency of group V secretory phospholipase A2 (GV sPLA2) protects from experimental AAA. The impact of GV sPLA2 deficiency on angiotensin (Ang) II–induced cardiac fibrosis was also investigated. Apolipoprotein E (apoE)−/− mice and apoE−/− mice lacking GV sPLA2 (GV DKO) were infused with 1000 ng/kg per minute Ang II for up to 28 days. Increases in systolic blood pressure, plasma aldosterone level, and urinary and heart prostanoids were similar in apoE−/− and GV DKO mice after Ang II infusion. The incidence of aortic rupture in Ang II–infused GV DKO mice (10%) was significantly reduced compared with apoE−/− mice (29.4%). Although the incidence of AAA in GV DKO mice (81.3%) and apoE−/− mice (100%) was similar, the mean percentage increase in maximal luminal diameter of abdominal aortas was significantly smaller in GV DKO mice (68.5% ± 7.7%) compared with apoE−/− mice (92.6% ± 8.3%). Deficiency of GV sPLA2 resulted in increased Ang II–induced cardiac fibrosis that was most pronounced in perivascular regions. Perivascular collagen, visualized by picrosirius red staining, was associated with increased TUNEL staining and increased immunopositivity for macrophages and myofibroblasts and nicotinamide adenine dinucleotide phosphate oxidase (NOX)-2 and NOX-4, respectively. Our findings indicate that GV sPLA2 modulates pathological responses to Ang II, with different outcomes for AAA and cardiac fibrosis.
Cardiovascular diseases are the number one killer worldwide (World Health Organization, http://www.who.int/mediacenter/factsheets/fs310/en/index.html, updated June 2011, last accessed June 19, 2012), and a role for the renin-angiotensin-aldosterone system as a contributor to cardiovascular disease is clear.1–3 Inhibition of the renin-angiotensin-aldosterone system with either angiotensin-converting enzyme inhibitors or angiotensin (Ang) II receptor blockers is effective in reducing cardiovascular morbidity and mortality in humans.1,4 Furthermore, the beneficial effects of Ang II receptor blockers in the treatment of congestive heart failure and myocardial fibrosis have been demonstrated in both animal models and human trials.1,4,5 Recent studies suggest that, in addition to its action as a potent vasoconstrictor, Ang II directly mediates pathological responses in the heart and vasculature, including endothelial activation and reactive oxygen species (ROS) generation, through activation of the Ang II receptor 1.1,2,6 However, the mechanisms and pathways underlying these effects are not fully understood.
We recently reported that deficiency of group X secretory phospholipase A2 (GX sPLA2) ameliorates abdominal aortic aneurysm (AAA) formation in Ang II–infused apolipoprotein E (apoE)−/− mice.7 AAA is recognized as an inflammatory disorder characterized by localized extracellular matrix degradation, leading to dilation of the abdominal aorta. Aortic rupture due to AAA is a major cause of mortality in men aged 65 to 85 years,8 and therapeutic interventions that have been proved to prevent AAA progression and aortic rupture in humans are not available. Aneurysms produced in apoE−/− mice that are chronically infused with Ang II exhibit many features of human AAA, including medial degeneration, inflammation, thrombus formation, and rupture of the abdominal aorta.9 Thus, the Ang II infusion model has been widely used as a relevant model for mechanistic studies of AAA. The decreased incidence of AAA in apoE−/− mice deficient in GX sPLA2 (GX DKO mice) was associated with decreased aortic expression of several genes implicated in AAA, including matrix metalloproteinase-2, IL-6, and cyclooxygenase-2 (COX-2). Furthermore, analysis by gelatin zymography demonstrated that both the latent and active forms of matrix metalloproteinase-2 were decreased in abdominal aortas of Ang II–infused GX DKO mice compared with apoE−/− mice.7 These findings suggest that GX sPLA2 promotes Ang II–induced pathological responses, leading to AAA formation in this animal model. Notably, we also reported that GX sPLA2 is present in human aneurysmal tissue, suggesting that this enzyme may contribute to human AAA.7
GX sPLA2 is one member of a family of 10 isozymes that catalyze the hydrolysis of the sn-2 ester of glycerophospholipids to produce free fatty acids and lysophospholipids. Determining the biological functions of specific sPLA2s is challenging, given the relatively large number of family members, their overlapping tissue distribution, and their distinct phospholipid substrate specificity. Numerous important biological and pathological functions, such as host immunity and inflammation,10,11 atherosclerosis,12,13 apoptosis,14–16 and ROS generation,15,17 have been mediated or enhanced by sPLA2s. One mechanism by which sPLA2s are thought to exert their biological effects is through the generation of free fatty acids and lysophospholipids that have pleiotropic effects on cell function. By liberating arachidonic acid, sPLA2s hold the potential to produce a wide variety of bioactive lipid mediators, including prostaglandins, thromboxanes, leukotrienes, and lipoxins. In vitro studies using artificial phospholipid substrates indicate that group V (GV) and GX sPLA2s are the most potent in hydrolyzing phosphatidylcholine, the major phospholipid on mammalian cell membranes. However, the extent to which these two enzymes perform redundant functions in vivo is not clear. In this study, we investigated whether deficiency of GV sPLA2, such as GX, protects apoE−/− mice from Ang II–induced AAA. We also investigated whether either GX or GV sPLA2 deficiency modulates the development of cardiac fibrosis that occurs in Ang II–infused mice.18
Materials and Methods
Experimental Animals
Male C57BL/6 and apoE-−/− mice backcrossed 10 times on a C57BL/6 background were originally obtained from the Jackson Laboratory (Bar Harbor, ME). GV sPLA2-deficient mice, also backcrossed >10 times with C57BL/6 mice, were a gift from Jonathan Arm (Harvard Medical School, Brigham and Women's Hospital, Boston, MA).19 GV sPLA2−/− mice were crossed with apoE−/− mice to obtain GV sPLA2−/− × apoE−/− (GV DKO) mice. All mice were housed in microisolator cages with a normal rodent diet and water provided ad libitum. Ang II (1000 ng/kg per minute; Sigma, St. Louis, MO) or saline was administered s.c. into male mice (aged 8 to 10 weeks) at 3, 7, 10, or 28 days, as indicated in the figure legends, via Alzet osmotic minipumps (model 2004; Sigma) using established techniques.20 Where indicated, losartan (25 mg/kg per day) was delivered, along with Ang II, via osmotic minipumps, as previously described.21 Studies were performed with the approval of the University of Kentucky, Lexington, Institutional Animal Care and Use Committee.
Blood Pressure Measurements
Mean systolic blood pressure levels were measured in conscious mice using a computerized tail-cuff method (BP-2000; Visitech Systems Inc., Apex, NC). Mice were acclimatized to the system for 1 week before the initiation of studies, and systolic blood pressure was measured 5 days per week until study termination.
Quantification of AAAs
AAAs were assessed in anesthetized mice by high-frequency ultrasonography using a Vevo 660 high-resolution system (VisualSonics Inc., Toronto, ON, Canada). Maximal luminal diameters of suprarenal abdominal aortas were determined 1 day before the initiation of Ang II infusion and 1 day before study termination. Necropsy was performed on all animals that died during the Ang II infusion, and macroscopic photographs were taken of the abdominal cavities. All of the animals that died before study termination succumbed within the first 7 days after pump implantation because of aortic rupture.
Histological Analyses of Heart Sections
Hearts were collected from PBS-perfused mice, fixed in 10% formaldehyde, and embedded in paraffin. Cross sections (5 μm thick) were collected at the level of the papillary muscles and stained with picrosirius red, as previously described.22 Collagen deposition was visualized under polarized light as red birefringent fluorescence and quantified by two independent investigators in a blinded manner using ImagePro software. Apoptosis was detected using a commercially available kit for the TUNEL assay (Roche Diagnostics Operations Inc., Indianapolis, IN). Collagen was also visualized using Gomori trichrome staining. Briefly, samples were incubated in Bouin solution for 1 hour at 56°C, washed in distilled H2O, counterstained in Weigert hematoxylin for 10 minutes at room temperature, washed again, and incubated in Gomori stain for 20 minutes at room temperature. Frozen sections from AAA were subjected to immunohistochemistry (IHC) using chicken anti-mouse GV,13 GX sPLA2 antibodies (10 μg/mL), or rabbit anti-macrophage serum (Accurate Chemical and Scientific Corp, Westbury, NY) in 1:1000 dilution. IHC staining was also performed on paraffin-embedded sections of mouse hearts using rat anti-mouse CD68 antibody for macrophages and rabbit anti-mouse α-actin antibody for smooth muscle cells (SMCs) and myofibroblasts, along with biotinylated goat anti-rat or goat anti-rabbit secondary antibodies, respectively. Samples were visualized with the ABC system (Vector Laboratories, Inc., Burlingame, CA) and AEC (3-amino-9-ethylcarbazole) as chromogen.
RNA Isolation and Quantitative RT-PCR
Abdominal aortas, defined as the part of the vessel between the diaphragm and the aortic bifurcation, were cleaned of adhering fat tissues, placed in RNAlater (Ambion, Valencia, CA), and then homogenized in RNeasy Fibrous Mini Kit solution (Qiagen). The heart tissues were homogenized, and the RNA was extracted by the TRIzol method (Sigma). For real-time RT-PCR, 0.2 to 0.5 μg of RNA was reverse transcribed using the Reverse Transcription System (Promega, Madison, WI). Real-time RT-PCR was performed using Power SYBR Green Master Mix (Applied Biosystems, University Park, IL) on a DNA iQ5 Multicolor Real-time PCR Detection system (Life Technologies, Grand Island, NY). Quantification was performed using the standard curve method and normalized with 18S. Sequences of PCR primers are available on request.
Indirect Immunofluorescence
Hearts were collected from PBS-perfused mice, fixed in 10% formaldehyde, and embedded in paraffin. Cross sections (5 μm thick) cut at the level of the papillary muscles were mounted on slides. Paraffin was removed by two washes in xylene. Samples were rehydrated in decreasing concentrations of ethanol (100%, 95%, and 70%) and then washed two times with PBS. Antigen retrieval was performed by gentle heating in 10 mmol/L citrate. Samples were blocked for 1 hour at room temperature with PBS containing 1.5% normal goat or rabbit serum. Sections were then incubated for 1 hour at room temperature with a combination of primary antibodies, as indicated. The primary antibodies were chicken anti-mouse GV sPLA2 IgG (10 μg/mL)13 and rabbit anti-smooth muscle α-actin IgG (1:100; Abcam, Cambridge, MA) for visualization of vascular SMCs and myofibroblasts. After four washes with 0.2% Tween-20 in PBS, GV sPLA2 was detected using Alexa Fluor 568–labeled goat anti-chicken IgG (1:200; Molecular Probes, Cambridge, MA). Vascular SMCs were detected using Alexa Fluor 488–labeled goat anti-rabbit IgG (1:200; Molecular Probes). NOX-2 and NOX-4 were detected using rabbit antibodies (Abcam) and Alexa Fluor 568–labeled goat anti-rabbit IgG (1:200; Molecular Probes). Slides were mounted using fluorescence-protecting medium containing DAPI (Vectashield; Vector Laboratories). Confocal microscopy was performed using an Olympus laser-scanning microscope with UV, argon (488-nm), and krypton (568-nm) lasers.
Aldosterone Quantification
Aldosterone was measured in plasma using a commercially available kit (Diagnostic Systems Labs, Webster, TX).
Quantification of Prostanoid Metabolites
Mice were individually housed for 24 hours in metabolic cages for collection of urine. Lipids were extracted from urine using acidified organic solvents with addition of internal recovery standard. Lipids were extracted from heart tissue after homogenization, using acidified organic solvents with addition of internal recovery standard.23 Liquid chromatography–tandem mass spectrometry analyses of eicosanoids were performed using an ABI 4000-Qtrap hybrid linear ion-trap triple-quadruple mass spectrometer (International Equipment Trading Ltd, Vernon Hills, IL) in multiple reaction monitoring mode. Eicosanoids were separated using a Zorbax Eclipse XDB C8 column (5 μm thick, 4.6 × 150 mm) (Agilent, Santa Clara, CA). The mobile phase consisted of 63:39:0.5 v/v/v water/acetonitrile/formic acid as solvent A and 50:50 v/v acetonitrile/iso-propaqnol as solvent B. For the analysis of eicosanoids, the separation was achieved using a linear gradient of 0% to 100% solvent B in 6 minutes, maintained at 100% solvent B for the next 4 minutes, and equilibrated back to the initial conditions in 3 minutes. The flow rate was 0.3 mL/minute, with a column temperature of 30°C. The sample injection volume was 10 μL. The mass spectrometer was operated in the negative electrospray ionization mode, with optimal ion source settings determined by synthetic standards with a declustering potential of −35 V, an entrance potential of −10 V, a collision energy of −18 V, a collision cell exit potential of −7 V, curtain gas of 30 psi, ion-spray voltage of −4500 V, ion source gas of 40 psi, and temperature of 550°C. Multiple reaction monitoring transitions were as follows: dextro (d)6-tetranor prostaglandin E2 metabolite, mass/charge ratio (m/z) 333.5/314.6, 333.5/296.6; tetranor prostaglandin E2 metabolite, m/z 327.5/308.6, 327.5/290.6; d4-prostaglandin D2 (PGD2), m/z 355.7/318.8, 355.7/236.8; PGD2, m/z 351.7/314.7, 351.7/232.5; d4-thromboxane B2, m/z 373.7/172.8, 373.7/198.7; thromboxane B2, m/z 369.7/168.7, 369.7/194.7; d4-6-keto prostaglandin F1α, m/z 373.7/166.7, 373.7/248.8; and 6-keto-prostaglandin F1α, m/z 369.7/162.8, 369.7/244.7. Liquid chromatography–tandem mass spectrometry calibrations were obtained with R2 ≥ 0.998, except for tetranor prostaglandin E2 metabolite R2 ≥ 0.996.
Statistical Analysis
For comparison of two groups on a continuous response variable, a two-sample Student's t-test was used. A one-way analysis of variance, followed by Tukey's multiple comparison test, was used to compare more than two groups. The percentage incidence of AAAs was analyzed by Fisher's exact test. P < 0.05 was considered to be statistically significant. All data were represented as mean ± SE.
Results
GV sPLA2 Deficiency Modulated Ang II–Induced AAA in ApoE−/− Mice
We recently reported that GX sPLA2 deficiency significantly reduced the incidence of Ang II–induced AAA in apoE−/− mice.7 We also determined that GX sPLA2 was present in AAA of Ang II–infused apoE−/− mice, most prominently in regions containing macrophages.7 By using an isoform-specific antibody, we demonstrated that GV sPLA2 was also present in AAA of Ang II–infused apoE−/− mice; as expected, GV sPLA2 was not detected in AAA of GV DKO mice (see Supplemental Figure S1 at http://ajp.amjpathol.org). GV sPLA2 appeared to be more diffusely distributed in AAA compared with what we previously described for GX sPLA2,7 although staining was evident in regions immunopositive for the macrophage marker CD68 (see Supplemental Figure S2 at http://ajp.amjpathol.org).
To investigate whether GV sPLA2 contributed to experimental AAA, we assessed AAA formation in apoE−/− and GV DKO mice infused with Ang II for 7, 10, or 28 days (1000 ng/kg per minute). During these studies, some of the mice died during the first 7 days of Ang II infusion because of aortic rupture (see Supplemental Figure S3 at http://ajp.amjpathol.org). This early event was attributed to initial effects of Ang II that led to medial disruption and aortic wall weakening.24 Interestingly, aortic rupture during the first 7 days of Ang II infusion was significantly less prevalent in GV DKO mice (10% incidence) compared with apoE−/− mice (29.4% incidence; Figure 1A), indicating that GV sPLA2 contributed to the processes leading to aortic rupture. AAA progression was assessed in all surviving animals by determining the maximal luminal diameters of abdominal aortas before and after Ang II infusion by in vivo ultrasonography. For both strains of mice, Ang II infusion resulted in an increase in luminal diameter that advanced with longer durations of infusion (Figure 1B). However, at each time point, the expansion of abdominal aortas in GV DKO mice was less pronounced compared with apoE−/− mice, with the difference reaching statistical significance in mice infused for 10 or 28 days (Figure 1, B and C; see also Supplemental Figure S4 at http://ajp.amjpathol.org). Although the overall expansion of the abdominal aorta was reduced in GV DKO mice, the total number of mice that either developed an aneurysm (defined as a >50% increase in the maximal diameter of the abdominal aorta lumen) or died from aortic rupture during the 28-day Ang II infusion was not significantly different in apoE−/− mice (100%) compared with GV DKO mice (81.25%). We interpreted our data to suggest that GV sPLA2 deficiency did not influence the incidence of Ang II–induced AAA, but might have alleviated its severity, as evidenced by a reduced rupture incidence and a decreased expansion of the abdominal aorta.
Figure 1.
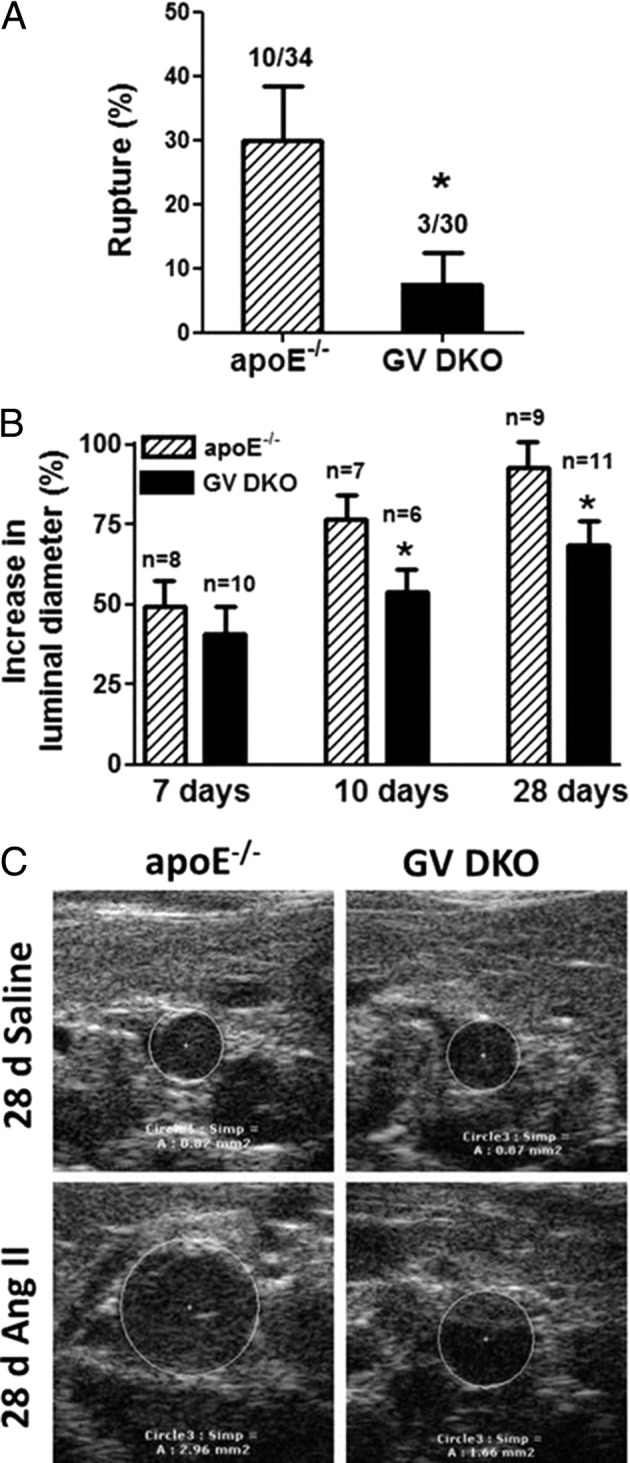
A: Rupture rate in apoE−/− and GV sPLA2−/− × apoE−/− (GV DKO) mice during the first 7 days of Ang II infusion. The number of animals that had an aortic rupture and the total number of animals in five separate experiments are indicated (two experiments were terminated at 7 days, one experiment was terminated at 10 days, and two experiments were terminated at 28 days). B: Mean ± SEM percentage increase in maximal luminal diameter of the abdominal aorta compared with baseline after the indicated duration of Ang II infusion. C: Representative images showing the lumens of abdominal aortas of mice infused with saline or Ang II for 28 days. *P < 0.05 versus apoE−/− mice.
We reported that the decreased incidence of AAA in GX DKO mice was associated with an altered inflammatory response to Ang II, as evidenced by significantly reduced induction of specific matrix metalloproteinases and inflammatory mediators in aortas of GX DKO mice compared with apoE−/− mice after 10-day Ang II infusion.7 Similar to findings in GX DKO mice, the induction of IL-6 was significantly blunted in aortas of GV DKO mice compared with apoE−/− mice after a 10-day Ang II infusion (see Supplemental Figure S5 at http://ajp.amjpathol.org). However, in contrast to GX DKO mice, Ang II–induced increases in the abundance of F4/80, COX-2, and matrix metalloproteinase-2 mRNAs were not significantly reduced in GV DKO mice compared with apoE−/− mice (see Supplemental Figure S5 at http://ajp.amjpathol.org). Interestingly, basal expression of COX-2 in GV DKO mice was significantly lower compared with apoE−/− mice, perhaps reflecting the known cross talk between COX-2 and GV sPLA2.25,26 Taken together, our data indicated that GV sPLA2 augmented Ang II–induced inflammatory responses and AAA formation in apoE−/− mice, although to a lesser extent than we previously reported for GX sPLA2.7
GV sPLA2 Deficiency Increases Ang II–Induced Heart Fibrosis in ApoE−/− Mice
GV sPLA2 was expressed in the human27 and mouse28 heart, where it was implicated in the pathogenesis of myocardial ischemia-reperfusion injury.29 Thus, it was of interest to determine whether deficiency of this enzyme modulated Ang II–induced cardiac injury, leading to heart fibrosis. Accordingly, heart sections were prepared from apoE−/− and GV DKO mice infused with either saline or Ang II for 7 or 28 days and stained with picrosirius red to visualize collagen fibrils. Somewhat surprisingly, Ang II–infused GV DKO mice exhibited a significant increase in collagen deposition compared with Ang II–infused apoE−/− mice that was evident in interstitial areas, but most prominent in perivascular regions of both the right and left ventricles (Figure 2, A and B; see also Supplemental Figure S6A at http://ajp.amjpathol.org). The enhanced fibrotic response in Ang II–infused GV DKO mice was not associated with a significant difference in the extent of cardiac hypertrophy (Figure 2C). The increase in collagen deposition was associated with 11-fold higher collagen I mRNA expression in hearts of GV DKO mice compared with apoE−/− mice after 7 days of Ang II infusion, a difference that was not statistically significantly different because of large variability in the GV DKO mice (see Supplemental Figure S6B at http://ajp.amjpathol.org).
Figure 2.

Collagen content in hearts of apoE−/− and GV DKO mice infused with Ang II for 28 days. A: Picrosirius red staining of representative heart sections visualized under light microscopy (top panels) and polarized light microscopy (bottom panels). The images are composed from four individual pictures. Original magnification, ×4. B: Collagen quantification is performed under polarized light using ImagePro software (n = 6 to 8). *P < 0.05 versus Ang II–infused apoE−/− mice. C: Heart/body ratio. *P < 0.05 versus corresponding saline-infused mice. n.s., not significant. D: Collagen content is quantified as in B in hearts from GV DKO mice infused for 7 days with saline, Ang II, or Ang II plus losartan (Lo; n = 4 to 6). *P < 0.05 versus saline-infused mice; †P < 0.05 versus mice infused with Ang II alone.
To investigate whether the fibrotic response in Ang II–infused GV DKO mice required angiotensin receptor subtype 1a (AT1aR) signaling, the mice were co-infused with losartan (25 mg/kg per day), along with Ang II for 7 days. As previously published,21 losartan prevented the development of Ang II–induced AAAs (data not shown). Losartan also significantly reduced cardiac fibrosis in Ang II–infused GV DKO mice, demonstrating that the AT1aR signaling pathway was necessary for the pathogenic effect observed in these mice (Figure 2D). On the other hand, hyperlipidemia did not appear to play a role in this pathological characteristic because the profibrotic effect was also observed in C57BL/6 mice lacking GV sPLA2 (see Supplemental Figure S7 at http://ajp.amjpathol.org). Studies in GX DKO mice indicated that, unlike GV sPLA2, GX sPLA2 did not protect the heart from Ang II–induced cardiac fibrosis (see Supplemental Figure S8 at http://ajp.amjpathol.org).
GV sPLA2 Is Up-Regulated in Hearts of Ang II–Infused ApoE−/− Mice
We performed real-time RT-PCR and IHC staining to assess whether Ang II altered the expression of GV sPLA2 in apoE−/− mouse hearts. Our data indicated a trend for increased GV sPLA2 mRNA abundance after 3 days of Ang II infusion, but this difference did not reach statistical significance (Figure 3A). Because it was possible that GV sPLA2 expression was selectively increased only in certain cell types, we performed immunofluorescence analysis to assess GV sPLA2 expression in the heart after 7 days of Ang II infusion. GV sPLA2 was detected in cardiomyocytes in control apoE−/− hearts, consistent with previous reports that this isozyme was localized to cardiomyocytes in human hearts, after myocardial infarction,30 and BALB/c mouse hearts.28 The intensity of staining was not dramatically altered in cardiomyocytes in response to Ang II (Figure 3B). We also detected positive immunostaining for GV sPLA2 that colocalized with factor VIII, an endothelial marker, in control and Ang II–infused hearts. However, this staining was sporadic, probably because of loss of endothelial cells during the processing of the slides (data not shown). Notably, GV sPLA2–immunopositive staining also colocalized with vascular SMCs in apoE−/− mouse hearts and appeared to be more pronounced after 7-day Ang II infusion (Figure 3B). As expected, GV sPLA2 was not detected in hearts of GV DKO mice (Figure 3C).
Figure 3.

Expression of GV sPLA2 in saline and Ang II–infused apoE−/− mouse hearts. A: Real-time RT-PCR showing increased mRNA abundance in apoE−/− hearts after 3 days of Ang II infusion. B: IHC analysis of heart sections from an apoE−/− mouse infused with saline or Ang II for 7 days. GV sPLA2 (red fluorescence) and SMC α-actin (green fluorescence) are visualized by confocal microscopy. Arrows indicate smooth muscle cells, and asterisks indicate cardyomyocytes. Original magnification, ×40. C: GV sPLA2 is not detected in the GV DKO mouse heart.
Ang II Infusion Resulted in Severe Perivascular Fibrosis, Apoptosis, and Macrophage Infiltration in the Myocardium of GV DKO Mice
Results from picrosirius red staining indicated extensive collagen deposition in the perivascular region of GV DKO hearts in response to long-term Ang II infusion. This finding was substantiated by additional histological examination of hearts stained with Gomori trichrome, which revealed the presence of many mononuclear cells (Figure 4A: the region of congregated mononuclear cells is evident on the high-magnification image), a damaged intimal layer (Figure 4A), and extensive connective tissue deposition in the region surrounding the vessel wall in GV DKO animals that was not observed in apoE−/− mice. TUNEL staining revealed an abundance of apoptotic nuclei in the myocardium of Ang II–infused GV DKO mice that was not apparent in apoE−/− mice (Figure 4B). In addition, IHC staining revealed the presence of myofibroblasts and macrophages in the perivascular regions of Ang II–infused hearts that was particularly evident in GV DKO mice (Figure 5). Taken together, these data suggested that GV sPLA2 protected the heart from Ang II–induced damage to the vessel wall and neighboring myocardium.
Figure 4.

A: Gomori trichrome staining of representative heart sections from apoE−/− and GV DKO mice infused with saline or Ang II, showing perivascular damage in Ang II–infused mice that is most pronounced in GV DKO mice. The box in the bottom middle panel indicates a region shown at higher magnification in the bottom right panel. Arrow indicates damaged endothelium. B: TUNEL staining (green fluorescence) showing multiple apoptotic nuclei surrounding vessels of an Ang II–infused GV DKO mouse. Original magnification, ×40.
Figure 5.

IHC of mouse hearts infused with Ang II for 7 days. The SMCs/myofibroblasts (A and C) and macrophages (B and D) in apoE−/− (A and B) and GV DKO (C and D) mice, respectively, visualized by immunostaining using anti-α-actin or anti-CD68 antibodies. Original magnification, ×20.
GV sPLA2 Deficiency Does Not Result in General Hypersensitivity to Ang II Infusion
To exclude the possibility that the enhanced fibrotic response in GV DKO mice was due to an overall hyperresponsiveness to Ang II, we measured blood pressures and plasma aldosterone levels in apoE−/− and GV DKO mice infused with saline or Ang II (1000 ng/kg per minute) for 28 days. These analyses showed that systemic renin-angiotensin-aldosterone system activation in response to Ang II infusion, as assessed by elevated systolic blood pressure and plasma aldosterone levels, was similar for the two strains (see Supplemental Figure S9, A and B, at http://ajp.amjpathol.org). In addition, the effect of Ang II infusion to augment whole body prostanoid synthesis,31 as reflected by 24-hour urinary excretion of the major metabolites of prostaglandin E2 (PGE2), prostacyclin (PGI2), PGD2, and thromboxane A2 (TxA2) measured 7 days after initiation of Ang II infusion, was not altered in GV DKO mice (see Supplemental Figure S9, C–F, at http://ajp.amjpathol.org). Furthermore, analysis of heart tissue from apoE−/− and GV DKO mice indicated that both PGI2 and PGE2 metabolites, but not TxA2 or PGI2 metabolites, were significantly increased after Ang II infusion, and GV sPLA2 deficiency did not alter these increases (Figure 6). Taken together, our data indicated that GV sPLA2 deficiency did not result in a generally altered response to Ang II.
Figure 6.
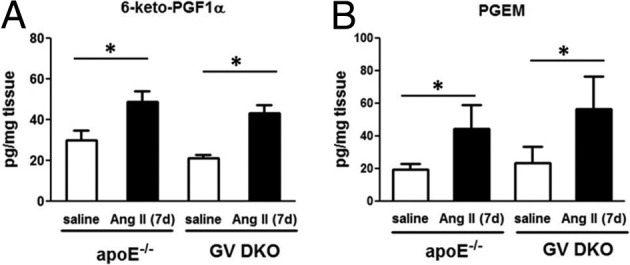
Prostanoid metabolites 6-keto-PGF1α, (prostacyclin metabolite, A) and; PGEM (PGE metabolite, B) in hearts from mice after 7-day saline or Ang II infusion measured by reversed-phase electrospray ionization tandem mass spectrometry (n = 8 to 10). No significant differences are detected between apoE−/− and GV DKO mice within a treatment group. *P < 0.05 versus saline-infused mice.
Increased NOX-2 and NOX-4 Immunostaining Is Detected in Ang II–Infused GV DKO Hearts in Regions of Perivascular Fibrosis
Considerable evidence suggested that NADPH-dependent production of ROS plays a key role in Ang II–mediated cardiovascular pathological conditions.32,33 Most studies pointed to NOX-2 as the major isoform responsible for heart hypertrophy, endothelial dysfunction, and apoptosis.33–37 Another study, however, reported that NADPH oxidase activity was elevated in NOX-2−/− mice after aortic banding, which was attributed to increased NOX-4 expression.38 To investigate the possibility that GV sPLA2 reduced oxidative stress in the heart vasculature, NOX-2 and NOX-4 expression was assessed by IHC in hearts of apoE−/− and GV DKO mice after 7-day saline or Ang II infusion. NOX-2 in the myocardium of both apoE−/− and GV DKO mice infused with saline was detected at low levels (Figure 7), and NOX-4 was not detected (data not shown). Interestingly, both NOX-2 and NOX-4 immunostaining appeared to be markedly increased in the hearts of Ang II–infused GV DKO mice compared with apoE−/− mice in regions of perivascular fibrosis (Figure 7).
Figure 7.

NOX-2 and NOX-4 expression in hearts of apoE−/− and GV DKO mice infused with saline or Ang II for 7 days, as indicated. Hearts isolated from apoE−/− and GV DKO mice are subjected to immunofluorescence using anti-NOX-2 or NOX-4 antibodies and visualized under confocal microscopy using red fluorescence–labeled secondary IgGs. Original magnification, ×60.
Discussion
In this study, we report the novel finding that GV sPLA2 modulates the vascular remodeling that occurs in mice in response to long-term Ang II infusion. By using apoE−/− mice with targeted disruption of GV sPLA2 (GV DKO mice), we determined the following: i) GV sPLA2 contributes to the progression of Ang II–induced AAA, as evidenced by a reduced incidence of aortic rupture and decreased expansion of the abdominal aorta in GV DKO mice compared with apoE−/− mice; and ii) GV sPLA2 protects mice from cardiac fibrosis, particularly perivascular fibrosis induced by Ang II. The finding that GV sPLA2 deficiency protects mice from Ang II–induced AAA is consistent with the previous report that the nonselective sPLA2 inhibitor, varespladib, significantly reduces AAA in this same animal model.39 The effect of varespladib on the development of Ang II–induced heart fibrosis has not been addressed. A possible explanation that could account for the beneficial effect of GV sPLA2 deficiency toward AAA development, while having a detrimental effect on heart fibrosis, is that, in both cases, there is an effect on tissue remodeling that has opposite outcomes for the two pathological conditions. Our findings highlight that the physiological/pathophysiological consequence of GV sPLA2 activity may be multidirectional, potentially influenced by the tissue where it is expressed and the context driving its expression. The importance of our study relates to the development of sPLA2 inhibitors for clinical use,40 which may not prove to be universally beneficial.
We recently reported that deficiency of a related enzyme, GX sPLA2, reduces the incidence of AAA in Ang II–infused apoE−/− mice by approximately 50%.7 Whether GV and GX sPLA2 contribute to AAA formation through common mechanism(s) remains to be determined. It is possible that these enzymes play redundant functions in the abdominal aorta, and that deficiency of both GV and GX sPLA2 would completely abolish Ang II–induced AAA. The finding that the induction of IL-6 is blunted in GV DKO mice, as we reported previously for GX DKO mice,7 is consistent with other evidence that IL-6 is a key mediator of AAA.41 An association between serum sPLA2 activity and the presence of aneurysm, but not progression, has been reported.42 However, serum sPLA2 activity most likely reflects GIIA sPLA2 activity,43 whereas GV and GX sPLA2 are thought to exert localized actions. Thus, a role for GV or GX sPLA2 as a serum marker for aneurysm progression seems unlikely. Whether systemic GIIA sPLA2 contributes to Ang II–induced AAA in mice merits another study.
In contrast to its beneficial effect on AAA, deficiency of GV sPLA2 resulted in significantly increased cardiac fibrosis that was evident in mice infused with Ang II for 7 or 28 days. Although there was evidence of interstitial collagen deposition in Ang II–infused GV DKO mice, the most pronounced fibrosis localized to perivascular regions. This fibrotic effect occurred in the absence of any alteration in the hypertensive response to Ang II. Notably, studies in C57BL/6 mice indicated that deficiency of GV sPLA2 leads to increased Ang II–induced cardiac fibrosis, even in the absence of hyperlipidemia. Interestingly, the extent of cardiac fibrosis was not significantly altered in Ang II–infused GX DKO mice compared with the control strain, despite a pronounced effect, as previously noted, of GX sPLA2 to augment Ang II–induced AAA.7
Increased collagen deposition in hearts of Ang II–infused GV DKO mice was significantly reduced by losartan administration, pointing to AT1aR signaling in the fibrotic effect. There is substantial evidence implicating Ang II in cardiac fibrosis,44 and several studies in humans have suggested that angiotensin-converting enzyme inhibitors or AT1aR antagonists are effective in preventing secondary cardiovascular events and chronic heart failure.31 Whether these beneficial outcomes are attributable to reductions in blood pressure or other pleiotropic effects of Ang II has been difficult to determine. Accumulating evidence indicates that Ang II acts as a circulating vasoconstrictor affecting blood pressure and as a paracrine and autocrine hormone to locally induce cell growth, oxidative stress, and inflammation, leading to tissue damage and, ultimately, cardiac hypertrophy, fibrosis, and heart failure.3 The recent development of genetically modified mouse strains has provided insights differentiating the local effects of Ang II from its hemodynamic functions. For example, transgenic mice with cardiac-specific overexpression of Ang II have 10- to 15-fold higher Ang II expression in cardiac tissue compared with nontransgenic littermates, whereas plasma Ang II levels and systolic blood pressure are similar.45 Under basal conditions, these mice develop mild cardiac fibrosis without cardiac hypertrophy.45 More recently, Xu et al46 developed a mouse model with low circulating renin–Ang II and high cardiac-specific Ang II expression and demonstrated that Ang II in the context of hypertension acts locally via AT1aR to increase macrophage infiltration, oxidative stress, apoptosis, and fibrosis in the heart. In another study, transgenic knock-in mice expressing a constitutively active AT1aR exhibited early and progressive cardiac fibrosis without cardiomyocyte hypertrophy or inflammation.47 The fibrosis, which was predominantly perivascular, was associated with only mild hypertension. Although these mouse models point to a direct action of Ang II via the AT1aR in regulating fibrotic responses in the heart, additional studies are needed to precisely define how AT1aR activation leads to fibrosis. Our findings suggest that GV sPLA2 may counteract AT1aR-dependent pathways leading to cardiac remodeling through a mechanism that appears to be independent of the hemodynamic effects of Ang II, a conclusion that is supported by the fact that both ventricles were affected as a result of GV sPLA2 deficiency (data not shown).
GV sPLA2 mRNA is expressed at high levels in the human heart,48 and GV sPLA2 protein has been associated with ischemic cardiomyocytes during the acute phase of infarction, supporting the concept that GV sPLA2 plays a role in cardiac remodeling in humans.30 In mice, deficiency of GV sPLA2 reduces myocardial injury and echocardiographic left ventricular dysfunction after ischemia-reperfusion.29 The pathological effect of GV sPLA2 in myocardial ischemia-reperfusion injury was attributed to the coordinated action of GV sPLA2, along with activated cPLA2, to amplify the local production of leukotriene B4 and thromboxane A2.29 Although the previous study indicated that GV sPLA2, in conjunction with cPLA2, plays a causative role in acute myocardial ischemia-reperfusion injury, our results suggest that GV sPLA2 plays a cardioprotective role in the setting of long-term Ang II infusion. Although fibrosis may develop as a direct consequence of cardiomyocyte death produced by acute ischemia, injuries that trigger chronic cardiomyocyte damage and gradual fibrous tissue deposition are also common. The mechanisms underlying cardiac remodeling in these two scenarios may be distinct, with GV sPLA2 activity having seemingly divergent consequences.
Numerous studies have documented the ability of GV sPLA2 to generate arachidonic acid as a substrate for eicosanoid biosynthesis.10,19,49–51 The eicosanoid profile present in tissues orchestrates the inflammatory response to acute and chronic injury and varies substantially in different cell types and on different inflammatory stimuli, depending on the combination of prostanoid and leukotriene synthases that are expressed at the site of injury.52 With inflammation, both the amount and the profile of eicosanoids produced by cells may be altered dramatically. For example, although proportionally more TxA2 is produced by resting macrophages compared with PGE2, this ratio changes when the cells are activated by lipopolysaccharide stimulation.53 Locally generated eicosanoids can either promote or restrain inflammation, depending on the presence of a diverse range of cellular receptors that are coupled to intracellular signaling pathways regulating divergent effects on cell function. Interestingly, the same bioactive mediator may contribute both to the initiation and resolution of inflammation, depending on the context.54 Ang II enhances systemic biosynthesis of prostaglandins in mice, as reflected by increased urinary secretion of the major PGE2, PGD2, PGI2, and TxA2 metabolites31 (see Supplemental Figure S9, C–F, at http://ajp.amjpathol.org). Evidence suggests that modulation of the prostanoid profile can affect both the development and extent of Ang II–induced AAA.3,31,55 Arachidonic acid metabolites have also been linked to cardiac fibrosis associated with inflammation, with PGE2 generally thought to augment and PGI2 thought to attenuate fibrotic responses.35,36 It is tempting to speculate that, in the setting of Ang II infusion, the absence of GV sPLA2 tips the heart to a more fibrotic eicosanoid profile, an effect that may be manifested in a localized and transient manner. We were unable to detect significant differences in the eicosanoid content of hearts from Ang II–infused apoE−/− and GV DKO mice, perhaps because of insufficient sensitivity to detect changes confined to small regions of the heart or restricted to certain cell types.
A striking finding from our study was the marked induction of NOX-2 and NOX-4 that colocalized with areas of collagen deposition in GV DKO hearts. A major mechanism by which Ang II exerts its detrimental effects in the heart is through NADPH-driven generation of ROS. Numerous studies have shown that the primary source of ROS in the heart is NADPH oxidase, with NOX-2 and NOX-4, but not NOX-1, being the main isoforms responsible.33 In addition to stimulating ROS formation and increasing oxidase activity, Ang II also up-regulates the expression of NADPH oxidase subunits.32 The importance of NOX-2 in Ang II–induced cardiac fibrosis has been demonstrated in NOX-2–null mice, which have significantly reduced NF-κB activation, NADPH oxidase activity, interstitial fibrosis, and expression of fibronectin and procollagen I mRNA, compared with wild-type mice, in response to Ang II infusion.37
Aldosterone may have a direct role in cardiac remodeling independent of sodium retention.46 This hypothesis is supported by reports that low-dose spironolactone has clear beneficial effects in patients with heart failure who are independent of alterations in blood pressure or fluid retention.54 Whether GV sPLA2 modulates the response to Ang II by altering aldosterone-mediated effects merits further investigation. Another potential mechanism underlying our findings may relate to the fact that GV sPLA2-deficient macrophages show impaired phagocytosis.56 Thus, the observed accumulation of macrophages and TUNEL-positive cells around the vessel wall might be the result of defective clearance of dead cells, leading to increased expression of inflammatory markers and ROS generation. Defining the mechanisms underlying GV sPLA2-mediated protection against heart fibrosis may provide novel therapeutic approaches for treating human fibrotic diseases.
Acknowledgments
We thank Kathy Forrest for her technical support and Manjula Sunkara and Dr. Andrew J. Morris for the quantification of prostanoid metabolites.
Footnotes
Supported by a Training Program at the University of Kentucky (T32 HL072743 to L.D.), grants from the NIH (RO1 DK082419 and P01 HL080100 to N.R.W.), and grants from the NIH National Center for Research Resources (5P20RR021954-05) and the National Institute of General Medical Sciences (8 P20 GM103527-05), which provided resources used by this project.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at http://dx.doi.org/10.1016/j.ajpath.2012.05.037.
Supplementary data
Representative immunostaining of cross sections of abdominal aortas from apoE−/− (A) and GV DKO (B) mice infused with 1000 ng/kg per minute of Ang II for 28 days using chicken anti-mouse GV sPLA2 antibody. Original magnification, ×20.
Representative immunostaining of cross sections of abdominal aortas from apoE−/− mice infused with 1000 ng/kg per minute of Ang II for 28 days stained with anti-CD68 (macrophages) (A) or chicken anti-mouse GV sPLA2 antibodies (B). Original magnification, ×20.
Representative pictures of five different apoE−/− (A–E) and one GV DKO (F) mouse showing the cause of death during the first 7 days of Ang II infusion to be aortic rupture, followed by acute hemorrhage into the abdominal cavity.
Representative pictures of three aortas from apoE−/− (A) and GV DKO (B) mice after 28 days of Ang II infusion.
Results from real-time RT-PCR analyses of aortas from apoE−/− and GV DKO mice infused with saline or Ang II for 10 days (n = 6 to 7 per group). *P < 0.05, **P < 0.01 versus corresponding saline-infused mice; †P < 0.05 versus similarly treated apoE−/− mice. MMP-2, matrix metalloproteinase-2; ns, not significantly different versus Ang II–infused apoE−/− mice.
A: Collagen deposition in hearts of apoE−/− and GV DKO mice infused with Ang II for 7 days (n = 4) is assessed by staining sections with picrosirius red and visualizing them under polarized light, as described in Materials and Methods. B: Relative abundance of collagen I mRNA in hearts of apoE−/− and GV DKO mice after 7 days of saline or Ang II infusion (n = 4). *P < 0.05 compared to saline-infused mice or Ang II-infused apoE−/− mice.
A: Heart/body ratio in saline and Ang II–infused mice. n.s., no significant difference between apoE−/− and GV sPLA2−/− mice. *P < 0.05 versus saline-infused mice. B: Gomori staining of representative hearts from C57BL/6 and GV sPLA2−/− mice infused with Ang II for 7 days. The boxes in the two top images indicate the regions that are shown at higher magnification in the two bottom images. White arrows, characteristic perivascular lesions.
Collagen content in hearts of apoE−/− and GX DKO mice infused with Ang II for 28 days. Collagen quantification of picrosirius red–stained hearts is performed under polarized light using ImagePro software (n = 5). *P < 0.05 versus corresponding saline-infused mice. n.s., no significant difference between apoE−/− and GX DKO mice.
A: Systolic blood pressure in mice infused with saline or Ang II for 28 days. B: Plasma aldosterone levels in mice infused with saline or Ang II for 28 days. C–F: Prostanoid metabolites in urine collected from mice during the final 24-hour period of 7-day saline or Ang II infusion measured by reversed-phase electrospray ionization tandem mass spectrometry. *P < 0.05, **P < 0.01 versus corresponding saline-infused mice. No significant differences were detected between apoE−/− and GV DKO mice within a treatment group. PGEM, prostaglandin E2 metabolite; PGD, prostaglandin D; TxB2, thromboxane B2 6-keto-PGF1a, prostacyclin metabolite.
References
- 1.Ueda S. New approaches to blockade of the renin-angiotensin-aldosterone system: evidence from randomized controlled trials (RCTs) of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: questions remain unsolved. J Pharm Sci. 2010;113:292–295. doi: 10.1254/jphs.10r08fm. [DOI] [PubMed] [Google Scholar]
- 2.Mehta P.K., Griendling K.K. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 3.Marchesi C., Paradis P., Schiffrin E.L. Role of the renin–angiotensin system in vascular inflammation. Trends Pharmacol Sci. 2008;29:367–374. doi: 10.1016/j.tips.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 4.Shah R.V., Desai A.S., Givertz M.M. The effect of renin-angiotensin system inhibitors on mortality and heart failure hospitalization in patients with heart failure and preserved ejection fraction: a systematic review and meta-analysis. J Card Fail. 2010;16:260–267. doi: 10.1016/j.cardfail.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Yoshida H., Takahashi M., Tanonaka K., Maki T., Nasa Y., Takeo S. Effects of ACE inhibition and angiotensin II type 1 receptor blockade on cardiac function and G proteins in rats with chronic heart failure. Br J Pharmacol. 2001;134:150–160. doi: 10.1038/sj.bjp.0704219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Touyz R.M. Reactive oxygen species as mediators of calcium signaling by angiotensin II: implications in vascular physiology and pathophysiology. Antioxid Redox Signal. 2005;7:1302–1314. doi: 10.1089/ars.2005.7.1302. [DOI] [PubMed] [Google Scholar]
- 7.Zack M., Boyanovsky B.B., Shridas P., Bailey W., Forrest K., Howatt D.A., Gelb M.H., de Beer F.C., Daugherty A., Webb N.R. Group X secretory phospholipase A2 augments angiotensin II-induced inflammatory responses and abdominal aortic aneurysm formation in apoE-deficient mice. Atherosclerosis. 2011;214:58–64. doi: 10.1016/j.atherosclerosis.2010.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakalihasan N., Limet R., Defawe O.D. Abdominal aortic aneurysm. Lancet. 2005;365:1577–1589. doi: 10.1016/S0140-6736(05)66459-8. [DOI] [PubMed] [Google Scholar]
- 9.Daugherty A., Cassis L.A. Mouse models of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2004;24:429–434. doi: 10.1161/01.ATV.0000118013.72016.ea. [DOI] [PubMed] [Google Scholar]
- 10.Lambeau G., Gelb M.H. Biochemistry and physiology of mammalian secreted phospholipases A2. Annu Rev Biochem. 2008;77:495–520. doi: 10.1146/annurev.biochem.76.062405.154007. [DOI] [PubMed] [Google Scholar]
- 11.Boyanovsky B.B., Webb N.R. Biology of secretory phospholipase A2. Cardiovasc Drugs Ther. 2009;23:61–72. doi: 10.1007/s10557-008-6134-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ivandic B., Castellani L.W., Wang X.-P., Qiao J.-H., Mehrabian M., Navab M., Fogelman A.M., Grass D.S., Swanson M.E., de Beer M.C., de Beer F., Lusis A.J. Role of group II secretory phospholipase A2 in atherosclerosis: increased atherogenesis and altered lipoproteins in transgenic mice expressing group IIa secretory phospholipase A2. Arterioscler Thromb Vasc Biol. 1999;19:1284–1290. doi: 10.1161/01.atv.19.5.1284. [DOI] [PubMed] [Google Scholar]
- 13.Bostrom M.A., Boyanovsky B.B., Jordan C.T., Wadsworth M.P., Taatjes D.J., de Beer F.C., Webb N.R. Group V secretory phospholipase A2 promotes atherosclerosis: evidence from genetically altered mice. Arterioscler Thromb Vasc Biol. 2007;27:600–606. doi: 10.1161/01.ATV.0000257133.60884.44. [DOI] [PubMed] [Google Scholar]
- 14.Taketo M.M., Sonoshita M. Phospolipase A2 and apoptosis. Biochim Biophys Acta. 2002;1585:72–76. doi: 10.1016/s1388-1981(02)00326-8. [DOI] [PubMed] [Google Scholar]
- 15.Giri S., Khan M., Rattan R., Singh I., Singh A.K. Krabbe disease: psychosine-mediated activation of phospholipase A2 in oligodendrocyte cell death. J Lipid Res. 2006;47:1478–1492. doi: 10.1194/jlr.M600084-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Zhao X., Wang D., Zhao Z., Xiao Y., Sengupta S., Xiao Y., Zhang R., Lauber K., Wesselborg S., Feng L., Rose T.M., Shen Y., Zhang J., Prestwich G., Xu Y. Caspase-3-dependent activation of calcium-independent phospholipase A2 enhances cell migration in non-apoptotic ovarian cancer cells. J Biol Chem. 2006;281:29357–29368. doi: 10.1074/jbc.M513105200. [DOI] [PubMed] [Google Scholar]
- 17.Muralikrishna Adibhatla R., Hatcher J.F. Phospholipase A2, reactive oxygen species, and lipid peroxidation in cerebral ischemia. Free Radic Biol Med. 2006;40:376–387. doi: 10.1016/j.freeradbiomed.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 18.Matsusaka T., Katori H., Homma T., Ichikawa I. Mechanism of cardiac fibrosis by angiotensin: new insight revealed by genetic engineering. Trends Cardiovasc Med. 1999;9:180–184. doi: 10.1016/s1050-1738(00)00018-9. [DOI] [PubMed] [Google Scholar]
- 19.Satake Y., Diaz B.L., Balestrieri B., Lam B.K., Kanaoka Y., Grusby M.J., Arm J.P. Role of group V phospholipase A2 in zymosan-induced eicosanoid generation and vascular permeability revealed by targeted gene disruption. J Biol Chem. 2004;279:16488–16494. doi: 10.1074/jbc.M313748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daugherty A., Cassis L. Chronic angiotensin II infusion promotes atherogenesis in low density lipoprotein receptor −/− mice. Ann N Y Acad Sci. 1999;892:108–118. doi: 10.1111/j.1749-6632.1999.tb07789.x. [DOI] [PubMed] [Google Scholar]
- 21.Daugherty A., Manning M.W., Cassis L.A. Antagonism of AT2 receptors augments angiotensin II-induced abdominal aortic aneurysms and atherosclerosis. Br J Pharmacol. 2001;134:865–870. doi: 10.1038/sj.bjp.0704331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wadsworth M., Sobel B., Schneider D., Taatjes D. Delineation of the evolution of compositional changes in atheroma. Histochem Cell Biol. 2002;118:59–68. doi: 10.1007/s00418-002-0419-x. [DOI] [PubMed] [Google Scholar]
- 23.Su W., Yeku O., Olepu S., Genna A., Park J.-S., Ren H., Du G., Gelb M.H., Morris A.J., Frohman M.A. 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol Pharmacol. 2009;75:437–446. doi: 10.1124/mol.108.053298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saraff K., Babamusta F., Cassis L.A., Daugherty A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:1621–1626. doi: 10.1161/01.ATV.0000085631.76095.64. [DOI] [PubMed] [Google Scholar]
- 25.Balsinde J., Balboa M., Dennis E. Functional coupling between secretory phospholipase A2 and cyclooxygenase-2 and its regulation by cytosolic group IV phospholipase A2. Proc Natl Acad Sci U S A. 1998;95:7951–7956. doi: 10.1073/pnas.95.14.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shinohara H., Balboa M.A., Johnson C.A., Balsinde J., Dennis E.A. Regulation of delayed prostaglandin production in activated P388D1 macrophages by group IV cytosolic and group V secretory phospholipase A2s. J Biol Chem. 1999;274:12263–12268. doi: 10.1074/jbc.274.18.12263. [DOI] [PubMed] [Google Scholar]
- 27.Cupillard L., Koumanov K., Mattei M.-G., Lazdunski M., Lambeau G. Cloning, chromosomal mapping, and expression of a novel human secretory phospholipase A2. J Biol Chem. 1997;272:15745–15752. doi: 10.1074/jbc.272.25.15745. [DOI] [PubMed] [Google Scholar]
- 28.Valentin E., Ghomashchi F., Gelb M., Lazdunski M., Lambeau G. On the diversity of secreted phospholipases A2. J Biol Chem. 1999;274:31195–31202. doi: 10.1074/jbc.274.44.31195. [DOI] [PubMed] [Google Scholar]
- 29.Yano T., Fujioka D., Saito Y., Kobayashi T., Nakamura T., Obata J.E., Kawabata K., Watanabe K., Watanabe Y., Mishina H., Tamaru S., Kugiyama K. Group V secretory phospholipase A2 plays a pathogenic role in myocardial ischaemia-reperfusion injury. Cardiovasc Res. 2011;90:335–343. doi: 10.1093/cvr/cvq399. [DOI] [PubMed] [Google Scholar]
- 30.Ishikawa Y., Komiyama K., Masuda S., Murakami M., Akasaka Y., Ito K., Akishima-Fukasawa Y., Kimura M., Fujimoto A., Kudo I., Ishii T. Expression of type V secretory phospholipase A2 in myocardial remodelling after infarction. Histopathology. 2005;47:257–267. doi: 10.1111/j.1365-2559.2005.02227.x. [DOI] [PubMed] [Google Scholar]
- 31.Werner C.M., Böhm M. Review: the therapeutic role of RAS blockade in chronic heart failure. Ther Adv Cardiovasc Dis. 2008;2:167–177. doi: 10.1177/1753944708091777. [DOI] [PubMed] [Google Scholar]
- 32.Griendling K.K., Sorescu D., Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 33.Nabeebaccus A., Zhang M., Shah A. NADPH oxidases and cardiac remodelling. Heart Fail Rev. 2011;16:5–12. doi: 10.1007/s10741-010-9186-2. [DOI] [PubMed] [Google Scholar]
- 34.Hingtgen S.D., Tian X., Yang J., Dunlay S.M., Peek A.S., Wu Y., Sharma R.V., Engelhardt J.F., Davisson R.L. Nox2-containing NADPH oxidase and Akt activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy. Physiol Genomics. 2006;26:180–191. doi: 10.1152/physiolgenomics.00029.2005. [DOI] [PubMed] [Google Scholar]
- 35.Cave A.C., Brewer A.C., Narayanapanicker A., Ray R., Grieve D.J., Walker S., Shah A.M. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006;8:691–728. doi: 10.1089/ars.2006.8.691. [DOI] [PubMed] [Google Scholar]
- 36.Bendall J.K., Cave A.C., Heymes C., Gall N., Shah A.M. Pivotal role of a gp91phox-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation. 2002;105:293–296. doi: 10.1161/hc0302.103712. [DOI] [PubMed] [Google Scholar]
- 37.Murdoch C., Alom-Ruiz S., Wang M., Zhang M., Walker S., Yu B., Brewer A., Shah A. Role of endothelial Nox2 NADPH oxidase in angiotensin II-induced hypertension and vasomotor dysfunction. Basic Res Cardiol. 2011;106:527–538. doi: 10.1007/s00395-011-0179-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuroda J., Ago T., Matsushima S., Zhai P., Schneider M.D., Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A. 2010;107:15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fraser H., Hislop C., Christie R.M., Rick H.L., Reidy C.A., Chouinard M.L., Eacho P.I., Gould K.E., Trias J. Varespladib (A-002), a secretory phospholipase A2 inhibitor, reduces atherosclerosis and aneurysm formation in ApoE−/− mice. J Cardiovasc Pharmacol. 2009;53:60–65. doi: 10.1097/FJC.0b013e318195bfbc. [DOI] [PubMed] [Google Scholar]
- 40.Rosenson R.S. Phospholipase A2 inhibition and atherosclerotic vascular disease: prospects for targeting secretory and lipoprotein-associated phospholipase A2 enzymes. Curr Opin Lipidol. 2010;21:473–480. doi: 10.1097/MOL.0b013e32833eb581. [DOI] [PubMed] [Google Scholar]
- 41.Tieu B.C., Lee C., Sun H., LeJeune W., Recinos A., Ju X., Spratt H., Guo D.-C., Milewicz D., Tilton R.G., Brasier A.R. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest. 2009;119:3637–3651. doi: 10.1172/JCI38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Golledge J., Mallat Z., Tedgui A., Norman P.E. Serum secreted phospholipase A2 is associated with abdominal aortic aneurysm presence but not progression. Atherosclerosis. 2011;216:458–460. doi: 10.1016/j.atherosclerosis.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 43.Nevalainen T.J., Eerola L.I., Rintala E., Laine V.J.O., Lambeau G., Gelb M.H. Time-resolved fluoroimmunoassays of the complete set of secreted phospholipases A2 in human serum. Biochim Biophys Acta. 2005;1733:210–223. doi: 10.1016/j.bbalip.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 44.Leask A. Potential therapeutic targets for cardiac fibrosis: TGFβ, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010;106:1675–1680. doi: 10.1161/CIRCRESAHA.110.217737. [DOI] [PubMed] [Google Scholar]
- 45.van Kats J.P., Methot D., Paradis P., Silversides D.W., Reudelhuber T.L. Use of a biological peptide pump to study chronic peptide hormone action in transgenic mice. J Biol Chem. 2001;276:44012–44017. doi: 10.1074/jbc.M106132200. [DOI] [PubMed] [Google Scholar]
- 46.Xu J., Carretero O.A., Liao T.-D., Peng H., Shesely E.G., Xu J., Liu T.S., Yang J.J., Reudelhuber T.L., Yang X.-P. Local angiotensin II aggravates cardiac remodeling in hypertension. Am J Physiol. 2010;299:H1328–H1338. doi: 10.1152/ajpheart.00538.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Billet S., Bardin S., Verp S., Baudrie V., Michaud A., Conchon S., Muffat-Joly M., Escoubet B., Souil E., Hamard G., Bernstein K.E., Gasc J.M., Elghozi J.-L., Corvol P., Clauser E. Gain-of-function mutant of angiotensin II receptor, type 1A, causes hypertension and cardiovascular fibrosis in mice. J Clin Invest. 2007;117:1914–1925. doi: 10.1172/JCI28764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen J., Engle S.J., Seilhamer J.J., Tischfield J.A. Cloning and recombinant expression of a novel human low molecular weight Ca(2+)-dependent phospholipase A2. J Biol Chem. 1994;269:2365–2368. [PubMed] [Google Scholar]
- 49.Murakami M., Shimbara S., Kambe T., Kuwata H., Winstead M.V., Tischfield J.A., Kudo I. The functions of five distinct mammalian phospholipase A2s in regulating arachidonic acid release: type IIa and type V secretory phospholipase A2s are functionally redundant and act in concert with cytosolic phospholipase A2. J Biol Chem. 1998;273:14411–14423. doi: 10.1074/jbc.273.23.14411. [DOI] [PubMed] [Google Scholar]
- 50.Diaz B.L., Satake Y., Kikawada E., Balestrieri B., Arm J.P. Group V secretory phospholipase A2 amplifies the induction of cyclooxygenase 2 and delayed prostaglandin D2 generation in mouse bone marrow culture-derived mast cells in a strain-dependent manner. Biochim Biophys Acta. 2006;1761:1489–1497. doi: 10.1016/j.bbalip.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balsinde J., Shinohara H., Lefkowitz L.J., Johnson C.A., Balboa M.A., Dennis E.A. Group V phospholipase A(2)-dependent induction of cyclooxygenase-2 in macrophages. J Biol Chem. 1999;274:25967–25970. doi: 10.1074/jbc.274.37.25967. [DOI] [PubMed] [Google Scholar]
- 52.Wang D., Patel V.V., Ricciotti E., Zhou R., Levin M.D., Gao E., Yu Z., Ferrari V.A., Lu M.M., Xu J., Zhang H., Hui Y., Cheng Y., Petrenko N., Yu Y., FitzGerald G.A. Cardiomyocyte cyclooxygenase-2 influences cardiac rhythm and function. Proc Natl Acad Sci U S A. 2009;106:7548–7552. doi: 10.1073/pnas.0805806106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gitlin J.M., Loftin C.D. Cyclooxygenase-2 inhibition increases lipopolysaccharide-induced atherosclerosis in mice. Cardiovasc Res. 2009;81:400–407. doi: 10.1093/cvr/cvn286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ricciotti E., FitzGerald G.A. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.King V.L., Trivedi D.B., Gitlin J.M., Loftin C.D. Selective cyclooxygenase-2 inhibition with celecoxib decreases angiotensin II-induced abdominal aortic aneurysm formation in mice. Arterioscler Thromb Vasc Biol. 2006;26:1137–1143. doi: 10.1161/01.ATV.0000216119.79008.ac. [DOI] [PubMed] [Google Scholar]
- 56.Balestrieri B., Hsu V.W., Gilbert H., Leslie C.C., Han W.K., Bonventre J.V., Arm J.P. Group V secretory phospholipase A2 translocates to the phagosome after zymosan stimulation of mouse peritoneal macrophages and regulates phagocytosis. J Biol Chem. 2006;281:6691–6698. doi: 10.1074/jbc.M508314200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative immunostaining of cross sections of abdominal aortas from apoE−/− (A) and GV DKO (B) mice infused with 1000 ng/kg per minute of Ang II for 28 days using chicken anti-mouse GV sPLA2 antibody. Original magnification, ×20.
Representative immunostaining of cross sections of abdominal aortas from apoE−/− mice infused with 1000 ng/kg per minute of Ang II for 28 days stained with anti-CD68 (macrophages) (A) or chicken anti-mouse GV sPLA2 antibodies (B). Original magnification, ×20.
Representative pictures of five different apoE−/− (A–E) and one GV DKO (F) mouse showing the cause of death during the first 7 days of Ang II infusion to be aortic rupture, followed by acute hemorrhage into the abdominal cavity.
Representative pictures of three aortas from apoE−/− (A) and GV DKO (B) mice after 28 days of Ang II infusion.
Results from real-time RT-PCR analyses of aortas from apoE−/− and GV DKO mice infused with saline or Ang II for 10 days (n = 6 to 7 per group). *P < 0.05, **P < 0.01 versus corresponding saline-infused mice; †P < 0.05 versus similarly treated apoE−/− mice. MMP-2, matrix metalloproteinase-2; ns, not significantly different versus Ang II–infused apoE−/− mice.
A: Collagen deposition in hearts of apoE−/− and GV DKO mice infused with Ang II for 7 days (n = 4) is assessed by staining sections with picrosirius red and visualizing them under polarized light, as described in Materials and Methods. B: Relative abundance of collagen I mRNA in hearts of apoE−/− and GV DKO mice after 7 days of saline or Ang II infusion (n = 4). *P < 0.05 compared to saline-infused mice or Ang II-infused apoE−/− mice.
A: Heart/body ratio in saline and Ang II–infused mice. n.s., no significant difference between apoE−/− and GV sPLA2−/− mice. *P < 0.05 versus saline-infused mice. B: Gomori staining of representative hearts from C57BL/6 and GV sPLA2−/− mice infused with Ang II for 7 days. The boxes in the two top images indicate the regions that are shown at higher magnification in the two bottom images. White arrows, characteristic perivascular lesions.
Collagen content in hearts of apoE−/− and GX DKO mice infused with Ang II for 28 days. Collagen quantification of picrosirius red–stained hearts is performed under polarized light using ImagePro software (n = 5). *P < 0.05 versus corresponding saline-infused mice. n.s., no significant difference between apoE−/− and GX DKO mice.
A: Systolic blood pressure in mice infused with saline or Ang II for 28 days. B: Plasma aldosterone levels in mice infused with saline or Ang II for 28 days. C–F: Prostanoid metabolites in urine collected from mice during the final 24-hour period of 7-day saline or Ang II infusion measured by reversed-phase electrospray ionization tandem mass spectrometry. *P < 0.05, **P < 0.01 versus corresponding saline-infused mice. No significant differences were detected between apoE−/− and GV DKO mice within a treatment group. PGEM, prostaglandin E2 metabolite; PGD, prostaglandin D; TxB2, thromboxane B2 6-keto-PGF1a, prostacyclin metabolite.