

1. Introduction
Peroxisome proliferator-activated receptors (PPARs) belong to a family of ligand-activated nuclear receptors that includes the estrogen, thyroid hormone, and glucocorticoid receptors1. The PPAR family consists of three subtypes encoded by three separate genes: PPAR-α (NR1C1), PPAR-β/δ (NR1C2), and PPAR-γ (NR1C3). Distinct PPAR subtype tissue distributions2,3 and unique ligand-binding pockets drive separate but often complementary patterns of gene expression in response to PPAR ligands4. PPAR activation occurs upon cognate synthetic or endogenous ligand binding to the ligand-binding domain (LBD). Activated PPARs heterodimerize with retinoid X receptors (RXRs), another class of nuclear receptor, which subsequently bind to the hexameric direct repeat peroxisome proliferator response elements (PPRE)5,6 and recruit co-activator protein complexes to positively regulate expression of target genes (Figure 1A). PPARs also mediate ligand-dependent repression of inflammatory gene expression through the association with co-repressor protein complexes7.
Figure 1.

PPAR transcriptional regulation and the production of endogenous ligands. A) Endogenous lipid ligand precursors undergo enzymatic conversion to active lipids, leading to their binding to PPAR/RXR heterodimers on target genes and recruitment of co-activator complexes that activate transcription. B) Cell-specific PPAR activation is regulated by the expression of metabolizing enzymes 15-hydroxyprostaglandin dehydrogenase (15-PGDH) expressed in colonic epithelial cells and prostaglandin D2 synthase (PGD2S) in macrophages, leading to the production of endogenous ligands 15-keto-prostaglandin E2 and 15-deoxy-Δ12,14-prostaglandin J2 respectively.
Nearly all nuclear receptors share structural similarity consisting of a conserved DNA-binding domain (DBD) and LBD1. The PPAR subtype structural similarities contribute to the partial overlapping function of PPARs across different tissues. In hepatocytes, PPAR-α positively regulates fatty acid β-oxidation, ketogenesis, and gluconeogenesis, while suppressing amino acid catabolism and inflammatory responses8. PPAR-α plays anti-inflammatory roles in smooth muscle cells and vascular endothelial cells9,10. PPAR-β/δ (PPAR-δ) plays roles in lipid metabolism11, fatty acid oxidation and energy dissipation12, anti-inflammation13, and colon cancer14. PPAR-γ is an essential modulator of fat cell differentiation15-17 and lipid storage and plays important anti-inflammatory roles in macrophages18,19 and other tissues such as the colon20. PPAR-γ also contributes to insulin sensitivity21, in part through the regulation of adiponectin, an adipo(cyto)kine that enhances insulin sensitivity22.
PPAR-γ is activated by synthetic ligand thiazolidinediones (TZDs)7. TZDs, including rosiglitazone and pioglitazone, are potent insulin sensitizers that have a myriad of potential benefits for patients with cardiovascular disease including improvements in endothelial function, lipid profiles and atherosclerosis23-25. TZDs, however, augment renal sodium reabsorption, leading to fluid retention that can exacerbate heart failure26-28. Recent meta-analyses have raised questions surrounding the safety of TZDs, linking the drugs to the occurrence of myocardial infarction and death29,30. While some studies suggest that the relative risks of rosiglitazone are higher than pioglitazone, the possibility that all TZDs may have adverse risk profiles has not been excluded. The unwanted side effects of TZDs have raised the prospects for the development of newer and safer PPAR ligands, such as the therapeutic usage of natural PPAR ligands. Recent studies have identified physiologically relevant endogenous PPAR ligands linked to the expression of their endogenous synthetic enzymes in specific tissues. Examples include 15-keto-prostaglandin E2 produced by 15-hydroxyprostaglandin dehydrogenase (15-PGDH) in colonic epithelial cells and 15-deoxy-Δ12,14-prostaglandin J2 produced by prostaglandin D2 synthase (PGD2S) in macrophages (Figure 1B). Further study of the effectiveness of natural PPAR ligands or synthetic molecules that mimic the actions of natural ligands is needed to determine their potential as clinical therapeutics.
This review will characterize the structural and functional relationships of PPARs, define the regulatory mechanisms that control PPAR activities, and review the candidate natural ligands of PPARs to provide a framework for understanding the roles of PPARs as anti-inflammatory therapeutics.
2. PPAR Structure
PPARs share similar structural features with other nuclear receptors5, including a poorly conserved amino-terminal domain, a highly conserved DBD, a connecting hinge region (also referred to as the C-terminal extension; CTE), and a discrete LBD31. The central DBD is highly conserved among PPAR isoforms. The LBD contains an interior binding pocket specific for the cognate ligand. The domain also carries the moderately conserved ligand-regulated transcriptional activation function-2 (AF-2)32 that forms part of the ligand-binding pocket and is required for recruitment of co-activators such as NCoA-1/SRC-133. The N-terminal region (the A/B domain) is variable in length between receptors and contains a poorly conserved transcriptional activation function domain (AF-1), the activity of which is controlled by the cognate ligand34. The AF-1 region of the PPAR family members plays a role in determining PPAR isotype-selective gene expression differences35. The activity of the A/B domain is regulated by post-translational modifications.
3. Ligand-binding affinity
Resolution of the crystal structure of ligand-free (apo) or ligand-bound (holo) nuclear receptor LBDs with the associated co-activator fragments36-39 has provided the molecular details of ligand-induced transcriptional activation by nuclear receptors. Nuclear receptor LBDs are folded into three layers of α-helices that allow the formation of a ligand-binding pocket buried within the core of the α-helices. A globular domain consisting of 11-13 α-helices is arranged in anti-parallel helical sheets that combine to make what is described as an α-helical sandwich5. Three long helices (helices 3, 7, and 10) form the two outer layers of the sandwich, while the middle layer of helices (helices 4, 5, 8, and 9) is present in only half of the globular domain, creating a cavity in the structure for binding of the ligand in most of the receptors40.
The first step of nuclear receptor activation is initiated by ligand binding. The specificity of the LBD-ligand complex is largely based on hydrophobic interactions, hydrogen-bonding networks, and the steric size and shape of the binding pocket5. The ligand-binding pocket varies greatly in size and shape between nuclear receptors as do the binding affinities toward their respective ligands. For example, estrogen receptor, retinoic acid receptor and vitamin D receptor have smaller ligand-binding pockets and bind their respective ligands, 17β-estradiol, retinoic acid, and vitamin D3, with high affinities (subnanomolar)41. The PPAR-γ LBD is composed of 13 α-helices and a small four-stranded β-sheet that forms a large (approximately 1440 Å3) hydrophobic ligand-binding pocket typical of the promiscuous nuclear receptors, including the PPARs and the pregnane X receptor (PXR), that bind many different ligands with low affinities42. The LBD pockets of PPARs have a distinct three-arm T shape, allowing them to bind ligands with multiple branches such as phospholipids and synthetic fibrates in addition to singly-branched fatty acids43. Similarly, the LBD pocket of PXR is large (1200Å3) with an elliptical shape and structural plasticity allowing the recognition of a broad range of xenobiotic compounds and endogenous C21 steroids (pregnanes)44. The LBD of PPAR-δ is narrower than those of PPAR-γ and PPAR-α, prohibiting binding of many large ligands that activate the other receptors45. PPAR-α contains the most lipohilic ligand-binding pocket, explaining the affinity for a variety of saturated fatty acids43. As a result of the LBD pocket structures, PPARs possess a relatively low binding affinity for a broad range of putative ligands including fatty acids, phospholipids, eicosanoids and prostaglandin metabolites7.
Activation by ligand-binding modulates transcriptional activity of PPARs. Ligand-triggered activation stabilizes the interaction between PPAR and RXR and enhances the formation of functional PPAR-RXR heterodimers46. Evaluation of the crystal structure of the PPAR-γ-RXR-α-DNA complex demonstrated that the LBD assists the DBDs of both heterodimeric partners in binding to DNA. In addition, the LBD forms the centerpiece of the complex around which the other domains from PPAR-γ and RXR-α are arranged47. The LBD-ligand interaction, therefore, plays a key role leading to complex formation that results in gene activation or repression.
4. Transcriptional Control by PPARs
4.1 Peroxisome Proliferator Response Elements
PPARs, like most nuclear receptors, function as ligand-gated transcription factors. In contrast to steroid hormone receptors, which bind to DNA as homodimers in a ligand-dependent manner, PPAR/RXR heterodimers can bind to DNA in the presence or absence of ligands for either heterodimeric partner. However, interaction of PPARs with lipid or synthetic ligands enhances heterodimerization with RXR and increases DNA binding48. Early studies of PPARs demonstrated that the nuclear receptor PPAR-α heterodimerizes with RXR in response to natural ligand polyunsaturated fatty acids and the fibrate Wy14,64349. The PPAR-RXR heterodimer subsequently binds to regulatory DNA elements called PPREs and initiates transcription.
The PPRE is a direct repeat type 1 (DR1) consisting of a hexameric nucleotide recognition motif (AGGTCA) spaced by one nucleotide. An additional AACT consensus motif is positioned 5′ to the DR150. The PPAR-RXR-DNA crystal structure revealed that the PPAR-γ hinge region (CTE portion) makes an extensive DNA interaction, binding to the element 5′ upstream of the hexameric repeat47. Genome wide chromatin immunoprecipitation sequencing (ChIP-seq) studies to determine the DNA elements bound specifically by PPAR-γ confirmed the localization of the receptor to DR1-like element PPAR-γ consensus motifs under physiologic conditions in macrophages and adipocytes and demonstrated that PPRE-binding sites may be either shared between cell types or cell-type specific51.
4.2 PPAR expression
Individual PPAR receptor expression levels underlie the tissue-specific activation profiles of PPAR ligands. PPAR-α may bind to a given PPRE in tissues where it predominates, such as the liver, while PPAR-γ may regulate the same response element in other cells types such as adipocytes or macrophages where PPAR-γ expression is higher. The lipoprotein lipase (LPL) gene, for example, contains a PPRE that is regulated by both PPAR-α and PPAR-γ depending upon the cell type. Treatment with PPAR-α agonists such as fenofibrate induces LPL exclusively in liver, while the PPAR-γ agonist BRL49653 induces expression in adipose tissue without affecting transcription in liver52. Similarly, a shared bi-directional PPRE located in the region between the murine PEX11α and Perilipin genes is regulated by both PPAR-α and PPAR-γ, however, the direction of activation determining which gene is activated is cell type specific. In the liver, PPAR-α binds to this PPRE and selectively activates the PEX11α gene, leading to peroxisome proliferation. In adipose tissue, PPAR-γ binds the same PPRE and selectively activates the Perilipin gene which promotes triglyceride accumulation by shielding lipid droplets53. Despite shared PPRE utilization in some instances, PPARs maintain a high degree of subtype specificity, and one PPAR isotype does not necessarily complement the function of another when expressed in a given cell type. Adenoviral expression of PPAR-γ1 in mouse liver, for example, leads to induction of genes involved in lipid accumulation and adipogenesis that are not readily activated by PPAR-α present in the liver54. The data suggests that PPAR subtype expression levels contribute but do not fully account for different PPAR responses across cell types.
Differences in PPAR-γ isoform expression also contribute to tissue specific differences in PPAR-mediated gene expression. Alternative splicing and differential promoter usage result in expression of two PPAR-γ isoforms, PPAR-γ1 and PPAR-γ2. PPAR-γ2 carries an additional 30 amino acids at the N-terminus and is largely expressed in adipocytes where it drives a higher ligand-independent basal activity compared to PPAR-γ1. The N-terminal ligand-independent activation function 1 (AF-1) domain is responsible for the difference in basal transcription between the two isoforms. The activation function of the N-terminal domain of PPAR-γ2 is 5-6 fold greater than that for PPAR-γ155. Deleting PPAR-γ2 confirmed a role of the receptor in the adipose proliferative response56. Using chimeric PPAR-γ/PPAR-δ proteins, the AF-1 region of PPAR-γ was shown to be essential for adipogenesis35,57 while comparison of the PPAR-γ1 and PPAR-γ2 isoforms confirmed that PPAR-γ2 was more effective at inducing adipogenesis58. These data imply that the relative cell specific distribution of PPAR subtypes and isoforms contributes to differences in PPAR-mediated target gene expression and ligand responsiveness among cell types.
4.3 Post-translation Modification of PPARs
PPAR activity can be modulated by post-translational modification. In the case of PPAR-α and -γ, phosphorylation of the A/B segment has been found to play an important role in modulating receptor activation. Mitogen-activated protein (MAP) kinase mediated phosphorylation of serine 82 in the PPAR-γ1 A/B-domain (or the corresponding serine 112 in PPAR-γ2) has been shown to inhibit both ligand-dependent and independent PPAR transactivation59. PPAR-γ activity is reduced upon serine 82 phosphorylation following MAPK activation by growth factors (epidermal growth factor, platelet-derived growth factor, transforming growth factor-β, or insulin) as well as c-Jun N-terminal kinase 1/2 or p38 activation59-63. Conversely, modification of the same residue by cyclin-dependent kinases, cdk7 and cdk9, was found to increase PPAR-γ activity64,65. Phosphorylation of the PPAR-γ A/B-domain, therefore, may either inhibit or stimulate transcriptional activity, depending upon the cellular context and/or kinases involved. Additionally, recent work has demonstrated that reductions in insulin-sensitizing genes dysregulated during obestity, such as adiponectin and adipsin, are linked to the phosphorylation of serine 273 in the PPAR-γ2 induced by cyclin-dependent kinase 566. Synthetic ligands blocked cdk5-mediated phosphorylation at serine 273 in concert with improvements of insulin sensitivity. The results provide a mechanistic framework for the development of novel PPAR-γ ligands that have insulin-sensitizing but fewer adverse side effects.
Sumoylation of PPARs involving the covalent attachment of small ubiquitin-like modifier (SUMO) peptides also modifies PPAR activity. SUMO is a 100-amino acid polypeptide that is covalently attached to proteins through a process typically coordinated by E3 ligases. Four SUMO proteins have been described. The modification occurs at the lysine residue of the consensus motif φKXE/D (φ: large hydrophobic residue, X: any residue) in the substrate proteins. Sumoylation of transcription factors typically leads to repression of transcription. Androgen receptor67, glucocorticoid receptor68, liver X receptor69, PPARs and other nuclear receptors have been shown to be SUMO modified. Two functional sumoylation sites have been identified for PPAR-γ, lysine 107 in the AF-1 domain and lysine 395 in the AF-2 domain (lysine 77 and lysine 365 in PPAR-γ1, respectively). Conjugation of SUMO-1 or -2 to lysine 107 by PIAS1 or PIASxβ inhibits PPAR-γ activity. The sumoylation defective K107R mutant of PPAR-γ2 exhibited stronger transactivation than the wild type protein53,70,71. In contrast to K107 SUMO conjugation, SUMOylation of K395 contributes to the anti-inflammatory function PPAR-γ on inflammatory genes such as inducible nitric oxide synthase (Nos2) in macrophages72. Treatment with ligand resulted in sumoylation of K395, targeting PPAR-γ to the NCoR-containing corepressor complexes bound on NFκB target genes, sustaining inflammatory gene repression.
PPAR-γ activity can in part be regulated by ubiquitination. Ubiquitination is the covalent attachment of ubiquitin, a 76-amino-acid peptide to lysine residues in the substrate protein. The PPAR-γ protein is polyubiquitinated and targeted for degradation by the proteasome. Ligand-dependent activation of PPAR-γ is accompanied by ubiquitination and subsequent proteasomal degradation of the receptor, resulting in selective down-regulation of PPAR-γ expression after activation73. Treatment of adipocytes with interferon-γ also increased ubiquitination and led to degradation of PPAR-γ74,75.
PPAR-α activity is regulated by post-translational modifications including phosphorylation76, ubiquitination77 and SUMOylation. Murine PPAR-α is SUMOylated at K358, promoting interaction with GA-binding protein alpha bound to the Cyp7b1 promoter and down-regulating this gene78. SUMO-1 conjugation of human PPAR-α at K185 of the hinge region of the receptor down-regulates its transcriptional activity by recruiting the corepressor NCoR. Mutation of this lysine K185R increased transcriptional activity while ligand activation inhibited SUMOylation and preventing NCoR binding79.
4.4 Coactivators and Corepressors
Nuclear receptors can function as molecular switches, alternating between states of transcriptional repression and activation, depending on the presence or absence of ligand and association with coactivators or corepressors80. Coregulator proteins are often components of large multiprotein complexes that act sequentially and/or in combinatorial fashion to remodel chromatin, mobilize nucleosomes and recruit the transcription machinery81. Many of the common cofactors for the nuclear receptor family of proteins regulate PPAR activity, including the p160 co-activator members SRC-1/NCoA133,82, TIF2/GRIP1/NCoA2/SRC-283,84, and pCIP/ACTR/AIB1/SRC-385. The nuclear receptor interaction domain of these coactivators is highly conserved and contains one or more copies of the consensus motif LXXLL which can mediate ligand-dependent interaction with nuclear receptors86-88. In contrast, the nuclear receptor corepressor (NCoR) and silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) corepressor molecules contain an extended LXXXIXXXL motif that binds to the coactivator binding pocket in the absence of agonists or the presence of antagonists. Binding of agonists to the LBD induces a conformational change that displaces the LXXXIXXXL motif and generates a ‘charge clamp’ for LXXLL motifs42, 89.. The p160 proteins serve as foundations upon which coactivator complexes are assembled. The p160 proteins recruit factors such as cAMP responsive element binding protein (CREB) binding protein (CBP)/p300 which possesses the histone acetyltransferase (HAT) activity for remodeling chromatin and allowing transcriptional activation90. PPAR activity, therefore, depends on the presence of the associated coactivator complex proteins.
PPAR subtype specificity is partly imposed by differential affinity of the receptors towards their individual cofactors. The A/B domain (containing AF-1) plays an important role in defining the PPAR isotype-specific transcriptional responses. Three A/B-interacting proteins have been identified that display isotype-specific interactions: PPAR-γ co-activator 2/SCAN domain protein 1 (PGC-2/SDP1), HIV-1 Tat-interacting protein 60 (Tip60), and the corepressor tribbles homolog 3 (TRB3). PGC-2 and Tip60 are co-activators of PPAR-γ that promote while TRB3 is a corepressor of PPAR-γ that suppresses adipogenesis57,91,92. Additionally, a portion of the transcriptional activation of PPAR-γ depends upon the presence of an intact A/B domain. The A/B domain dependent genes are the highly selective PPAR-γ target genes, many of which are involved in lipid storage. Deletion of the PPAR-γ2 A/B-domain reduced the recruitment of CBP and p300 to the PPREs of genes that required this domain for full activation93. The results confirm that PPAR subtype specific domain-interacting cofactors, such as those interacting with the A/B-domain, help define PPAR-subtype specific functions.
The ligand-independent silencing activity of nuclear receptors is due to an ability to recruit NCoR/SMRT corepressors and establish a corepressor complex on target gene promoters that can affect chromatin structure through the associated histone deacetylase (HDAC) activity of the complex proteins94. PPAR-γ is capable of recruiting the corepressors SMRT and NCoR in 3T3-L1 cells, repressing basal PPAR-mediated transcriptional activity in the absence of ligand95. PPAR agonists therefore convert PPAR/RXR heterodimers from repressors of transcription to transcriptional activators at these sites by inducing a conformational change in the ligand binding domain that results in exchange of NCoR/SMRT corepressor complexes for coactivator complexes that contain components with LXXLL interaction motifs (Figure 2A) .
Figure 2.

Transcriptional activation and repression by PPARs. A) In the absence of agonist ligand, co-repressor complexes bind to PPAR/RXR heterodimers on response elements in target gene enhancers and promoters, inhibiting gene expression. Upon agonist binding, corepressor complexes dissociate and co-activator complexes are recruited, resulting in transcriptional activation of target genes. B) Transrepression of NFκB and AP-1 target genes by direct interactions of PPARs with NFκB and/or AP-1 proteins. Such interactions may prevent coactivator recruitement required for transcriptional activation. C) PPAR inhibition of AP-1-dependent gene expression by inhibition of c-Jun N-terminal kinase (JNK) activity required for activation of AP-1 target genes. D) PPAR inhibition of inflammatory response genes by competition for commonly used, rate-limiting coactivators. E) PPARγ repression of inflammatory response genes by inhibition of NCoR clearance. Ligand induced SUMOylation of PPAR-γ results in its docking to NCoR-containing co-repressor complexes bound to AP-1 elements via non-phosphorylated cJun. This interaction inhibits signal-dependent turnover of NCoR complexes required for transcriptional activation. Repression mechanisms may operate in a cell-specific and in some cases gene specific manner. The relative importance of specific mechanisms has not been established in vivo.
4.5 Permissive Chromatin
In addition to the role of PPAR isotype expression in defining tissue-specific activation by PPARs, the specificity and potency of activation by PPARs are highly dependent upon cell type96. Cofactor differences between cell types contribute to PPRE activity by altering the available binding sites in addition to roles they play in PPAR-containing activation or repression complexes. Genome-wide studies of adipocytes demonstrated that PPAR-γ localizes preferentially to lipid and carbohydrate metabolism genes, many of which are downregulated after the knockdown of PPAR-γ expression. Chromatin immunoprecipitation (ChIP) of PPAR-γ in adipocytes confirmed enrichment of PPRE binding sites (DR1-like elements) in the majority of the binding regions identified by sequencing PPAR-γ bound DNA regions (ChIP-seq)97,98. C/EBP-α and C/EBP-β binding motifs were enriched in the proximity of PPAR-γ binding regions, suggesting a cooperative role for the CCAAT/enhancer binding protein (C/EBP) family in the PPAR-γ-dependent target gene expression of adipocytes that was confirmed after C/EBP knockdown.
PPAR binding site specificity, however, is cell-type specific. The cell-specificity of binding is determined in part by the presence of heterochromatin in a non-permissive, transcriptionally silent or active (euchromatin) state. Dimethy lysine 9 of histone 3 (H3K9Me2) and trimethyl lysine 27 of histone 3 (H3K27Me3) markers are present on transcriptionally silent heterochromatin99,100. When comparing macrophages and adipocytes, the macrophage-unique PPAR-γ-binding sites were marked by these repressive histone methylation marks in adipocytes51 consistent with inactive chromatin in the adipocyte that prevented PPAR-γ from binding. The data suggest a model where cell specificity of PPAR binding is restricted by the presence of heterochromatin in permissive or nonpermissive states for a given PPAR binding site. Whereas C/EBP was an important factor defining functional binding sites in adipocytes, the hematopoietic transcription factor PU.1 co-localized with PPAR-γ in areas of open chromatin in the subset of immune genes unique to the macrophage51. The data supports the hypothesis that cell specific factors may establish open chromatin configurations that permit PPAR binding site accessibility. Co-factors such as PU.1, therefore, help to define the macrophage specialized functions of PPAR-γ, such as maintenance of the alternative macrophage phenotype101,102, cholesterol uptake and efflux in atherosclerotic plaques103,104, antigen cross-presentation to T lymphocytes105, and dendritic cell immunogenicity106 that are unique from the metabolic functions of the receptor presumably by allowing additional PPAR binding sites that are unique to the macrophage.
4.6 Endogenous Ligand Regulation
PPARs are activated by fatty acids and naturally occurring fatty acid-derived molecules including eicosanoid and prostaglandin derivatives. The relative tissue quantity of endogenous ligands for PPARs is expected to play a role in defining the activation state of a given PPAR subtype in a given cell. Co-repressor displacement107 or co-activator recruitment108,109 along with histone acetylation110,111 in the setting of ligand-dependent PPAR-γ activation (either endogenous lipid species or exogenous ligand such as fibrates or thiazolidinediones) guides temporal or disease-state-specific PPAR-γ enhancer activation. A representative example is the loss of TRAP220 mediator complex component at PPAR-γ-specific binding regions in the cystic fibrosis transmembrane regulator (Cftr)-null mouse colonic epithelial cells associated with a concomitant reduction of target gene expression and the endogenous PPAR-γ ligand 15-keto-prostaglandin E2(15-keto-PGE2)112. Measurable differences in endogenous PPAR-γ ligand quantities defined the disease-state difference in PPAR-γ activation between the Cftr-null and wild type colonic cells. The relevant endogenous PPAR ligands in a given cell type, therefore, can control the cell-type and PPAR-isotype specific transcription activity under physiologic conditions.
Specific PPAR ligand species are present in different tissue types, accounting for a component of cell-specific PPAR activation. Prostaglandin D2 synthase (PGD2S), the enzyme responsible for producing PGD2, the precursor of 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), is expressed predominantly in macrophages and specialized antigen-presenting cells and is absent in epithelial cells113. As a result, 15d-PGJ2 can be found in monocytes after IL-13 treatment114 and is present in the cytoplasm of foamy or spindle macrophages of human atherosclerotic plaque115 but was not detected by liquid chromatography/tandem mass spectrometry from colonic epithelial cells (author’s unpublished observation). 15d-PGJ2 production increases during the inflammatory process coordinate with increased expression of PGD2S. Similarly, 15-hydroxyprostaglandin dehydrogenase (15-PGDH), the enzyme responsible for producing 15-keto-PGE2, is highly expressed in pulmonary and colonic epithelial cells but is not expressed in macrophages. As a result,15-keto-prostaglandin E2 was found to be an important physiologically relevant PPAR-γ ligand in colonic epithelial cells112. The regulation of cell-specific or tissue-specific ligand production, therefore, contributes to cell-specific PPAR isoform activation states (Figure 1A).
5. PPARs and Transrepression
5.1 PPARs and inflammatory responses
NF-κB is a critical activator of genes involved in the inflammatory process116. Pro-inflammatory cytokines activate the IκB kinase (IKK) complex that phosphorylates the NF-κB inhibitors, triggering their degradation and freeing NF-κB to translocate to the nucleus where it induces target genes such as the inducible cyclooxygenase (COX2) gene117. Additionally, AP-1, STAT and IRF transcription factors contribute to inflammatory responses. AP-1 is composed of homo- or heterodimers among members within the Jun and Fos families. c-Jun in association with c-Fos is the major AP-1 heterodimer118,119.
Several mechanisms have been described for the negative regulation of inflammatory gene expression by nuclear receptors. In addition to the roles of PPARs in lipid and glucose metabolism, the three PPAR isoforms have emerged as key regulators of inflammatory and immune responses. PPARs are expressed in vascular and immunological cell types such as monocytes/macrophages, endothelial cells, lymphocytes and dendritic cells. PPARs can inhibit inflammatory gene expression by several mechanisms, including direct interaction with NF-κB or AP-1 subunits, modulation of kinase activities, competitive binding for limiting pools of coactivators, and interaction with co-repressors. PPAR-γ has been demonstrated to mediate anti-inflammatory signaling in a number of inflammatory model systems, antagonizing the production of cytokines induced by gamma interferon (IFN-γ) and lipopolysaccharides (LPS) through inhibition of STAT1, NF-kB, and AP-118,19. Additional research has suggested that PPAR ligands display anti-inflammatory effects in several disease models including atherosclerosis, obesity-induced insulin resistance, autoimmune encephalomyelitis, psoriasis, inflammatory bowel disease, and arthritis7.
Some of these models have been described using the ligand 15d-PGJ2 which has been demonstrated to show PPAR-γ independent anti-inflammatory effects. It is probable that some of these mechanisms only operate in the case of specific cell types or PPAR isoforms. Consistent with this, PPAR-γ agonists do not interfere with NF-κB nuclear entry in macrophages and specifically regulate a subset of LPS-inducible genes120-122, indicating promoter-specific mechanisms underlying transrepression in the macrophage rather than a global effect of antagonizing inflammatory signaling.
5.2 Direct Interactions Between PPARs and Inflammatory Transcription Factors
Many nuclear receptors can exhibit inhibitory effects that do not involve direct sequence-specific DNA binding by the DNA binding domain. One established mechanism is the tethering of nuclear receptors to negatively regulated transcription factors, resulting in inhibition of DNA-binding and/or transactivating functions. Studies of the anti-inflammatory effects of glucocorticoids initially described this model. Following glucocorticoid treatment, the glucocorticoid receptor (GR) interacts directly with AP-1123-125 and NF-κB126,127 subunits to mediate an anti-inflammatory effect. Similar inhibition of AP-1 has been seen with the RXR-α ligand 9-cis-retinoic acid128.
Several reports have indicated that PPARs may exert a component of their anti-inflammatory effects in certain cell types through mechanisms involving direct interactions with inflammatory transcription factors including NF-κB, AP-1, or STAT, resulting in inhibition of coactivator recruitment (Figure 2B) or recruitment of corepressors. Ligand-activated PPAR-α has been shown to interact with p65 and c-Jun and prevent inflammatory activation by IL-1α129. PPAR-α ligands have also been shown to induce expression of the inhibitory protein inhibitor of kappa B (IκBα) in liver and smooth muscle cells, which sequesters NF-κB in the cytoplasm and reduces activity130-132. PPAR-γ has similarly been shown to interfere with c-Jun promoter binding in vascular endothelial cells10 and to interfere with NF-κB nuclear localization in macrophages133 and colonic epithelial cells134 under specific experimental conditions. PPAR-γ agonists including ciglitazone and 15d-PGJ2 also stabilized IκBα levels in lungs, resulting in reduced activation of NF-κB135. The overall contribution of these mechanisms to the anti-inflammatory role of PPARs is unclear since PPAR ligands appear to have promoter-specific anti-inflammatory functions, particularly in the low micromolar range of concentration that is receptor dependent, rather than general anti-inflammatory effects on all promoters.
5.3 Regulation of Kinase Activity
A second mechanism for transrepression involves inhibition of signal transduction pathways necessary for transcriptional activation. c-Jun transcriptional activity is enhanced by amino-terminal phosphorylation on Ser63 or Ser73 by members of the Jun amino-terminal kinase (JNK) and mitogen-activated protein-serine (MAP) families136-138. Activation of c-Jun, for example, is greatly increased by phosphorylation of Ser63 or Ser73 by members of the Jun amino-terminal kinase (JNK) superfamily. Glucocorticoids, retinoic acid, and T3 have been shown to inhibit JNK, blocking c-Jun phosphorylation and inhibiting AP-1 dependent activation mediated by inflammatory stimuli such as TNF-α139,140. Inhibiting JNK and other signaling mediated phosphorylation of c-Jun, reduces the induction of AP-1-related inflammatory target genes.
A role for the anti-inflammatory effects of PPAR-γ ligands in regulating kinase activity has been demonstrated in the colon, where ligand activation of PPAR-γ and RXR-α reduced JNK and p38 MAPK activation induced by the chemical colitis agent 2,4,6-trinitrobenzene sulfonic acid (TNBS) (Figure 2C). The study confirmed an anti-inflammatory effect of PPAR-γ ligands, although an impact on c-Jun phosphorylation or AP-1 activation was not confirmed141. PPAR-α agonists have been shown to antagonize the phosphorylation of c-Jun in microglial BV-2 cells in response to whole body radiation142. Other studies failed to show that PPAR-α and/or - γ agonists inhibit c-Jun or p38 phosphorylation and that they primarily act on transcription events in the nucleus143,144.
5.4 Competitive Interference with Coactivators
One studied model of repression by nuclear receptors describes that inhibition of inflammatory gene activation is mediated through competitive binding of limiting quantities of the general coactivators CREB-binding protein (CBP) and p300. A recruitment model has been proposed for proteins that interact with CBP/p300, such as the c-Jun and p65, demonstrating that interference or squelching of one another can occur when the intracellular levels of CBP are limiting. CBP/p300 is recruited to the promoters of activated genes by association with the stress-inducible factors NF-κB, IRF, and AP-1 where it contributes to transcriptional activation145-147. Consistent with this, recent work has confirmed LPS-inducible p300 chromatin association with transcriptional enhancers in LPS-inducible transcripts on a genome-wide scale148. Activation of alternative parallel pathways that utilize the same co-activators (CBP/p300) might diminish the inflammatory stimulus response if the co-activator concentration is limiting. Competition for CBP/p300 has been proposed to contribute to the anti-inflammatory effects of glucocorticoids147, although this mechanism did not explain glucocorticoid repression of the IL-6 promoter in human embryonic kidney cells149,150.
Some evidence suggests that a portion of the anti-inflammatory effects of PPAR ligands may be mediated through coactivator competition (Figure 2D). Upon stimulation of PPAR-γ with ligand, the interaction between PPAR-γ and CBP is increased151. In the case of the inducible nitric oxide synthase (Nos2) promoter, PPAR-γ mediated repression occurred only in the presence of wild type receptor that was capable of recruiting CBP152, while transrepression did not occur with the ‘charge clamp’ containing mutant PPAR-γ that fails to interact with coactivators. Similar studies in epithelial cells found that overexpression of CBP/p300 could partially block the anti-inflammatory effects of PPAR-γ agonists on COX2 expression153. The data supports a model where PPAR-mediated transrepression is correlated with transactivation. Other studies of PPAR-α have shown that PPAR-dependent repression was independent of the CBP co-expression in the cell129. The basis for the differences between these experimental conclusions is unclear, although many of the experiments performed have been completed using overexpression systems, and it is suspected that the earlier work may not fully reflect the native levels of co-regulators in the cell.
5.5 Co-repressor Model of Transrepression
Two distinct models for corepressor-dependent transrepression have been proposed by PPARs. In the case of PPAR-δ, an association with the transcriptional repressor BCL-6 occurred in the absence of PPAR-δ ligands, antagonizing the BCL-6 repressor function. In the presence of ligand, PPAR-δ released BCL-6, which exerted anti-inflammatory effects by repressing transcription from NF-κB-dependent promoters13. In the second model, recent studies have highlighted the role of PPAR-γ in stabilizing corepressor nuclear receptor corepressor (NCoR)-histone deacetylase-3 (HDAC3) containing complexes at target promoters72. In addition to mediating repressive functions of unliganded nuclear receptors, NCoR corepressor complexes also confer repression functions to other classes of transcription factors. Recent studies indicate that NCoR complexes occupy subsets of inflammatory response genes in macrophages and other cell types and play roles in maintaining these genes in a repressed state under basal conditions. In the presence of an inflammatory stimulus such as LPS, the NCoR-containing repressor complex is rapidly cleared from the promoter, resulting in de-repression as prerequisite for subsequent binding of activating factors such as NF-κB and subsequent transcriptional activation. Treatment with PPAR-γ ligands reduced NCoR clearance from the promoter, resulting in maintenance of repression (Figure 2E). This mechanism was linked to SUMOylation of lysine 365 of PPAR-γ. Mutation of the K365 residue, prevented SUMOylation of the receptor, eliminated PPAR-γ recruitment to the inflammatory promoter, and allowed signal-dependent NCoR clearance. It is important to note at this point that all mechanistic studies of anti-inflammatory actions of PPARs have been carried out in cell culture systems and the biological roles of any specific mechanism remains to be determined in an in vivo context.
6. Synthetic and Endogenous PPAR Ligands
Because of the broad roles of PPARs in regulating metabolism, inflammation, differentiation, and cellular growth, a large number of specific and potent synthetic ligands have been generated (Figures 3-5). While PPAR-α is activated by the fibrate drugs such as clofibrate and Wy14,643, PPAR-γ is the receptor for the thiazolidinedione (TZD) class of antidiabetic drugs. TZDs stimulate pre-adipocyte differentiation154 and induce insulin sensitization155, leading to their usefulness in treating diabetes. TZD drugs were recognized to activate the aP2 (FABP4) gene transcription156 through an enhancer element identified to bind the PPAR-γ/RXR heterodimer157. Subsequent studies proved direct binding of the TZD BRL49653 (rosiglitazone) to the PPAR-γ protein158 and demonstrated that the adipogenic program of TZDs were mediated though ligand-dependent PPAR-γ activation. The affinity of common PPAR-γ agonists measured by competition binding is 40 nM for rosiglitazone and 4.8 μM for pioglitazone159. GW2433 is a synthetic agonist for PPAR-δ. Synthetic agonists and antagonists for PPARs have served as the basis for countless studies of the effects of PPARs.
Figure 3.

Natural and synthetic PPAR-γ ligands. PPAR-γ ligands include the synthetic agonist BRL49653, the endogenous agonists 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), 15S-hydroxy-5Z,8Z,11Z,13E-eicosatetraenoic acid (15-HETE), and 15-keto-prostaglandin E2, and the endogenous antagonist 1-acyl-2,3-cyclic-glycerophosphate.
Figure 5.

Natural and synthetic PPAR-δ ligands. PPAR-δ ligands include the synthetic agonist GW2433 and the endogenous agonists 15S-hydroxy-5Z,8Z,11Z,13E-eicosatetraenoic acid (15-HETE) and 5Z,8Z,11Z,14Z,17Z-eicosapentaenoic acid (EPA).
Natural ligands for PPARs consist of fatty acids and cyclooxygenase-derived eicosanoids and prostaglandins that bind to PPARs with relatively low affinity in many cases (i.e. binding constants in the micromolar range) (Tables 1-3). Identification of the potential endogenous ligands for PPARs has often rested on screening of candidate molecules rather than mass spectroscopy. This approach combined with the promiscuous nature of the LBD structure of PPARs has led to claims that numerous natural lipid species are PPAR agonists. The importance of many of these compounds have not been fully established in vivo, although several recent studies have characterized specific lipid species that appear to be relevant and at the appropriate ambient tissue concentrations to act as ligands in specific cell types.
Table 1.
PPAR-γ ligands
| Ligand | Activity | Biological Activity | Ref |
|---|---|---|---|
| 15d-PGJ2 | agonist | In murine sepsis model, administration of ligand reduced lung injury and PMN trafficking to lung and intestine, blocked expression of adhesion molecules, and improved survival |
176 |
| Blocks inflammatory signaling through NF-κB, AP- 1, and STAT-1, thus reducing expression of Nos2, Mmp9, and Msr1 |
18, 19 |
||
| Covalently interacts with PPARγ LBD at Cys285 | 168 | ||
| Inhibits NF-κB activation by preventing degradation of IkBα in a PPAR-γ independent fashion |
174 | ||
| Inhibits MMP9 activity in human monocytes | 216 | ||
| 13-HODE | agonist | Decreases Nos2 expression in macrophages | 197 |
| Decreases CCR2 expression in THP1 human monocytes |
214 | ||
| Found in atherosclerotic plaques | 187 | ||
| 15-HETE | agonist | Decreases Nos2 expression in macrophages | 197 |
| Decreases superoxide production and degranulation of PMN stimulated by fMLP, LTB4, and PAF |
208 | ||
| Inhibits PMN migration across cytokine-activated endothelium in cell culture by reducing LTB4 receptors |
209, 210 |
||
| 9-HODE | agonist | Found in atherosclerotic plaques | 188 |
| Decreases CCR2 expression in THP1 human monocytes |
214 | ||
| Nitroalkene fatty acids |
Induce adipocyte differentiation and glucose uptake |
227, 228 |
|
| Block cytokine release and NF-κB activation in HUVECS stimulated by TNFα or atherogenic response |
231 | ||
| Decrease Nos2 and Mcp1 expression in RAW264.7 macrophages. Effect was not blocked by PPARγ synthetic antagonist |
232 | ||
| Covalently interact with PPARγ LBD in Cys285 at micromolar concentrations |
230 | ||
| 15-keto-PGE2 | agonist | Induces adipogenesis of 3T3L1 cells | 243 |
| Abundance of 15-keto-PGE2 was reduced in Cftr- null colonic epithelial cells, which exhibit defective PPAR-γ mediated gene expression |
112 | ||
| 5-methoxy-indole acetate |
agonist | serotonin metabolite that is bound to the AF-2 subpocket of PPAR-γ LBD. PPAR macrophage and stimulate 3T3-L1 adipogenesis. |
244 |
| PGF2a | antagonist | Inhibits adipocyte differentiation | 250 |
| Inhibits PPAR-γ activity by inducing phosphorylation of PPAR-γ at serine 112 |
250 | ||
| PPAR-γS112A knock-in mice maintained insulin sensitivity in model of diet-induced obesity |
251 | ||
| Cyclic phosphatidic acid |
antagonist | Stabilizes PPAR-γ-SMRT interaction | 252 |
| Blocks TZD-stimulated adipogenesis and lipid accumulation in macrophages |
|||
| Production induced by insulin, PMA, LPS, or H2O2 |
Table 3.
PPAR-δ ligands
Many of the endogenous ligands are derived from arachidonic acid and linoleic acid as products of the lipoxygenase and cyclooxygenase pathways (Figure 6). Prostaglandin-endoperoxide synthase (PTGS), also known as cyclooxygenase, is the key rate-limiting enzyme in prostaglandin biosynthesis. Two isoforms of the enzyme exist. COX1 (PTGS1) is constitutively expressed in most tissues while COX2 (PTGS2) is inducible and is considered a pro-inflammatory enzyme. Phospholipase A2-derived arachidonic acid is first converted to an unstable endoperoxide intermediate by cyclooxygenases and subsequently to one of several related prostanoids including PGD2, PGE2, PGF2a, prostacyclin (PGI2) or thromboxane A2117. While synthesis of prostaglandins and release of arachidonic acid is regulated by activation of phospholipases, prostanoid production is dependent on the expression levels of the metabolizing synthases that vary in a cell-specific and stimulus-specific manner. Prostaglandins, for example, are generally present physiologically in body fluids at picomolar to low nanomolar concentrations160, however, local prostaglandin concentrations in the micromolar range have been detected at sites of inflammation161.
Figure 6.

Biosynthetic pathways leading to generation of some of the naturally occurring PPAR ligands. A. Ligands generated from arachidonic acid. B. Ligands generated from linoleic acid.
The endogenous ligands for PPAR-γ in various tissues have not been firmly established. Physiological agonists of PPAR-γ include the lipoxygenase products 13-HODE and 15-HETE, the prostaglandins 15d-PGJ2 (15-deoxy-Δ12-14-prostaglandin J2) and 15-keto-prostaglandin E2, as well as nitroalkene fatty acids. 16:0/18:1-GPC (1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine) and possibly leukotriene B4 (LTB4) serve as endogenous ligands for PPAR-α. Eicosapentaenoic acid and possibly 15-HETE are ligands for PPAR-δ. Species of lysophosphatidic acid (1-acyl-2-hydroxy-sn-glycero-3-phosphate, LPA) have been proposed to act via PPAR-γ162,163. LPA has been shown to bind PPAR-γ, activate transient reporter assays, and stimulate lipid accumulation in human monocytes. LPA, however, generally mediates cell signaling through the activation of surface LPA receptors and pro-inflammatory functions through the activation of NF-κB, AP-1 and C/EBPβ164, and its role as an endogenous PPAR-γ ligand has not been fully clarified. Dietary conjugated linoleic isomers have also been proposed to act through PPARs.
7. PPAR-γ endogenous ligands
7.1. PPAR-γ agonists
7.1.1 Cyclopentenone prostaglandin 15d-PGJ2
The J-series cyclopentenone prostaglandins are metabolites of prostaglandin D2. Prostaglandin D2 synthase (PGD2S) metabolizes cyclooxygenase (COX)-derived PGH2 to the PGD2 that is spontaneously converted to 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) by non-enzymatic dehydration (Figures 3 and 6). The J2 series of prostaglandins differ from other classes of prostanoids by the presence of an electrophilic α,β-unsaturated carbonyl group in the cyclopentenone ring, a structure proposed to account for some of their receptor-independent biological actions. 15d-PGJ2 was recognized in 1983 as a degradation product of PGD2, formed in the presence of albumin165. Subsequently, 15d-PGJ2 was identified as the first natural ligand agonist for PPAR-γ, inducing the transcription of PPAR target genes166,167 (Figure 3). 15d-PGJ2 induced the interaction between PPAR-γ and all coactivators tested (SRC-1, TIF2, AIB-1, p300, TRAP220/DRIP205)151 consistent with its function as an activating ligand. 15d-PGJ2 was found to inhibit synthetic ligand binding for PPAR-γ, with IC50 of approximately ~1 μM for [3H]-BRL49653167. It was subsequently demonstrated to covalently bind to the sulfhydryl group of cysteine 285 in the PPAR-γ ligand binding pocket through a Michael addition reaction by the α,β-unsaturated carbonyl168 that readily reacts with substances containing nucleophilic groups such as cysteinyl thiols. Mutation of the cysteine residue in the PPAR-γ LBD resulted in the loss of activation by 15d-PGJ2, while the mutant was still activated by synthetic ligands. Based on the model of the α,β-unsaturated carbonyl of 15d-PGJ2 covalently bound to the cysteine residue of the PPAR-γ LBD through a Michael addition reaction, a number of potential activating endogenous ligands including several oxidized eicosanoid and keto-prostaglandins have been proposed168.
A major question in subsequent studies has been whether 15d-PGJ2 is present in sufficient quantities at physiological levels to act as an activating ligand in vivo. The fact that there is no enzymatic formation of cyclopentenone prostaglandins initially raised this concern. PGD2 undergoes chemical dehydration to form the cyclopentenone prostaglandin PGJ2, which is further dehydrated by loss of the 15-hydroxyl group and migration of the 13,14-double bond of PGJ2 to form 15d-PGJ2. This reaction proceeds at a slow rate compared to the formation of other prostaglandins.
In addition to a role in PPAR-γ activation, 15d-PGJ2 also functions to block inflammatory stimuli mediated through various transcription factors, including NF-κB, AP-1, and signal transducer and activator of transription-1 (STAT-1). Treatment of LPS-stimulated macrophages demonstrated that the compound was more potent than synthetic PPAR ligands at inhibiting the induction of inflammatory genes, including inducible nitric oxide synthase (Nos2), gelatinase B (Mmp9), and scavenger receptor (Msr1)18,19. This result led to the discovery that 15d-PGJ2 has anti-inflammatory effects that were independent of PPAR-γ169, a result that was confirmed in PPAR-γ null macrophages treated with 15d-PGJ2 in the micromolar (6-10 μM) concentration range170,171. These anti-inflammatory effects were mediated at least in part through the prevention of phosphorylation and degradation of the NF-κB inhibitor IκBα by IKK inhibition172,173. 15d-PGJ2 covalently binds to and inhibits IκB kinase, a key activator of NF-κB174. This covalent binding is mediated through a Michael addition reaction to the reactive cylopentenone ring.
The levels of cyclopentenone prostaglandins, PGD2 and 15-deoxy-Δ12,14-PGJ2, have been shown to increase in vivo in response to inflammatory stimulus, corresponding to the inflammatory resolution phase. In the rat carrageenin-induced pleurisy model, COX2 levels and PGE2 levels increase 2 hours after experiment onset, associated with a polymorphonuclear leukocyte (PMN) infiltrate. The PMNs are replaced by migrating mononuclear cells, which differentiate into macrophages. Later in the process, PGE2 levels fall and the cyclopentenone prostaglandin levels including 15d-PGJ2 peak, associated with the activation mechanisms that result in inflammatory resolution175. In other studies of a murine polymicrobial sepsis model, 15d-PGJ2 was shown to be an effective anti-inflammatory agent, where injecting 15d-PGJ2 following LPS treatment reduced lung injury and neutrophil trafficking to lung and small intestines, blocked the expression of adhesion molecules, and improved survival176. It was also found to be an endogenously produced anti-inflammatory mediator in the murine zymosan peritonitis model. The authors demonstrated production of both PGD2 and subsequently 15d-PGJ2 in vivo. 15d-PGJ2 contributed to the resolution phase of inflammation by dampening pro-inflammatory cytokines and augmenting the production of interleukin-10177. PGD2 synthase-null mice did not produce 15d-PGJ2 during zymosan peritonitis and developed prolonged macrophage and lymphocyte accumulation associated with increased TNF-α and reduced IL-10 levels. These experiments mechanistically link endogenous PGD2 production with 15d-PGJ2 and the resolution of acute inflammation.
Thus 15d-PGJ2, in addition to its role as an activating ligand for PPAR-γ, plays anti-inflammatory roles that contribute to the resolution and dampening of inflammation. While it clearly acts as a PPAR-γ ligand at micromolar concentrations, it is unclear to what extent 15d-PGJ2 plays anti-inflammatory or PPAR-regulating roles in vivo. Studies of adipocyte differentiating 3T3L1 cells by liquid chromatography/tandem mass spectrometry (LC-MS/MS) demonstrated that the cell-associated 15d-PGJ2 concentrations were three orders of magnitude lower (~1 nM) than concentrations required for PPARγ-dependent effects178. LC-MS/MS studies from zymosan-stimulated peritoneal macrophages demonstrated concentrations an order of magnitude higher (~ 5-15 nM), however this was still far below the levels previously used for PPAR-activation and NF-κB inhibition studies177. Furthermore, 15d-PGJ2 was not increased in the joint fluid of patients with arthritis nor in the urine of patients with diabetes or obesity178. These data provide evidence that while 15d-PGJ2 can be produced in vivo under specific experimental conditions where macrophages are involved, its levels may be too low to be compatible with roles as an endogenous activator of PPAR-γ. Regional differences in lipid concentrations occurring as a result of active transport or nuclear accumulation of 15d-PGJ2, however, might account for higher local concentrations than have been measured in these experiments. Cyclopentenone prostaglandins have been shown to be actively transported into cells and transferred to the nuclei through a process involving binding to the liver fatty acid binding proteins (L-FABP)179. Similarly, PGE2 has been shown to undergo active uptake, leading to cytoplasmic compartment concentrations 25-fold higher than that of the extracellular medium180. It is possible that under specific physiologic conditions, the nuclear concentrations may exceed that needed for mediating the anti-inflammatory or PPAR-activation effects. Since the interaction with the PPAR-γ LBD is covalent, regional and temporal concentration changes coupled to the receptor may be more capable of modulating PPAR receptor function than if the ligand binding was reversible. Future studies aimed at determining the physiological relevance of 15d-PGJ2 particularly in monocytes and macrophages where the production is inducible would be helpful to clarify the in vivo affects of the lipid while utilizing more specific measures of receptor-dependent gene activation such as chromatin immunoprecipitation to measure coactivator promoter recruitment.
7.1.2 15-Lipoxygenase metabolites
Lipid peroxides are formed by the action of lipoxygenases, a group of enzymes implicated in the pathogenesis of a number of inflammatory disorders including asthma181, diabetic vascular disease182, and atherosclerosis. Lipoxygenases catalyze the region- and stereospecific oxygenation of polyunsaturated fatty acids to their corresponding hydroperoxy derivatives183. These fatty acid hydroperoxides are further transformed to bioactive compounds such as the leukotrienes and lipoxins, which play pro-inflammatory or anti-inflammatory roles184. Three major classes of mammalian lipoxygenases (5-, 12-, and 15-LOX) exist which catalyze the oxygenation of their substrate. Some lipoxygenase enzymes may exhibit multiple positional specificities185. The role of oxidized lipids in the development of atherosclerosis has been studied since the presence of lipid peroxides was discovered in human atherosclerotic aorta in 1952186. The lipid peroxides 13-hydroxy-9Z,11E-octadecadienoic acid (13-HODE) and 9-hydroxy-10E,12Z-octadecadienoic acid (9-HODE) were found within atherosclerotic plaques187,188, and a role of peroxidized linoleic acid was postulated.
The human 15-lipoxygenase is expressed in the macrophages of atherosclerotic lesions and has been implicated in the pathogenesis of atherosclerosis based on the associated lipid peroxides189-191. Oxidative modification of cholesterol esters in low density lipoprotein (LDL) particles leads to the formation of oxLDL which is rapidly taken up by macrophages or vascular smooth muscle cells via scavenger receptors like SR-A or CD36, leading to foam cell formation. Analysis of the oxygenated lipids from aortas of rabbits fed an atherogenic diet and human atherosclerotic lesions confirmed the presence of 13-HODE and 9-HODE, the same lipid peroxides that are generated by lipoxygenase-treatment of LDL192,193. The S-isomer of 13-HODE was the predominant oxygenated fatty acid isolated. In 1998, 13-HODE and 9-HODE of oxidized low-density lipoproteins (oxLDL) were identified as endogenous PPAR ligands, mediating PPAR-γ-dependent transcription in macrophages194 (Figure 3). PPAR-γ was demonstrated to be expressed in foam cells of human atherosclerotic lesions, with the expression correlating with the oxidized lipid component195, and the receptor was found to promote the uptake of oxLDL by inducing scavenger receptor CD36196. IL-4 stimulated the production of endogenous PPAR ligands by up-regulating the murine 15-LOX homolog, 12/15-lipoxygenase197.
After the early studies suggested a pro-atherogenic role of 15-lipoxygenase, a possible anti-atherogenic role was demonstrated in a 15-lipoxygenase transgenic rabbit model. Transgenic expression of the human 15-LOX in rabbits fed a high fat, high cholesterol diet or regular diet on the Watanabe heritable hyperlipidemic (WHHL) genetic background, were less prone to atherosclerotic disease198. Following this work, however, several subsequent studies demonstrated a pro-atherogenic function of the 15-LOX enzyme. Pharmacological inhibition of 15-LOX in hypercholesterolemic rabbits attenuated atherosclerosis199 while genetic disruption of the 12/15-LOX in the background of two high risk mouse models of atherosclerosis (apoE and LDLR null) or in the hematopoietic cells of apoE null mice by transplantation of 12/15-LOX null bone marrow retarded the initiation and progression of atherosclerosis200-202. The latter model is interesting because it implicates macrophages rather than the vascular endothelial cells as the source of genetic protection of 12/15-LOX deletion in the mouse model.
15-Lipoxygenase dioxygenates arachidonic acid mainly to 15S-hydroperoxy-5Z,8Z,11Z,13E-eicosatetraenoic acid (15-HpETE). A minor reaction product 12S-hydroperoxy-5Z,8Z,10E,14Z-eicosatetraenoic acid (12-HpETE) is always detectable. The major product of metabolism of linoleic acid is 13S-hydroperoxy-9Z,11E-octadecadienoic acid (13-HpODE). A high degree of stereo-selectivity for the S enantiomers differentiates the enzymatic 15-lipoxygenase reaction from the non-enzymatic lipid peroxidation reaction.203 In mammalian cells, the hydroperoxy products 15-HpETE, 12-HpETE, and 13-HpODE, are rapidly reduced to their corresponding hydroxy-lipids (15-HETE, 12-HETE, and 13-HODE) by selenium-containing glutathione peroxidases (GPXs). In addition to 13-HODE and 9-HODE, 15-HETE is an endogenous ligand for PPAR-γ, mediating PPAR-dependent transcription194. Two human 15-LO isotypes have been demonstrated which display slightly different specificities. Human 15-lipoxygenase-1 (15-LOX-1) is expressed in reticulocytes, lung and colonic tissue and primarily converts linoleic acid to 13-HODE. The 15-lipoxygenase-2 (15-LOX-2) is expressed in epidermis, prostate, lung, and colonic tissue and primarily generates 15(S)-HETE from arichidonic acid204,205. 15(S)-HETE is also generated by 15-LOX-1. The oxo metabolite 13-oxoODE has also been shown to act as a ligand for PPAR-γ, showing greater affinity for the receptor than 13-HODE194.
In addition to the pro-atherogenic role of 12/15-LO in mouse models, evidence suggests that 15-LOX metabolites play mostly athero-protective and anti-inflammatory roles. 15-HETE was demonstrated to be increased in the atherosclerotic aortas of cholesterol-fed and WHHL rabbits and was the predominant eicosanoid present206,207. 15-HETE inhibits superoxide production and degranulation of PMNs when stimulated with agonists such as the granulocyte activator N-formylmethionylleucylphenylalanine (fMLP), leukotriene B4 (LTB4), or platelet activating factor208. 15-HETE production also inhibits PMN migration across cytokine-activated endothelium in cell culture209 by reducing the presence of cell-surface LTB4 receptors210. Additionally, 15-LOX activity is linked to the anti-inflammatory lipoxins. 15-HpETE can be further metabolized by 5-lipoxygenase leading to the anti-inflammatory products 5S,6R,15S-trihydroxy-eicosatetraenoic acid (lipoxin A4) and 5S,14R,15S-trihydroxy-eicosatetraenoic acid (lipoxin B4). Lipoxins counteract the actions of pro-inflammatory factors like leukotrienes and inhibit neutrophil chemotaxis and transmigration of PMNs through epithelial cells211. Down-regulation of anti-inflammatory lipoxin A4 has recently been associated with several inflammatory diseases in human patients including asthma212, cystic fibrosis213, and ulcerative colitis205. Reduced expression of 15-LOX-2, the enzyme primarily responsible for production of the lipoxin precursor 15-HpETE, was confirmed in the case of patients with asthma and colitis. 15-HpETE and metabolites, therefore, play significant anti-inflammatory roles.
The 15-lipoxygenase lipid peroxide metabolites 9-HODE and 13-HODE as well as the arachidonic acid metabolite 15-HETE have been demonstrated to have anti-inflammatory effects through PPAR-γ. 13-HODE and 15-HETE (34 μM) down-regulated inducible nitric oxide synthase (Nos2) promoter activity in macrophages197. Both 9-HODE and 13-HODE down-regulated C-C chemokine receptor 2 (CCR2)214, a receptor for monocyte chemoattract protein-1 (MCP-1) in human monocyte Thp1 cells. MCP-1 is an important cytokine responsible for monocyte accumulation in areas of inflammation such as atherosclerotic plaques215. PPAR-γ agonists have been shown to antagonize gelatinase B (MMP-9), potentially contributing to atherosclerotic plaque stability216. Additionally, concomitant with the induction of the 15-lipoxygenase, monocytes treated with IL-4 become resistant to inflammatory stimulus by LPS. IL-4, which induces PPAR-γ and 15-LO expression, was found to exert an anti-inflammatory effect on monocytes, suppressing the secretion of proinflammatory cytokines including IL-1β, TNF-α, and IL-6 upon stimulation by LPS or IFN-γ217.
15-LOX metabolites have been demonstrated to show cell cycle effects in several cell types. While macrophage treatment with IL-4 was shown to be anti-inflammatory, IL-4 treatment of A549 lung adenocarcinoma cells increased 15-LOX activity, 15(S)-HETE production, and apoptosis induction218. 15-LOX-2 mediated 15(S)-HETE was found to mediate PPAR-γ-dependent transcription in PC-3 prostate cancer cells, while reducing proliferation and clonigenic capacity of the cells219. Additionally, 15-LOX-2 expression and 15(S)-HETE formation have been shown to be reduced in prostate carcinoma and absent in colon cancer220. Finally, the 15-LOX metabolite, 13(S)-HODE, was found to bind to and suppress activation and expression of PPAR-δ, leading to apoptosis in DLD colorectal cancer cell lines221.
Recent studies of the role of 12/15-lipoxygenase have raised questions about the anti-inflammatory role of 12/15-LOX metabolites. 12/15-LOX null mice, for example, were resistant to the streptozotocin inflammatory model of type 1 diabetes222 as well as the inflammatory effects of diet-induced obesity223. Macrophages from 12/15-LOX null mice were defective in interleukin-12 production but not TNF-α or nitric oxide release in response to LPS224, suggesting a gene-specific anti-inflammatory effect of the knockout. Select studies of the human 15-LOX-1 have also shown a pro-inflammatory phenotype. Over-expressing the human 15-LOX-1 in bronchial epithelial A549 cells, for example, increased inflammatory chemokine expression including the macrophage inflammatory protein 1-α (MIP-1α), RANTES, and IP-10 and increased the chemotaxis of monocyte-derived immature dendritic cells. Exogenous treatment of 15-HETE (50 μM) to control cells, however, did not increase MIP-1α expression, and PPAR-dependent transcriptional events were not assessed225. The mechanisms of the pro-inflammatory effects in these studies have not been resolved.
Study of the differences between the murine 12/15-LOX and human 15-LOX isotypes revealed that the murine 12/15-LO produces more 12-HETE than 15-HETE (3:1) while the human reticulocyte 15-LO produces less 12-HETE and more 15-HETE (1:12)185. The ratio of HETEs and the respective lipoxygenase activities may also vary depending upon the physiologic stimulus of the experimental condition and the specific 15-LOX isozyme expressed in a given cell type. 15-HETE was the predominant eicosanoid produced upon arachidonic acid incubation of aortas from rabbits receiving a high cholesterol diet while 12-HETE was produced in rabbits feeding on normal chow207, suggesting that the hypercholesterolemic state selectively induced 15-lipoxygenase activity in aortic tissue of rabbits. Furthermore, 12-HETE increases monocyte adhesion to endothelial cells226, an important early step in the formation of the early atherogenic lesion. The loss of 12-HETE in the 12/15-LOX null mouse may over-ride a possible role of the anti-inflammatory 15-lipoxygenase peroxides in murine atherogenic models due to a predominance of 12-lipoxygenase activity. Discrepancies between experimental data surrounding the murine 12/15-LOX and human 15-LOX genes, therefore, may potentially be explained by the differences in the lipoxygenase activities of the respective enzymes. Careful quantitative study of the lipoxygenase enzymes and lipid species by mass spectrometry in future studies to clarify the roles of lipid peroxides would help resolve conflicting data surrounding the 12/15-lipoxygenases and solidify their role in mediating protection against biological inflammatory diseases.
7.1.3 Nitroalkene fatty acids
Nitro fatty acids are also candidate natural ligands for PPAR-γ. The nitroalkene derivatives of linoleic acid (nitrolinoleic acid, LNO2) and oleic acid (nitro-oleic acid, OA-NO2) are formed via nitric oxide-dependent oxidative inflammatory reactions. LNO2 and OA-NO2 are endogenous ligands for PPAR-γ, robustly activating PPAR-γ-dependent reporter assays, endogenous target gene expression, and cell functions including adipocyte differentiation and glucose uptake227,228. Despite these functions in cell models, nitroalkene fatty acids would be expected to be short-lived inside the cell due to their capacity to undergo non-enzymatic Michael addition reactions. Both the nitroalkene derivatives of oleic acid (OA-NO2) and linoleic acid (LNO2) have been demonstrated to react readily with glutathione (GSH) and protein cysteine residues via Michael addition reactions229. Covalent modification of the PPAR-γ LBD at cysteine 285 has been demonstrated at micromolar concentrations230. Because glutathione is present at high concentrations in vivo, it is unclear whether nitro fatty acids are sufficiently stable to undergo this reaction at physiologic levels in order to activate PPAR-dependent transcription.
Nitro fatty acids have been demonstrated to show anti-inflammatory effects in several cell models. LNO2 and OA-NO2 prevent TNF-α-stimulated inflammatory and atherogenic responses in human umbilical vein endothelial cells (HUVECs) by blocked cytokine release and NF-κB activation231. In murine macrophage RAW264.7 cells, LNO2 and OA-NO2 exhibited anti-inflammatory effects by inhibiting LPS-induced STAT1 activation and suppressing Nos2 and Mcp-1 expression. This effect was not blocked by the addition of glutathione or GW9662, a PPAR-γ synthetic antagonist, suggesting a mechanism independent of thio-nitroalkylation or PPAR-γ232.
Whereas the first studies of LNO2 and OA-NO2 in the plasma of healthy subjects demonstrated concentrations of ~500 nM for free/nonesterified fatty acids as measured by LC-MS/MS228, other investigators demonstrated substantially lower concentrates by gas chromatography tandem mass spectrometry (GC-MS/MS). The two OA-NO2 isomers, 9-NO2-OA and 10-NO2-OA, were found at mean plasma concentrations of 880 and 940 pM, respectively233. A second study by the same group eliminating the high-performance liquid chromatography (HPLC) step to resolve the discrepancy between data sets by using a modified protocol demonstrated plasma OA-NO2 concentrations that were even lower but still in the pM range234. Furthermore, LNO2 occurred at lower concentrations than OA-NO2 and was found to be in the lower end of the pM range. While OA-NO2 is the most abundant nitrated unsaturated fatty acid in human plasma, the concentrations of both isomers in the pM range are far below those used in studies demonstrating anti-inflammatory and PPAR-γ-dependent effects. These findings raise questions about the endogenous roles of nitroalkene fatty acids. While nitroalkene fatty acids appear to act as potent agonists of PPAR-γ and have anti-inflammatory effects at high concentrations, their physiologic relevance remains to be established. Future studies in PPAR-γ and NO synthase deficient macrophages or other cell types would be helpful to establish the in vivo relevance and receptor-dependent effects of LNO2 and OA-NO2.
7.1.4 15-keto-prostaglandin E2
PGE2 is a short-lived mediator that is produced through the action of constitutive and inducible cyclooxygenase enzymes. The primary catabolic pathway of prostaglandins such as PGE2 is initiated by the oxidation of the 15(S)-hydroxyl group catalyzed by NAD+-dependent 15-hydroxyprostaglandin dehydrogenase (15-PGDH), generating 15-keto-prostaglandin E2 (15-keto-PGE2). Subsequent enzymatic metabolism of 15-keto-PGE2 takes place through the reduction of the Δ13 double bond catalyzed by the NADPH/NADH-dependent Δ13-15-ketoprostaglandin reductase (13-PGR)235. This later enzyme also exhibits NADP+-dependent leukotriene B4 12-hydroxydehydrogenase (12-LTB4DH) activity.
15-PGDH expression has been demonstrated in the prostate, uterus, liver, lung, adipocyte and gastrointestinal epithelium. 15-PGDH acts as a tumor suppressor and its expression is down-regulated in lung236, gastric237, colon238, and bladder cancers239. Over-expressing 15-PGDH induced apoptosis of A549 lung cancer cells236 and slowed the growth of SGC7901 gastric cancer cells240. Furthermore, mice with a null deletion of 15-PGDH were more susceptible to models of colon carcinogenesis241. It has not been determined which physiological effects of 15-PGDH deletion result from up-regulation of prostaglandins or concomitant down-regulation of oxidized prostaglandins.
While 15-keto-prostaglandins were generally thought to contain significantly reduced biological activity compared to their non-oxidized prostaglandins242, the dehydrogenase reaction produces an α,β-unsaturated ketone similar to that seen in 15d-PGJ2 that can undergo Michael addition with thiol groups. The oxidized prostaglandins 15-keto-PGE1, 15-keto-PGE2, 15-keto-PGF1, and 15-keto-PGF2 were all demonstrated to act as ligands for PPAR-γ in transient transfection assays168. The oxidized prostaglandins were more capable of activating the PPAR reporter when compared to their prostaglandin counterparts. 15-keto-PGE2 but not PGE2, 13,14-dihydro-15-keto-PGE2, nor 15-keto-PGF2a was capable of displacing rosiglitazone from binding the PPAR-γ LBD, suggesting that the former is the more effective endogenous ligand243 (Figure 3). 15-keto-PGE2 in the micromolar range (10 μM) was also demonstrated to induce adipogenesis of 3T3L1 cells in a similar manner as the synthetic PPAR-γ ligand rosiglitazone.
Recent work from our group confirms a role of 15-keto-PGE2 as a bona-fide endogenous ligand for PPAR-γ in the colon112. We found a defect in PPAR-γ mediated gene expression in the colonic epithelial cells and lungs of mice with a deletion in the cystic fibrosis transmembrane regulator (Cftr). While PPAR-γ protein levels were normal, recruitment of the co-activator TRAP220 to PPRE sites was reduced in Cftr-null colonic epithelial cells. We speculated that a defect in endogenous ligand levels might account for the reduced coactivator recruitment. We quantified 94 lipid analytes by LC-MS/MS and found that 15-HETE and 15-keto-PGE2 were the two most abundant species in the wild-type colonic epithelial cells that were previously implicated to act as PPAR-γ ligands in the low micromolar range. While 15-HETE abundance was unchanged between the wild type and Cftr-null cells, 15-keto-PGE2 abundance was reduced by 65% in Cftr-null cells. In concert with these results, we demonstrated 70% reduced expression of 15-PGDH in the Cftr-null cells.
We found levels of 15-keto-PGE2 in colonic tissue of 90 pg/mg tissue (~250 nM). While this is slightly below the micromolar range tested to be effective in transient assays, we demonstrated that the reduction of 15-keto-PGE2 correlated with reduced PPAR-γ target gene expression and co-activator TRAP220 recruitment at PPRE sites in Cftr-null cells. The results are consistent with a role of 15-keto-PGE2 as a physiologically relevant PPAR-γ ligand in the colon. Because 15-keto-PGE2 presumably interacts with PPAR-γ through the LBD cysteine via a Michael addition reaction (based on the presence of the α,β-unsaturated ketone), covalent LBD modification may account for the effectiveness of 15-keto-PGE2 in the sub-micromolar range. Alternatively, 15-keto-PGE2 may be concentrated in the nuclear compartment by binding to fatty acid binding proteins (e.g. L-FABP) where it activates PPAR-γ as has been demonstrated for cyclopentenone prostaglandins. Future research should establish the mechanism of PPAR-activation by 15-keto-PGE2 in the colon and other tissues where 15-PGDH is expressed.
7.1.5 Serotonin metabolites
Serotonin metabolites also act as endogenous agonists for PPAR-γ. The serotonin metabolite 5-methoxy-indole acetate (100 μM) was found to activate PPAR-γ mediated expression constructs in THP-1 macrophages and stimulate 3T3-L1 adipogenesis. 244 This study demonstrated that the 5-methoxy-indole acetate is bound to the AF-2 subpocket within the PPAR-γ LBD, while the other subpocket is simultaneously bound by a fatty acid metabolite. This suggests that PPAR-γ may function as a sensor for both the serotonin and fatty acid metabolic pathways. Serotonin levels are present in micromolar concentrations within the gastrointestinal tract and central nervous system, suggesting that the ligand may be present in these tissues. Future studies are needed to confirm the presence and functional activity of endogenous 5-methoxy-indole acetate in native tissues.
7.2 PPAR-γ antagonists
7.2.1 Prostaglandin F2α
PGF2α is synthesized by preadipocytes. It does not activative PPARs4,245 but potently inhibits adipocyte differentiation246,247. Endogenous production of PGF2α is lower in differentiating compared to uninduced preadipocytes. PGF2α utilizes the G-protein-coupled prostaglandin F receptors at the cell surface, initiating signal transduction through inositol triphosphate248,249. PGF2α was found to stimulate MAP kinase activation leading to phosphorylation of the PPAR-γ1 and PPAR-γ2 at serine 112 while indirectly antagonizing PPAR-γ induction and limiting adipocyte differentiation250. The data implicates PGF2α as a cellular control mechanism regulating the phosphorylation state of PPAR-γ. While PPAR-γ-S112A knock-in mice did not show increased weight or adipose mass compared to wild type mice, they maintained insulin sensitivity in the setting of diet-induced obesity251. The result suggests that compounds inhibiting PPAR-γ phosphorylation or regulating PGF2α may enhance insulin sensitivity without increasing body weight or adiposity, although additional investigations are needed to characterize the role of PGF2α in vivo.
7.2.2 Cyclic Phosphatidic Acid
Recent work established the first evidence of a direct endogenously produced PPAR-γ antagonist, cyclic phosphatidic acid (1-acyl-2,3-cyclic-glycerophosphate)252 (Figure 3). Cyclic phosphatidic acid (CPA) is generated in mammalian cells in a stimulus-coupled manner by phospholipase D2 (PLD2). CPA binds to a site within the PPAR-γ LBD with Kd in the hundred nanomolar range and prevents activation by PPAR-γ agonists such as TZDs. Upon binding CPA, the interaction with the corepressor SMRT is stabilized, reducing PPAR-γ activity, while the homologous PPAR-α and PPAR-δ receptors were neither inhibited nor activated by CPA. Although it is nearly undetectable in resting cells, CPA production was stimulated in human monocytes by insulin, phorbol myristate acetate (PMA), LPS or H2O2 through phospholipase D2. CPA was also shown to block TZD-stimulated adipogenesis and lipid accumulation in RAW264.7 and primary macrophages. CPA has been detected at concentrations of ~10 nM in human serum253. It is unknown how a given cell coordinates PPAR-γ activity through simultaneous production of endogenous ligands such as 15-keto-PGE2 in addition to ligand-binding antagonists such as CPA.
8. PPAR-α endogenous agonists
8.1 Conjugated linoleic acid
Conjugated linoleic acid (CLA) refers to a group of geometric and positional dienoic isomers of linoleic acid [18:2(n-6)] that is a natural fatty acid found in meats, cheeses and dairy products. The two predominant isomers of CLA found commonly in food and commercial preparations are 9Z,11E and 10E,12Z CLA. CLA has been shown to have potential beneficial effects in models of carcinogenesis, diabetes, colitis and atherosclerosis254. The 9Z,11E isomer constitutes up to 90% of total CLA and is thought to be responsible for the positive health benefits associated with CLA. These benefits include inhibition of allergic airway inflammation255, suppression of NF-κB activation in dendritic cells256, macrophages257, and colonic epithelial cells258, and suppression of inflammation in models of colitis259,260. These effects are at least in part mediated through enhanced production of IL-10256.
CLA isomers have also been proposed to act as ligands for PPARs. In hepatoma cells, for example, conjugated linoleic acid isomers were found to be more effective at inducing PPAR target genes and PPRE-reporter constructs than linoleate. Using a scintillation proximity assay to assess PPAR-α binding, a rank order of potency of (9Z,11E)>(10E,12Z)>(9E,11E)>furan-CLA was demonstrated, with IC50 ranging from 140 nM to 400 nM. While CLA isomers appear to be among the most avid fatty acid ligands for PPAR-α (IC50 = 140nM for the 9Z,11E isomer), the EC50 for activation of the PPAR-α target gene Cyp4a1 was >2 orders of magnitude higher (~ 50 μM)261, indicating that CLA may not be biologically available to act as a ligand due to metabolism effects such as excessive incorporation in phospholipid membranes or that it may be inefficient in recruiting co-activators required for gene activation despite binding to the receptor.
Conflicting results with respect to the CLA isomers and PPAR-activation have been demonstrated. While initial work suggested that CLA might activate PPAR-γ, as demonstrated by the effect of CLA on a PPAR-dependent reporter in rat CV-1 cells over-expressing PPAR-γ262, subsequent work demonstrated CLA isomer specific effects and antagonism of PPAR-γ. The 10E,12Z CLA isomer antagonized ligand-dependent activation of PPAR-γ-dependent target genes aP2 and Perilipin and reduced PPAR-γ expression in differentiating 3T3L1 adipocytes while the 9Z,11E isomer had no effect263 (possibly resulting from the low expression of PPAR-α in adipocytes). The authors went on to demonstrate that 10E,12Z CLA promoted NF-κB activation and IL-6 induction in adipocytes which was in part responsible for the down-regulation of PPAR-γ activity264. In contrast to this, CLA demonstrated anti-inflammatory effects in models of colitis. A mixture of CLA isomers reduced disease activity index scores, NF-κB activation and colonic inflammatory cytokine TNF-α production260. While the authors suggested that the effects were dependent upon PPARs, mechanistic data linking CLA isomers to PPAR-dependent transcription events was not clarified in epithelial or immune cells. Furthermore, recent epidemiological data has suggested that diets rich in linoleic acid were not protective and actually increased the risk of colitis in human patients265. However, ingested linoleic acid may largely be metabolized to arachidonic acid, forming substrates for cyclooxygenase (COX) and lipoxygenase (LOX) enzyme systems, rather than undergoing conversion to conjugated linoleic acid. Future mechanistic studies of the effects of specific CLA isomers on PPAR-dependent transcription and receptor binding in epithelial and immune cells would be helpful to unravel the roles of PPARs in mediating the effects of CLA on inflammation and gene activation.
8.2 16:0/18:1-GPC
PPAR-α is highly expressed in liver, where it regulates lipid transport, gluconeogenesis and fatty acid oxidation. PPAR-α has been demonstrated to respond to a variety of unsaturated and saturated fatty acids in reporter assays4. Because of its function in metabolism and data demonstrating binding to several fatty acids, PPAR-α was hypothesized to act as a sensor of nutritional status. Recently, a novel high affinity endogenous ligand for PPAR-α was identified in liver that links PPAR-α to the dietary carbohydrate responsive fatty acid synthase (FAS) responsible for palmitate (16:0) biosynthesis. A significant step towards this discovery was uncovered when it was demonstrated that liver-specific inactivation of fatty acid synthase (FAS) resulted in mice with decreased PPAR-α-dependent gene expression and a metabolic phenotype (hypoglycemia and steatohepatitis) resembling PPAR-α deficiency266. Subsequently, the authors identified the phospholipid 16:0/18:1-GPC (1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine) as the relevant PPAR-α-bound ligand generated by FAS using electrospray ionization mass spectrometry (Figure 4). They confirmed binding and co-activator recruitment to PPAR-α in vitro as well as acyl CoA oxidase (Acox1) and carnitine palmitoyl transferase (Cpt1a) induction in wild type but not PPAR-α-null mouse liver upon portal vein infusion of 16:0/18:1-GPC267. Binding affinity determined by scintillation proximity assay using 3H-Wy14,643 demonstrated IC50 = 33.25 μM. 16:0/18:1-GPC accounted for 4.4±1.2% of all nuclear phospholipids in liver.
Figure 4.
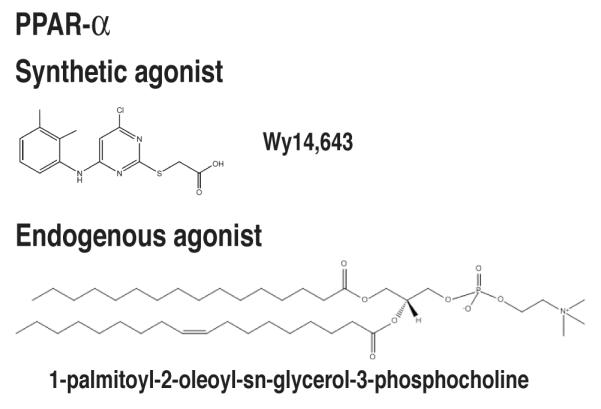
Natural and synthetic PPAR-α ligands. PPAR-α ligands include the synthetic agonist Wy14,643 and the endogenous agonist 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine.
In addition to CLA isomers and 16:0/18:1-GPC, the eicosanoid derivatives 8(S)-hydroxyeicosatetraenoic acid (HETE) and leukotriene B4 may act as PPAR-α ligands. 8(S)-HETE is the murine 8-lipoxygenase (8-LOX) product of arachidonic acid. The human 15-LOX-2 is regarded as the human ortholog of the mouse 8-LOX, sharing 78% identity in amino acid sequence. There is no overlap in the positional specificities of the two enzymes. The human 15-LOX-2 enzyme largely produces 15(S)-HETE rather than 8(S)-HETE268.
8.3 LTB4
Leukotriene B4 (LTB4) acts as a chemoattractant and secretagogue for PMNs, eosinophils and T lymphocytes269. Several reports have suggested that LTB4 might act as an endogenous PPAR-α agonist with affinities of Kd = 60-90 nM270,271 under specific conditions. One study suggested that LTB4 binds Xenopus PPAR-α with an affinity of 100 nM, although the IC50 for displacement was 10-50 mM of unlabeled LTB4270. A second group demonstrated that calcium-ionophore-treatment of Jurkat T cells activated a PPAR reporter construct in a process dependent upon 5-lipoxygenase, the enzyme responsible for LTB4 biosynthesis. Activation of the PPRE reporter by 10 μM LTB4 occurred in CV-1 cells as well, although PPAR target gene activation was not assessed272. In contrast to these findings, others have failed to show activation or binding to PPAR-α4. The differences in these studies have not been resolved such that LTB4 may play a role in PPAR activation under specific physiological conditions.
9. PPAR-δ endogenous agonists
Several synthetic agonists with high subtype selectivity have been described for PPAR-δ (Figure 5). The regulation of PPAR-δ by endogenous ligands, however, remains uncertain, largely due to the propensity of the receptor to bind a large number of lipid species with low affinity. Prostacyclin (PGI2) 273,274 and all-trans retinoic acid275 have been proposed to act as PPAR-δ ligands, however, conflicting results were found by other investigators4,276-278. Several fatty acids have been reported to induce PPAR-δ activity, although receptor interaction and specificity has not been established. The crystal structure of PPAR-δ has been solved recently in interaction with the hydrophobic lipids 5Z,8Z,11Z,14Z,17Z-eicosapentaenoic acid (EPA) and the synthetic agonist GW2433. The structure demonstrated that the ligand-binding pocket of PPAR-δ is sufficiently large to allow the hydrophobic tail of EPA to bind in multiple configurations. The carboxylic acid of EPA interacts with the AF-2 helix in a similar fashion of rosiglitzone interacting to PPAR-γ43. A second study demonstrated PPAR-δ-dependent effects of 15(S)-HETE treatment in fibroblasts. 15(S)-HETE induced the PPAR target gene and coactivator recruitment to the Angptl4 PPRE in fibroblasts in a PPAR-δ-dependent manner279. It is unclear if this effect is specific to fibroblasts. Whether anti-inflammatory effects of these putative PPAR-δ ligands are PPAR-dependent also remains to be established.
10. Conclusion
A number of recent studies have highlighted the increasing complexity of PPAR-mediated signaling. PPARs mediate transcriptional activation and repression of gene expression. Multiple molecular mechanisms contribute to the modulation of PPAR activity in a given cell type that regulate PPAR function. Lipid species have emerged as key regulators of PPAR activity. Comparisons between receptor-ligand affinities and the physiological concentrations of candidate ligands as measured by liquid chromatography/tandem mass spectrometry may serve as a guide for determining the relevance of putative ligands for a given PPAR receptor in a particular tissue. Additional studies of the potential therapeutic applications of natural PPAR-γ ligands, such as 15-keto-prostaglandin E2 and 15-deoxy-Δ12,14-prostaglandin J2, are needed because of the side effect profile of synthetic TZD agonists. Modulation of the activity of the synthetic pathways of these endogenous ligands may also prove to be of therapeutic interest as anti-inflammatory modulators.
Table 2.
PPAR- ligands
| Ligand | Activity | Biological Activity | Ref |
|---|---|---|---|
| Congulated linoleic acid (CLA) |
agonist | Inhibition of allergic airway inflammation | 255 |
| Suppression of NF-κB activation in dendritic cells, macrophages, and colonic epithelial cells |
256-258 | ||
| Suppression of inflammation in colitis models | 258 | ||
| 16:0/18:1-glycerol-3- phosphocholine |
agonist | Mice liver-specific knockout of fatty acid synthase (FAS), responsible for palmitate (16:0) biosysnthesis, have hypoglycemia and steatohepatitis, phenotypes similar to PPAR-α deficiency. |
266 |
| Induces expression of Acox1 and Cpt1a | 267 | ||
| Leukotriene B4 | agonist | Induces expression of PPAR-α regulated gene acyl-Co-A oxidase in primary hepatocytes. |
270 |
| Implicated in activating its turnover in liver by promoting expression of catabolic genes through PPAR-α. In an ear-swelling model, knockout of PPAR-α prolonged inflammatory phased induced by arachidonic acid or LTB4. |
Author Biographies

Gregory S. Harmon received his B.S. in Molecular Biology in 1994 from the University of California, San Diego. He completed his M.D. degree from the University of Chicago, Pritzker School of Medicine in 1999 and medical residency training at Northwestern University in 2002.
In 2005, he completed gastroenterology fellowship training at the University of California, San Diego. His postdoctoral studies were completed in the laboratory of Professor Christopher Glass in the Cellular and Molecular Medicine department at UCSD. He is Assistant Professor in the Division of Digestive Diseases at the University of California, Los Angeles. His research involves the study of inflammatory gastrointestinal disorders and the roles of lipids as regulators of peroxisome proliferator-activated receptors (PPARs).

Michael T. Lam received his B.A. in Molecular and Cellular Biology in 2003 from the University of California, Berkeley. He is currently in the Medical Scientist Training Program in UCSD School of Medicine. His graduate study is being completed in the laboratory of Christopher Glass in UCSD. His research involves the study of nuclear receptors in regulation of macrophage gene expression.

Christopher K. Glass is Professor of Cellular and Molecular Medicine and Professor of Medicine at the University of California, San Diego School of Medicine. His laboratory investigates the mechanisms by which sequence-specific transcription factors and associated co-activator and co-repressor proteins regulate the development and function of macrophages. A major focus is on members of the nuclear receptor family of transcription factors, particularly PPARs, which are regulated by fatty acid metabolites, and LXRs, which are regulated by oxysterols. Dr. Glass utilizes a combination of biochemical, cellular and in vivo model systems, incorporating macrophage-specific knockouts, genomic technologies and bioinformatics approaches, to unravel the contributions of these transcription factors to the regulation of innate immune responses and the pathogenesis of inflammatory diseases.
Footnotes
Disclosures: None of the authors has a conflict of interest to disclose.
REFERENCES
- (1).Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P. Cell. 1995;83:835. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, Umesono K, Evans RM. Proc. Natl. Acad. Sci. U.S.A. 1994;91:7355. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Chawla A, Schwarz EJ, Dimaculangan DD, Lazar MA. Endocrinology. 1994;135:798. doi: 10.1210/endo.135.2.8033830. [DOI] [PubMed] [Google Scholar]
- (4).Forman BM, Chen J, Evans RM. Proc. Natl. Acad. Sci. U.S.A. 1997;94:4312. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bain DL, Heneghan AF, Connaghan-Jones KD, Miura MT. Annu Rev Physiol. 2007;69:201. doi: 10.1146/annurev.physiol.69.031905.160308. [DOI] [PubMed] [Google Scholar]
- (6).Osumi T, Wen JK, Hashimoto T. Biochem Biophys Res Commun. 1991;175:866. doi: 10.1016/0006-291x(91)91645-s. [DOI] [PubMed] [Google Scholar]
- (7).Straus DS, Glass CK. Trends Immuno. 2007;28:551. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- (8).Mandard S, Muller M, Kersten S. Cell Mol Life Sci. 2004;61:393. doi: 10.1007/s00018-003-3216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, Delerive P, Fadel A, Chinetti G, Fruchart JC, Najib J, Maclouf J, Tedgui A. Nature. 1998;393:790. doi: 10.1038/31701. [DOI] [PubMed] [Google Scholar]
- (10).Delerive P, Martin-Nizard F, Chinetti G, Trottein F, Fruchart JC, Najib J, Duriez P, Staels B. Circ Res. 1999;85:394. doi: 10.1161/01.res.85.5.394. [DOI] [PubMed] [Google Scholar]
- (11).Peters JM, Lee SS, Li W, Ward JM, Gavrilova O, Everett C, Reitman ML, Hudson LD, Gonzalez FJ. Mol Cell Biol. 2000;20:5119. doi: 10.1128/mcb.20.14.5119-5128.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, Evans RM. Cell. 2003;113:159. doi: 10.1016/s0092-8674(03)00269-1. [DOI] [PubMed] [Google Scholar]
- (13).Lee CH, Chawla A, Urbiztondo N, Liao D, Boisvert WA, Evans RM, Curtiss LK. Science. 2003;302:453. doi: 10.1126/science.1087344. [DOI] [PubMed] [Google Scholar]
- (14).Harman FS, Nicol CJ, Marin HE, Ward JM, Gonzalez FJ, Peters JM. Nat Med. 2004;10:481. doi: 10.1038/nm1026. [DOI] [PubMed] [Google Scholar]
- (15).Tontonoz P, Hu E, Spiegelman BM. Cell. 1994;79:1147. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- (16).Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM. Mol Cell. 1999;4:585. doi: 10.1016/s1097-2765(00)80209-9. [DOI] [PubMed] [Google Scholar]
- (17).Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM. Mol Cell. 1999;4:611. doi: 10.1016/s1097-2765(00)80211-7. [DOI] [PubMed] [Google Scholar]
- (18).Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. Nature. 1998;391:79. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- (19).Jiang C, Ting AT, Seed B. Nature. 1998;391:82. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- (20).Su CG, Wen X, Bailey ST, Jiang W, Rangwala SM, Keilbaugh SA, Flanigan A, Murthy S, Lazar MA, Wu GD. J Clin Invest. 1999;104:383. doi: 10.1172/JCI7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Deeb SS, Fajas L, Nemoto M, Pihlajamaki J, Mykkanen L, Kuusisto J, Laakso M, Fujimoto W, Auwerx J. Nat Genet. 1998;20:284. doi: 10.1038/3099. [DOI] [PubMed] [Google Scholar]
- (22).Yamauchi T, Kamon J, Waki H, Murakami K, Motojima K, Komeda K, Ide T, Kubota N, Terauchi Y, Tobe K, Miki H, Tsuchida A, Akanuma Y, Nagai R, Kimura S, Kadowaki T. J Biol Chem. 2001;276:41245. doi: 10.1074/jbc.M103241200. [DOI] [PubMed] [Google Scholar]
- (23).Diep QN, El Mabrouk M, Cohn JS, Endemann D, Amiri F, Virdis A, Neves MF, Schiffrin EL. Circulation. 2002;105:2296. doi: 10.1161/01.cir.0000016049.86468.23. [DOI] [PubMed] [Google Scholar]
- (24).Mazzone T, Meyer PM, Feinstein SB, Davidson MH, Kondos GT, D’Agostino RB, Sr., Perez A, Provost JC, Haffner SM. JAMA. 2006;296:2572. doi: 10.1001/jama.296.21.joc60158. [DOI] [PubMed] [Google Scholar]
- (25).Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK. J Clin Invest. 2000;106:523. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Guan Y, Hao C, Cha DR, Rao R, Lu W, Kohan DE, Magnuson MA, Redha R, Zhang Y, Breyer MD. Nat Med. 2005;11:861. doi: 10.1038/nm1278. [DOI] [PubMed] [Google Scholar]
- (27).Home PD, Pocock SJ, Beck-Nielsen H, Gomis R, Hanefeld M, Jones NP, Komajda M, McMurray JJ. N Engl J Med. 2007;357:28. doi: 10.1056/NEJMoa073394. [DOI] [PubMed] [Google Scholar]
- (28).Lincoff AM, Wolski K, Nicholls SJ, Nissen SE. JAMA. 2007;298:1180. doi: 10.1001/jama.298.10.1180. [DOI] [PubMed] [Google Scholar]
- (29).Nissen SE, Wolski K. Arch Intern Med. 2010;170:1191. doi: 10.1001/archinternmed.2010.207. [DOI] [PubMed] [Google Scholar]
- (30).Loke YK, Kwok CS, Singh S. BMJ. 2011;342:d1309. doi: 10.1136/bmj.d1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Khorasanizadeh S, Rastinejad F. Trends Biochem Sci. 2001;26:384. doi: 10.1016/s0968-0004(01)01800-x. [DOI] [PubMed] [Google Scholar]
- (32).Danielian PS, White R, Lees JA, Parker MG. EMBO J. 1992;11:1025. doi: 10.1002/j.1460-2075.1992.tb05141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Westin S, Kurokawa R, Nolte RT, Wisely GB, McInerney EM, Rose DW, Milburn MV, Rosenfeld MG, Glass CK. Nature. 1998;395:199. doi: 10.1038/26040. [DOI] [PubMed] [Google Scholar]
- (34).Bourguet W, Germain P, Gronemeyer H. Trends Pharmacol Sci. 2000;21:381. doi: 10.1016/s0165-6147(00)01548-0. [DOI] [PubMed] [Google Scholar]
- (35).Hummasti S, Tontonoz P. Mol Endocrinol. 2006;20:1261. doi: 10.1210/me.2006-0025. [DOI] [PubMed] [Google Scholar]
- (36).Bourguet W, Ruff M, Chambon P, Gronemeyer H, Moras D. Nature. 1995;375:377. doi: 10.1038/375377a0. [DOI] [PubMed] [Google Scholar]
- (37).Wagner RL, Apriletti JW, McGrath ME, West BL, Baxter JD, Fletterick RJ. Nature. 1995;378:690. doi: 10.1038/378690a0. [DOI] [PubMed] [Google Scholar]
- (38).Renaud JP, Rochel N, Ruff M, Vivat V, Chambon P, Gronemeyer H, Moras D. Nature. 1995;378:681. doi: 10.1038/378681a0. [DOI] [PubMed] [Google Scholar]
- (39).Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Nature. 1997;389:753. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- (40).Li Y, Lambert MH, Xu HE. Structure. 2003;11:741. doi: 10.1016/s0969-2126(03)00133-3. [DOI] [PubMed] [Google Scholar]
- (41).Noy N. Biochemistry. 2007;46:13461. doi: 10.1021/bi7018699. [DOI] [PubMed] [Google Scholar]
- (42).Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, Milburn MV. Nature. 1998;395:137. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- (43).Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, Brown PJ, Sternbach DD, Lehmann JM, Wisely GB, Willson TM, Kliewer SA, Milburn MV. Mol Cell. 1999;3:397. doi: 10.1016/s1097-2765(00)80467-0. [DOI] [PubMed] [Google Scholar]
- (44).Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. Science. 2001;292:2329. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- (45).Xu HE, Lambert MH, Montana VG, Plunket KD, Moore LB, Collins JL, Oplinger JA, Kliewer SA, Gampe RT, Jr., McKee DD, Moore JT, Willson TM. Proc. Natl. Acad. Sci. U.S.A. 2001;98:13919. doi: 10.1073/pnas.241410198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Gampe RT, Jr., Montana VG, Lambert MH, Miller AB, Bledsoe RK, Milburn MV, Kliewer SA, Willson TM, Xu HE. Mol Cell. 2000;5:545. doi: 10.1016/s1097-2765(00)80448-7. [DOI] [PubMed] [Google Scholar]
- (47).Chandra V, Huang P, Hamuro Y, Raghuram S, Wang Y, Burris TP, Rastinejad F. Nature. 2008;456:350. doi: 10.1038/nature07413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Kliewer SA, Umesono K, Mangelsdorf DJ, Evans RM. Nature. 1992;355:446. doi: 10.1038/355446a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Keller H, Dreyer C, Medin J, Mahfoudi A, Ozato K, Wahli W. Proc. Natl. Acad. Sci. U.S.A. 1993;90:2160. doi: 10.1073/pnas.90.6.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).IJpenberg A, Jeannin E, Wahli W, Desvergne B. J Biol Chem. 1997;272:20108. doi: 10.1074/jbc.272.32.20108. [DOI] [PubMed] [Google Scholar]
- (51).Lefterova MI, Steger DJ, Zhuo D, Qatanani M, Mullican SE, Tuteja G, Manduchi E, Grant GR, Lazar MA. Mol Cell Biol. 2010;30:2078. doi: 10.1128/MCB.01651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Schoonjans K, Peinado-Onsurbe J, Lefebvre AM, Heyman RA, Briggs M, Deeb S, Staels B, Auwerx J. EMBO J. 1996;15:5336. [PMC free article] [PubMed] [Google Scholar]
- (53).Shimizu M, Yamashita D, Yamaguchi T, Hirose F, Osumi T. Mol Cell Biochem. 2006;286:33. doi: 10.1007/s11010-005-9052-z. [DOI] [PubMed] [Google Scholar]
- (54).Yu S, Matsusue K, Kashireddy P, Cao WQ, Yeldandi V, Yeldandi AV, Rao MS, Gonzalez FJ, Reddy JK. J Biol Chem. 2003;278:498. doi: 10.1074/jbc.M210062200. [DOI] [PubMed] [Google Scholar]
- (55).Werman A, Hollenberg A, Solanes G, Bjorbaek C, Vidal-Puig AJ, Flier JS. J Biol Chem. 1997;272:20230. doi: 10.1074/jbc.272.32.20230. [DOI] [PubMed] [Google Scholar]
- (56).Medina-Gomez G, Gray SL, Yetukuri L, Shimomura K, Virtue S, Campbell M, Curtis RK, Jimenez-Linan M, Blount M, Yeo GS, Lopez M, Seppanen-Laakso T, Ashcroft FM, Oresic M, Vidal-Puig A. PLoS Genet. 2007;3:e64. doi: 10.1371/journal.pgen.0030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Castillo G, Brun RP, Rosenfield JK, Hauser S, Park CW, Troy AE, Wright ME, Spiegelman BM. EMBO J. 1999;18:3676. doi: 10.1093/emboj/18.13.3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Mueller E, Drori S, Aiyer A, Yie J, Sarraf P, Chen H, Hauser S, Rosen ED, Ge K, Roeder RG, Spiegelman BM. J Biol Chem. 2002;277:41925. doi: 10.1074/jbc.M206950200. [DOI] [PubMed] [Google Scholar]
- (59).Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. J Biol Chem. 1997;272:5128. doi: 10.1074/jbc.272.8.5128. [DOI] [PubMed] [Google Scholar]
- (60).Hu E, Kim JB, Sarraf P, Spiegelman BM. Science. 1996;274:2100. doi: 10.1126/science.274.5295.2100. [DOI] [PubMed] [Google Scholar]
- (61).Camp HS, Tafuri SR. J Biol Chem. 1997;272:10811. doi: 10.1074/jbc.272.16.10811. [DOI] [PubMed] [Google Scholar]
- (62).Camp HS, Tafuri SR, Leff T. Endocrinology. 1999;140:392. doi: 10.1210/endo.140.1.6457. [DOI] [PubMed] [Google Scholar]
- (63).Zhang B, Berger J, Zhou G, Elbrecht A, Biswas S, White-Carrington S, Szalkowski D, Moller DE. J Biol Chem. 1996;271:31771. doi: 10.1074/jbc.271.50.31771. [DOI] [PubMed] [Google Scholar]
- (64).Compe E, Drane P, Laurent C, Diderich K, Braun C, Hoeijmakers JH, Egly JM. Mol Cell Biol. 2005;25:6065. doi: 10.1128/MCB.25.14.6065-6076.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Iankova I, Petersen RK, Annicotte JS, Chavey C, Hansen JB, Kratchmarova I, Sarruf D, Benkirane M, Kristiansen K, Fajas L. Mol Endocrinol. 2006;20:1494. doi: 10.1210/me.2005-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, Griffin PR, Spiegelman BM. Nature. 2010;466:451. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Poukka H, Karvonen U, Janne OA, Palvimo JJ. Proc. Natl. Acad. Sci. U.S.A. 2000;97:14145. doi: 10.1073/pnas.97.26.14145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Tian S, Poukka H, Palvimo JJ, Janne OA. Biochem J. 2002;367:907. doi: 10.1042/BJ20021085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, Rosenfeld MG, Glass CK. Mol Cell. 2007;25:57. doi: 10.1016/j.molcel.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Yamashita D, Yamaguchi T, Shimizu M, Nakata N, Hirose F, Osumi T. Genes Cells. 2004;9:1017. doi: 10.1111/j.1365-2443.2004.00786.x. [DOI] [PubMed] [Google Scholar]
- (71).Ohshima T, Koga H, Shimotohno K. J Biol Chem. 2004;279:29551. doi: 10.1074/jbc.M403866200. [DOI] [PubMed] [Google Scholar]
- (72).Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. Nature. 2005;437:759. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Hauser S, Adelmant G, Sarraf P, Wright HM, Mueller E, Spiegelman BM. J Biol Chem. 2000;275:18527. doi: 10.1074/jbc.M001297200. [DOI] [PubMed] [Google Scholar]
- (74).Waite KJ, Floyd ZE, Arbour-Reily P, Stephens JM. J Biol Chem. 2001;276:7062. doi: 10.1074/jbc.M007894200. [DOI] [PubMed] [Google Scholar]
- (75).Floyd ZE, Stephens JM. J Biol Chem. 2002;277:4062. doi: 10.1074/jbc.M108473200. [DOI] [PubMed] [Google Scholar]
- (76).Diradourian C, Girard J, Pegorier JP. Biochimie. 2005;87:33. doi: 10.1016/j.biochi.2004.11.010. [DOI] [PubMed] [Google Scholar]
- (77).Blanquart C, Barbier O, Fruchart JC, Staels B, Glineur C. J Biol Chem. 2002;277:37254. doi: 10.1074/jbc.M110598200. [DOI] [PubMed] [Google Scholar]
- (78).Leuenberger N, Pradervand S, Wahli W. J Clin Invest. 2009;119:3138. doi: 10.1172/JCI39019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Pourcet B, Pineda-Torra I, Derudas B, Staels B, Glineur C. J Biol Chem. 2010;285:5983. doi: 10.1074/jbc.M109.078311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Torchia J, Glass C, Rosenfeld MG. Curr Opin Cell Biol. 1998;10:373. doi: 10.1016/s0955-0674(98)80014-8. [DOI] [PubMed] [Google Scholar]
- (81).Powell E, Kühn P, Xu W. PPAR Res. 2007;2007:53843. doi: 10.1155/2007/53843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Zhu Y, Qi C, Calandra C, Rao MS, Reddy JK. Gene Expr. 1996;6:185. [PMC free article] [PubMed] [Google Scholar]
- (83).Voegel JJ, Heine MJ, Zechel C, Chambon P, Gronemeyer H. EMBO J. 1996;15:3667. [PMC free article] [PubMed] [Google Scholar]
- (84).Hong H, Kohli K, Garabedian MJ, Stallcup MR. Mol Cell Biol. 1997;17:2735. doi: 10.1128/mcb.17.5.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG. Nature. 1997;387:677. doi: 10.1038/42652. [DOI] [PubMed] [Google Scholar]
- (86).Heery DM, Hoare S, Hussain S, Parker MG, Sheppard H. J Biol Chem. 2001;276:6695. doi: 10.1074/jbc.M009404200. [DOI] [PubMed] [Google Scholar]
- (87).Heery DM, Kalkhoven E, Hoare S, Parker MG. Nature. 1997;387:733. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- (88).Voegel JJ, Heine MJ, Tini M, Vivat V, Chambon P, Gronemeyer H. EMBO J. 1998;17:507. doi: 10.1093/emboj/17.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Hu X, Lazar MA. Nature. 1999;402:93. doi: 10.1038/47069. [DOI] [PubMed] [Google Scholar]
- (90).Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. Cell. 1996;87:953. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- (91).van Beekum O, Brenkman AB, Grontved L, Hamers N, van den Broek NJ, Berger R, Mandrup S, Kalkhoven E. Endocrinology. 2008;149:1840. doi: 10.1210/en.2007-0977. [DOI] [PubMed] [Google Scholar]
- (92).Takahashi Y, Ohoka N, Hayashi H, Sato R. J Lipid Res. 2008;49:880. doi: 10.1194/jlr.M700545-JLR200. [DOI] [PubMed] [Google Scholar]
- (93).Bugge A, Grontved L, Aagaard MM, Borup R, Mandrup S. Mol Endocrinol. 2009;23:794. doi: 10.1210/me.2008-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Glass CK, Rosenfeld MG. Genes Dev. 2000;14:121. [PubMed] [Google Scholar]
- (95).Yu C, Markan K, Temple KA, Deplewski D, Brady MJ, Cohen RN. J Biol Chem. 2005;280:13600. doi: 10.1074/jbc.M409468200. [DOI] [PubMed] [Google Scholar]
- (96).Nielsen R, Grontved L, Stunnenberg HG, Mandrup S. Mol Cell Biol. 2006;26:5698. doi: 10.1128/MCB.02266-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Nielsen R, Pedersen TA, Hagenbeek D, Moulos P, Siersbaek R, Megens E, Denissov S, Borgesen M, Francoijs KJ, Mandrup S, Stunnenberg HG. Genes Dev. 2008;22:2953. doi: 10.1101/gad.501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Lefterova MI, Zhang Y, Steger DJ, Schupp M, Schug J, Cristancho A, Feng D, Zhuo D, Stoeckert CJ, Jr., Liu XS, Lazar MA. Genes Dev. 2008;22:2941. doi: 10.1101/gad.1709008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. Cell. 2007;129:823. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- (100).Trojer P, Reinberg D. Mol Cell. 2007;28:1. doi: 10.1016/j.molcel.2007.09.011. [DOI] [PubMed] [Google Scholar]
- (101).Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, Staels B, Chinetti-Gbaguidi G. Cell Metab. 2007;6:137. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- (102).Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, Chawla A. Nature. 2007;447:1116. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, Liao D, Nagy L, Edwards PA, Curtiss LK, Evans RM, Tontonoz P. Mol Cell. 2001;7:161. doi: 10.1016/s1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- (104).Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, Teissier E, Minnich A, Jaye M, Duverger N, Brewer HB, Fruchart JC, Clavey V, Staels B. Nat Med. 2001;7:53. doi: 10.1038/83348. [DOI] [PubMed] [Google Scholar]
- (105).Klotz L, Hucke S, Thimm D, Classen S, Gaarz A, Schultze J, Edenhofer F, Kurts C, Klockgether T, Limmer A, Knolle P, Burgdorf S. J Immunol. 2009;183:129. doi: 10.4049/jimmunol.0804260. [DOI] [PubMed] [Google Scholar]
- (106).Nencioni A, Grunebach F, Zobywlaski A, Denzlinger C, Brugger W, Brossart P. J Immunol. 2002;169:1228. doi: 10.4049/jimmunol.169.3.1228. [DOI] [PubMed] [Google Scholar]
- (107).Guan HP, Ishizuka T, Chui PC, Lehrke M, Lazar MA. Genes Dev. 2005;19:453. doi: 10.1101/gad.1263305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Ge K, Guermah M, Yuan CX, Ito M, Wallberg AE, Spiegelman BM, Roeder RG. Nature. 2002;417:563. doi: 10.1038/417563a. [DOI] [PubMed] [Google Scholar]
- (109).Gelman L, Zhou G, Fajas L, Raspe E, Fruchart JC, Auwerx J. J Biol Chem. 1999;274:7681. doi: 10.1074/jbc.274.12.7681. [DOI] [PubMed] [Google Scholar]
- (110).Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV, Stewart R, Thomson JA, Crawford GE, Kellis M, Ren B. Nature. 2009;459:108. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. Nat Genet. 2007;39:311. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- (112).Harmon GS, Dumlao DS, Ng DT, Barrett KE, Dennis EA, Dong H, Glass CK. Nat Med. 2010;16:313. doi: 10.1038/nm.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Urade Y, Ujihara M, Horiguchi Y, Ikai K, Hayaishi O. J Immunol. 1989;143:2982. [PubMed] [Google Scholar]
- (114).Berry A, Balard P, Coste A, Olagnier D, Lagane C, Authier H, Benoit-Vical F, Lepert JC, Seguela JP, Magnaval JF, Chambon P, Metzger D, Desvergne B, Wahli W, Auwerx J, Pipy B. Eur J Immunol. 2007;37:1642. doi: 10.1002/eji.200636625. [DOI] [PubMed] [Google Scholar]
- (115).Shibata T, Kondo M, Osawa T, Shibata N, Kobayashi M, Uchida K. J Biol Chem. 2002;277:10459. doi: 10.1074/jbc.M110314200. [DOI] [PubMed] [Google Scholar]
- (116).Barnes PJ, Karin M. N Engl J Med. 1997;336:1066. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- (117).Smith WL. Biochem J. 1989;259:315. doi: 10.1042/bj2590315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Bohmann D, Bos TJ, Admon A, Nishimura T, Vogt PK, Tjian R. Science. 1987;238:1386. doi: 10.1126/science.2825349. [DOI] [PubMed] [Google Scholar]
- (119).Angel P, Allegretto EA, Okino ST, Hattori K, Boyle WJ, Hunter T, Karin M. Nature. 1988;332:166. doi: 10.1038/332166a0. [DOI] [PubMed] [Google Scholar]
- (120).Welch JS, Ricote M, Akiyama TE, Gonzalez FJ, Glass CK. Proc. Natl. Acad. Sci. U.S.A. 2003;100:6712. doi: 10.1073/pnas.1031789100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, Hoffmann A, Subramaniam S, David M, Rosenfeld MG, Glass CK. Cell. 2005;122:707. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Shu H, Wong B, Zhou G, Li Y, Berger J, Woods JW, Wright SD, Cai TQ. Biochem Biophys Res Commun. 2000;267:345. doi: 10.1006/bbrc.1999.1968. [DOI] [PubMed] [Google Scholar]
- (123).Jonat C, Rahmsdorf HJ, Park KK, Cato AC, Gebel S, Ponta H, Herrlich P. Cell. 1990;62:1189. doi: 10.1016/0092-8674(90)90395-u. [DOI] [PubMed] [Google Scholar]
- (124).Yang-Yen HF, Chambard JC, Sun YL, Smeal T, Schmidt TJ, Drouin J, Karin M. Cell. 1990;62:1205. doi: 10.1016/0092-8674(90)90396-v. [DOI] [PubMed] [Google Scholar]
- (125).Schüle R, Rangarajan P, Kliewer S, Ransone LJ, Bolado J, Yang N, Verma IM, Evans RM. Cell. 1990;62:1217. doi: 10.1016/0092-8674(90)90397-w. [DOI] [PubMed] [Google Scholar]
- (126).Ray A, Prefontaine KE. Proc. Natl. Acad. Sci. U.S.A. 1994;91:752. doi: 10.1073/pnas.91.2.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Scheinman RI, Gualberto A, Jewell CM, Cidlowski JA, Baldwin AS., Jr. Mol Cell Biol. 1995;15:943. doi: 10.1128/mcb.15.2.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Salbert G, Fanjul A, Piedrafita FJ, Lu XP, Kim SJ, Tran P, Pfahl M. Mol Endocrinol. 1993;7:1347. doi: 10.1210/mend.7.10.8264664. [DOI] [PubMed] [Google Scholar]
- (129).Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, Fruchart JC, Tedgui A, Haegeman G, Staels B. J Biol Chem. 1999;274:32048. doi: 10.1074/jbc.274.45.32048. [DOI] [PubMed] [Google Scholar]
- (130).Delerive P, Gervois P, Fruchart JC, Staels B. J Biol Chem. 2000;275:36703. doi: 10.1074/jbc.M004045200. [DOI] [PubMed] [Google Scholar]
- (131).Vanden Berghe W, Vermeulen L, Delerive P, De Bosscher K, Staels B, Haegeman G. Adv Exp Med Biol. 2003;544:181. doi: 10.1007/978-1-4419-9072-3_22. [DOI] [PubMed] [Google Scholar]
- (132).Kleemann R, Gervois PP, Verschuren L, Staels B, Princen HM, Kooistra T. Blood. 2003;101:545. doi: 10.1182/blood-2002-06-1762. [DOI] [PubMed] [Google Scholar]
- (133).Chung SW, Kang BY, Kim SH, Pak YK, Cho D, Trinchieri G, Kim TS. J Biol Chem. 2000;275:32681. doi: 10.1074/jbc.M002577200. [DOI] [PubMed] [Google Scholar]
- (134).Kelly D, Campbell JI, King TP, Grant G, Jansson EA, Coutts AG, Pettersson S, Conway S. Nat Immunol. 2004;5:104. doi: 10.1038/ni1018. [DOI] [PubMed] [Google Scholar]
- (135).Zingarelli B, Sheehan M, Hake PW, O’Connor M, Denenberg A, Cook JA. J Immunol. 2003;171:6827. doi: 10.4049/jimmunol.171.12.6827. [DOI] [PubMed] [Google Scholar]
- (136).Pulverer BJ, Kyriakis JM, Avruch J, Nikolakaki E, Woodgett JR. Nature. 1991;353:670. doi: 10.1038/353670a0. [DOI] [PubMed] [Google Scholar]
- (137).Smeal T, Binetruy B, Mercola DA, Birrer M, Karin M. Nature. 1991;354:494. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- (138).Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. Cell. 1994;76:1025. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- (139).Caelles C, Gonzalez-Sancho JM, Munoz A. Genes Dev. 1997;11:3351. doi: 10.1101/gad.11.24.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Gonzalez MV, Jimenez B, Berciano MT, Gonzalez-Sancho JM, Caelles C, Lafarga M, Munoz A. J Cell Biol. 2000;150:1199. doi: 10.1083/jcb.150.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Desreumaux P, Dubuquoy L, Nutten S, Peuchmaur M, Englaro W, Schoonjans K, Derijard B, Desvergne B, Wahli W, Chambon P, Leibowitz MD, Colombel JF, Auwerx J. J Exp Med. 2001;193:827. doi: 10.1084/jem.193.7.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Ramanan S, Kooshki M, Zhao W, Hsu FC, Robbins ME. Free Radic Biol Med. 2008;45:1695. doi: 10.1016/j.freeradbiomed.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Uchimura K, Nakamuta M, Enjoji M, Irie T, Sugimoto R, Muta T, Iwamoto H, Nawata H. Hepatology. 2001;33:91. doi: 10.1053/jhep.2001.21145. [DOI] [PubMed] [Google Scholar]
- (144).Goetze S, Xi XP, Kawano H, Gotlibowski T, Fleck E, Hsueh WA, Law RE. J Cardiovasc Pharmacol. 1999;33:798. doi: 10.1097/00005344-199905000-00018. [DOI] [PubMed] [Google Scholar]
- (145).Arias J, Alberts AS, Brindle P, Claret FX, Smeal T, Karin M, Feramisco J, Montminy M. Nature. 1994;370:226. doi: 10.1038/370226a0. [DOI] [PubMed] [Google Scholar]
- (146).Zhong H, Voll RE, Ghosh S. Mol Cell. 1998;1:661. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
- (147).Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose DW, Glass CK, Rosenfeld MG. Cell. 1996;85:403. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- (148).Ghisletti S, Barozzi I, Mietton F, Polletti S, De Santa F, Venturini E, Gregory L, Lonie L, Chew A, Wei CL, Ragoussis J, Natoli G. Immunity. 2010;32:317. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- (149).De Bosscher K, Vanden Berghe W, Vermeulen L, Plaisance S, Boone E, Haegeman G. Proc. Natl. Acad. Sci. U.S.A. 2000;97:3919. doi: 10.1073/pnas.97.8.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).De Bosscher K, Vanden Berghe W, Haegeman G. Mol Endocrinol. 2001;15:219. doi: 10.1210/mend.15.2.0591. [DOI] [PubMed] [Google Scholar]
- (151).Kodera Y, Takeyama K, Murayama A, Suzawa M, Masuhiro Y, Kato S. J Biol Chem. 2000;275:33201. doi: 10.1074/jbc.C000517200. [DOI] [PubMed] [Google Scholar]
- (152).Li M, Pascual G, Glass CK. Mol Cell Biol. 2000;20:4699. doi: 10.1128/mcb.20.13.4699-4707.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Subbaramaiah K, Lin DT, Hart JC, Dannenberg AJ. J Biol Chem. 2001;276:12440. doi: 10.1074/jbc.M007237200. [DOI] [PubMed] [Google Scholar]
- (154).Kletzien RF, Clarke SD, Ulrich RG. Mol Pharmacol. 1992;41:393. [PubMed] [Google Scholar]
- (155).Oakes ND, Kennedy CJ, Jenkins AB, Laybutt DR, Chisholm DJ, Kraegen EW. Diabetes. 1994;43:1203. doi: 10.2337/diab.43.10.1203. [DOI] [PubMed] [Google Scholar]
- (156).Kletzien RF, Foellmi LA, Harris PK, Wyse BM, Clarke SD. Mol Pharmacol. 1992;42:558. [PubMed] [Google Scholar]
- (157).Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. Genes Dev. 1994;8:1224. doi: 10.1101/gad.8.10.1224. [DOI] [PubMed] [Google Scholar]
- (158).Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. J Biol Chem. 1995;270:12953. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- (159).Young PW, Buckle DR, Cantello BC, Chapman H, Clapham JC, Coyle PJ, Haigh D, Hindley RM, Holder JC, Kallender H, Latter AJ, Lawrie KW, Mossakowska D, Murphy GJ, Roxbee Cox L, Smith SA. J Pharmacol Exp Ther. 1998;284:751. [PubMed] [Google Scholar]
- (160).Mitchell MD, Flint AP, Bibby J, Brunt J, Arnold JM, Anderson AB, Turnbull AC. J Clin Endocrinol Metab. 1978;46:947. doi: 10.1210/jcem-46-6-947. [DOI] [PubMed] [Google Scholar]
- (161).Offenbacher S, Odle BM, Van Dyke TE. J Periodontal Res. 1986;21:101. doi: 10.1111/j.1600-0765.1986.tb01443.x. [DOI] [PubMed] [Google Scholar]
- (162).McIntyre TM, Pontsler AV, Silva AR, St Hilaire A, Xu Y, Hinshaw JC, Zimmerman GA, Hama K, Aoki J, Arai H, Prestwich GD. Proc. Natl. Acad. Sci. U.S.A. 2003;100:131. doi: 10.1073/pnas.0135855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Zhang C, Baker DL, Yasuda S, Makarova N, Balazs L, Johnson LR, Marathe GK, McIntyre TM, Xu Y, Prestwich GD, Byun HS, Bittman R, Tigyi G. J Exp Med. 2004;199:763. doi: 10.1084/jem.20031619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Zhao Y, Natarajan V. Cell Signal. 2009;21:367. doi: 10.1016/j.cellsig.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Fitzpatrick FA, Wynalda MA. J Biol Chem. 1983;258:11713. [PubMed] [Google Scholar]
- (166).Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. Cell. 1995;83:803. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- (167).Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. Cell. 1995;83:813. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- (168).Shiraki T, Kamiya N, Shiki S, Kodama TS, Kakizuka A, Jingami H. J Biol Chem. 2005;280:14145. doi: 10.1074/jbc.M500901200. [DOI] [PubMed] [Google Scholar]
- (169).Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, Sengchanthalangsy LL, Ghosh G, Glass CK. Proc. Natl. Acad. Sci. U.S.A. 2000;97:4844. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. Nat Med. 2001;7:48. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- (171).Moore KJ, Rosen ED, Fitzgerald ML, Randow F, Andersson LP, Altshuler D, Milstone DS, Mortensen RM, Spiegelman BM, Freeman MW. Nat Med. 2001;7:41. doi: 10.1038/83328. [DOI] [PubMed] [Google Scholar]
- (172).Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. Nature. 2000;403:103. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- (173).Castrillo A, Diaz-Guerra MJ, Hortelano S, Martin-Sanz P, Bosca L. Mol Cell Biol. 2000;20:1692. doi: 10.1128/mcb.20.5.1692-1698.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Delhase M, Hayakawa M, Chen Y, Karin M. Science. 1999;284:309. doi: 10.1126/science.284.5412.309. [DOI] [PubMed] [Google Scholar]
- (175).Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Nat Med. 1999;5:698. doi: 10.1038/9550. [DOI] [PubMed] [Google Scholar]
- (176).Kaplan JM, Cook JA, Hake PW, O’Connor M, Burroughs TJ, Zingarelli B. Shock. 2005;24:59. doi: 10.1097/01.shk.0000167108.88376.f2. [DOI] [PubMed] [Google Scholar]
- (177).Rajakariar R, Hilliard M, Lawrence T, Trivedi S, Colville-Nash P, Bellingan G, Fitzgerald D, Yaqoob MM, Gilroy DW. Proc. Natl. Acad. Sci. U.S.A. 2007;104:20979. doi: 10.1073/pnas.0707394104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Bell-Parikh LC, Ide T, Lawson JA, McNamara P, Reilly M, FitzGerald GA. J Clin Invest. 2003;112:945. doi: 10.1172/JCI18012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Fukushima M. Prostaglandins Leukot Essent Fatty Acids. 1992;47:1. doi: 10.1016/0952-3278(92)90178-l. [DOI] [PubMed] [Google Scholar]
- (180).Chan BS, Satriano JA, Pucci M, Schuster VL. J Biol Chem. 1998;273:6689. doi: 10.1074/jbc.273.12.6689. [DOI] [PubMed] [Google Scholar]
- (181).Chu HW, Balzar S, Westcott JY, Trudeau JB, Sun Y, Conrad DJ, Wenzel SE. Clin Exp Allergy. 2002;32:1558. doi: 10.1046/j.1365-2222.2002.01477.x. [DOI] [PubMed] [Google Scholar]
- (182).Natarajan R, Nadler JL. Arterioscler Thromb Vasc Biol. 2004;24:1542. doi: 10.1161/01.ATV.0000133606.69732.4c. [DOI] [PubMed] [Google Scholar]
- (183).Kühn H, Schewe T, Rapoport SM. Adv Enzymol Relat Areas Mol Biol. 1986;58:273. doi: 10.1002/9780470123041.ch7. [DOI] [PubMed] [Google Scholar]
- (184).Samuelsson B, Dahlen SE, Lindgren JA, Rouzer CA, Serhan CN. Science. 1987;237:1171. doi: 10.1126/science.2820055. [DOI] [PubMed] [Google Scholar]
- (185).Wittwer J, Hersberger M. Prostaglandins Leukot Essent Fatty Acids. 2007;77:67. doi: 10.1016/j.plefa.2007.08.001. [DOI] [PubMed] [Google Scholar]
- (186).Glavind J, Hartmann S, Clemmesen J, Jessen KE, Dam H. Acta Pathol Microbiol Scand. 1952;30:1. doi: 10.1111/j.1699-0463.1952.tb00157.x. [DOI] [PubMed] [Google Scholar]
- (187).Brooks CJ, Steel G, Gilbert JD, Harland WA. Atherosclerosis. 1971;13:223. doi: 10.1016/0021-9150(71)90025-6. [DOI] [PubMed] [Google Scholar]
- (188).Harland WA, Gilbert JD, Steel G, Brooks CJ. Atherosclerosis. 1971;13:239. doi: 10.1016/0021-9150(71)90026-8. [DOI] [PubMed] [Google Scholar]
- (189).Yla-Herttuala S, Rosenfeld ME, Parthasarathy S, Glass CK, Sigal E, Witztum JL, Steinberg D. Proc. Natl. Acad. Sci. U.S.A. 1990;87:6959. doi: 10.1073/pnas.87.18.6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Yla-Herttuala S, Rosenfeld ME, Parthasarathy S, Sigal E, Sarkioja T, Witztum JL, Steinberg D. J Clin Invest. 1991;87:1146. doi: 10.1172/JCI115111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Belkner J, Wiesner R, Rathman J, Barnett J, Sigal E, Kühn H. Eur J Biochem. 1993;213:251. doi: 10.1111/j.1432-1033.1993.tb17755.x. [DOI] [PubMed] [Google Scholar]
- (192).Kühn H, Belkner J, Zaiss S, Fahrenklemper T, Wohlfeil S. J Exp Med. 1994;179:1903. doi: 10.1084/jem.179.6.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Kühn H, Heydeck D, Hugou I, Gniwotta C. J Clin Invest. 1997;99:888. doi: 10.1172/JCI119253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Cell. 1998;93:229. doi: 10.1016/s0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- (195).Ricote M, Huang J, Fajas L, Li A, Welch J, Najib J, Witztum JL, Auwerx J, Palinski W, Glass CK. Proc. Natl. Acad. Sci. U.S.A. 1998;95:7614. doi: 10.1073/pnas.95.13.7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. Cell. 1998;93:241. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- (197).Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, Witztum JL, Funk CD, Conrad D, Glass CK. Nature. 1999;400:378. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- (198).Shen J, Herderick E, Cornhill JF, Zsigmond E, Kim HS, Kühn H, Guevara NV, Chan L. J Clin Invest. 1996;98:2201. doi: 10.1172/JCI119029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Sendobry SM, Cornicelli JA, Welch K, Bocan T, Tait B, Trivedi BK, Colbry N, Dyer RD, Feinmark SJ, Daugherty A. Br J Pharmacol. 1997;120:1199. doi: 10.1038/sj.bjp.0701007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Cyrus T, Witztum JL, Rader DJ, Tangirala R, Fazio S, Linton MF, Funk CD. J Clin Invest. 1999;103:1597. doi: 10.1172/JCI5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).George J, Afek A, Shaish A, Levkovitz H, Bloom N, Cyrus T, Zhao L, Funk CD, Sigal E, Harats D. Circulation. 2001;104:1646. doi: 10.1161/hc3901.095772. [DOI] [PubMed] [Google Scholar]
- (202).Huo Y, Zhao L, Hyman MC, Shashkin P, Harry BL, Burcin T, Forlow SB, Stark MA, Smith DF, Clarke S, Srinivasan S, Hedrick CC, Pratico D, Witztum JL, Nadler JL, Funk CD, Ley K. Circulation. 2004;110:2024. doi: 10.1161/01.CIR.0000143628.37680.F6. [DOI] [PubMed] [Google Scholar]
- (203).Kühn H. Prog Lipid Res. 1996;35:203. doi: 10.1016/s0163-7827(96)00008-2. [DOI] [PubMed] [Google Scholar]
- (204).Brash AR, Boeglin WE, Chang MS. Proc. Natl. Acad. Sci. U.S.A. 1997;94:6148. doi: 10.1073/pnas.94.12.6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Mangino MJ, Brounts L, Harms B, Heise C. Prostaglandins Other Lipid Mediat. 2006;79:84. doi: 10.1016/j.prostaglandins.2005.10.004. [DOI] [PubMed] [Google Scholar]
- (206).Henriksson P, Hamberg M, Diczfalusy U. Biochim Biophys Acta. 1985;834:272. doi: 10.1016/0005-2760(85)90166-3. [DOI] [PubMed] [Google Scholar]
- (207).Simon TC, Makheja AN, Bailey JM. Atherosclerosis. 1989;75:31. doi: 10.1016/0021-9150(89)90204-9. [DOI] [PubMed] [Google Scholar]
- (208).Smith RJ, Justen JM, Nidy EG, Sam LM, Bleasdale JE. Proc. Natl. Acad. Sci. U.S.A. 1993;90:7270. doi: 10.1073/pnas.90.15.7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Takata S, Papayianni A, Matsubara M, Jimenez W, Pronovost PH, Brady HR. Am J Pathol. 1994;145:541. [PMC free article] [PubMed] [Google Scholar]
- (210).Takata S, Matsubara M, Allen PG, Janmey PA, Serhan CN, Brady HR. J Clin Invest. 1994;93:499. doi: 10.1172/JCI116999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Serhan CN. Prostaglandins Other Lipid Mediat. 2002;68:69, 433. doi: 10.1016/s0090-6980(02)00047-3. [DOI] [PubMed] [Google Scholar]
- (212).Planaguma A, Kazani S, Marigowda G, Haworth O, Mariani TJ, Israel E, Bleecker ER, Curran-Everett D, Erzurum SC, Calhoun WJ, Castro M, Chung KF, Gaston B, Jarjour NN, Busse WW, Wenzel SE, Levy BD. Am J Respir Crit Care Med. 2008;178:574. doi: 10.1164/rccm.200801-061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (213).Karp CL, Flick LM, Park KW, Softic S, Greer TM, Keledjian R, Yang R, Uddin J, Guggino WB, Atabani SF, Belkaid Y, Xu Y, Whitsett JA, Accurso FJ, Wills-Karp M, Petasis NA. Nat Immunol. 2004;5:388. doi: 10.1038/ni1056. [DOI] [PubMed] [Google Scholar]
- (214).Han KH, Chang MK, Boullier A, Green SR, Li A, Glass CK, Quehenberger O. J Clin Invest. 2000;106:793. doi: 10.1172/JCI10052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Nelken NA, Coughlin SR, Gordon D, Wilcox JN. J Clin Invest. 1991;88:1121. doi: 10.1172/JCI115411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (216).Marx N, Sukhova G, Murphy C, Libby P, Plutzky J. Am J Pathol. 1998;153:17. doi: 10.1016/s0002-9440(10)65540-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).te Velde AA, Huijbens RJ, Heije K, de Vries JE, Figdor CG. Blood. 1990;76:1392. [PubMed] [Google Scholar]
- (218).Shankaranarayanan P, Nigam S. J Immunol. 2003;170:887. doi: 10.4049/jimmunol.170.2.887. [DOI] [PubMed] [Google Scholar]
- (219).Shappell SB, Gupta RA, Manning S, Whitehead R, Boeglin WE, Schneider C, Case T, Price J, Jack GS, Wheeler TM, Matusik RJ, Brash AR, Dubois RN. Cancer Res. 2001;61:497. [PubMed] [Google Scholar]
- (220).Ikawa H, Kamitani H, Calvo BF, Foley JF, Eling TE. Cancer Res. 1999;59:360. [PubMed] [Google Scholar]
- (221).Shureiqi I, Jiang W, Zuo X, Wu Y, Stimmel JB, Leesnitzer LM, Morris JS, Fan HZ, Fischer SM, Lippman SM. Proc. Natl. Acad. Sci. U.S.A. 2003;100:9968. doi: 10.1073/pnas.1631086100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Bleich D, Chen S, Zipser B, Sun D, Funk CD, Nadler JL. J Clin Invest. 1999;103:1431. doi: 10.1172/JCI5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).Nunemaker CS, Chen M, Pei H, Kimble SD, Keller SR, Carter JD, Yang Z, Smith KM, Wu R, Bevard MH, Garmey JC, Nadler JL. Am J Physiol Endocrinol Metab. 2008;295:E1065. doi: 10.1152/ajpendo.90371.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Zhao L, Cuff CA, Moss E, Wille U, Cyrus T, Klein EA, Pratico D, Rader DJ, Hunter CA, Pure E, Funk CD. J Biol Chem. 2002;277:35350. doi: 10.1074/jbc.M205738200. [DOI] [PubMed] [Google Scholar]
- (225).Liu C, Xu D, Liu L, Schain F, Brunnstrom A, Bjorkholm M, Claesson HE, Sjoberg J. Am J Physiol Lung Cell Mol Physiol. 2009;297:L196. doi: 10.1152/ajplung.00036.2008. [DOI] [PubMed] [Google Scholar]
- (226).Patricia MK, Kim JA, Harper CM, Shih PT, Berliner JA, Natarajan R, Nadler JL, Hedrick CC. Arterioscler Thromb Vasc Biol. 1999;19:2615. doi: 10.1161/01.atv.19.11.2615. [DOI] [PubMed] [Google Scholar]
- (227).Schopfer FJ, Lin Y, Baker PR, Cui T, Garcia-Barrio M, Zhang J, Chen K, Chen YE, Freeman BA. Proc. Natl. Acad. Sci. U.S.A. 2005;102:2340. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Baker PR, Lin Y, Schopfer FJ, Woodcock SR, Groeger AL, Batthyany C, Sweeney S, Long MH, Iles KE, Baker LM, Branchaud BP, Chen YE, Freeman BA. J Biol Chem. 2005;280:42464. doi: 10.1074/jbc.M504212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Baker LM, Baker PR, Golin-Bisello F, Schopfer FJ, Fink M, Woodcock SR, Branchaud BP, Radi R, Freeman BA. J Biol Chem. 2007;282:31085. doi: 10.1074/jbc.M704085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Schopfer FJ, Cole MP, Groeger AL, Chen CS, Khoo NK, Woodcock SR, Golin-Bisello F, Motanya UN, Li Y, Zhang J, Garcia-Barrio MT, Rudolph TK, Rudolph V, Bonacci G, Baker PR, Xu HE, Batthyany CI, Chen YE, Hallis TM, Freeman BA. J Biol Chem. 2010;285:12321. doi: 10.1074/jbc.M109.091512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (231).Hwang J, Lee KE, Lim JY, Park SI. Biochem Biophys Res Commun. 2009;387:633. doi: 10.1016/j.bbrc.2009.07.030. [DOI] [PubMed] [Google Scholar]
- (232).Ichikawa T, Zhang J, Chen K, Liu Y, Schopfer FJ, Baker PR, Freeman BA, Chen YE, Cui T. Endocrinology. 2008;149:4086. doi: 10.1210/en.2007-1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Tsikas D, Zoerner A, Mitschke A, Homsi Y, Gutzki FM, Jordan J. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:2895. doi: 10.1016/j.jchromb.2008.12.062. [DOI] [PubMed] [Google Scholar]
- (234).Tsikas D, Zoerner AA, Mitschke A, Gutzki FM. Lipids. 2009;44:855. doi: 10.1007/s11745-009-3332-4. [DOI] [PubMed] [Google Scholar]
- (235).Tai HH, Ensor CM, Tong M, Zhou H, Yan F. Prostaglandins Other Lipid Mediat. 2002;68:69. doi: 10.1016/s0090-6980(02)00050-3. [DOI] [PubMed] [Google Scholar]
- (236).Ding Y, Tong M, Liu S, Moscow JA, Tai HH. Carcinogenesis. 2005;26:65. doi: 10.1093/carcin/bgh277. [DOI] [PubMed] [Google Scholar]
- (237).Thiel A, Ganesan A, Mrena J, Junnila S, Nykanen A, Hemmes A, Tai HH, Monni O, Kokkola A, Haglund C, Petrova TV, Ristimaki A. Clin Cancer Res. 2009;15:4572. doi: 10.1158/1078-0432.CCR-08-2518. [DOI] [PubMed] [Google Scholar]
- (238).Yan M, Rerko RM, Platzer P, Dawson D, Willis J, Tong M, Lawrence E, Lutterbaugh J, Lu S, Willson JK, Luo G, Hensold J, Tai HH, Wilson K, Markowitz SD. Proc. Natl. Acad. Sci. U.S.A. 2004;101:17468. doi: 10.1073/pnas.0406142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Tseng-Rogenski S, Gee J, Ignatoski KW, Kunju LP, Bucheit A, Kintner HJ, Morris D, Tallman C, Evron J, Wood CG, Grossman HB, Lee CT, Liebert M. Am J Pathol. 2010;176:1462. doi: 10.2353/ajpath.2010.090875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (240).Liu Z, Wang X, Lu Y, Du R, Luo G, Wang J, Zhai H, Zhang F, Wen Q, Wu K, Fan D. Cancer Biol Ther. 2010;10 doi: 10.4161/cbt.10.8.12896. [DOI] [PubMed] [Google Scholar]
- (241).Myung SJ, Rerko RM, Yan M, Platzer P, Guda K, Dotson A, Lawrence E, Dannenberg AJ, Lovgren AK, Luo G, Pretlow TP, Newman RA, Willis J, Dawson D, Markowitz SD. Proc. Natl. Acad. Sci. U.S.A. 2006;103:12098. doi: 10.1073/pnas.0603235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Anggard E. Acta Physiol Scand. 1966;66:509. doi: 10.1111/j.1748-1716.1966.tb03231.x. [DOI] [PubMed] [Google Scholar]
- (243).Chou WL, Chuang LM, Chou CC, Wang AH, Lawson JA, FitzGerald GA, Chang ZF. J Biol Chem. 2007;282:18162. doi: 10.1074/jbc.M702289200. [DOI] [PubMed] [Google Scholar]
- (244).Waku T, Shiraki T, Oyama T, Maebara K, Nakamori R, Morikawa K. EMBO J. 2010;29:3395. doi: 10.1038/emboj.2010.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).Yu K, Bayona W, Kallen CB, Harding HP, Ravera CP, McMahon G, Brown M, Lazar MA. J Biol Chem. 1995;270:23975. doi: 10.1074/jbc.270.41.23975. [DOI] [PubMed] [Google Scholar]
- (246).Casimir DA, Miller CW, Ntambi JM. Differentiation. 1996;60:203. doi: 10.1046/j.1432-0436.1996.6040203.x. [DOI] [PubMed] [Google Scholar]
- (247).Serrero G, Lepak NM, Goodrich SP. Biochem Biophys Res Commun. 1992;183:438. doi: 10.1016/0006-291x(92)90500-k. [DOI] [PubMed] [Google Scholar]
- (248).Sugimoto Y, Hasumoto K, Namba T, Irie A, Katsuyama M, Negishi M, Kakizuka A, Narumiya S, Ichikawa A. J Biol Chem. 1994;269:1356. [PubMed] [Google Scholar]
- (249).Sakamoto K, Ezashi T, Miwa K, Okuda-Ashitaka E, Houtani T, Sugimoto T, Ito S, Hayaishi O. J Biol Chem. 1994;269:3881. [PubMed] [Google Scholar]
- (250).Reginato MJ, Krakow SL, Bailey ST, Lazar MA. J Biol Chem. 1998;273:1855. doi: 10.1074/jbc.273.4.1855. [DOI] [PubMed] [Google Scholar]
- (251).Rangwala SM, Rhoades B, Shapiro JS, Rich AS, Kim JK, Shulman GI, Kaestner KH, Lazar MA. Dev Cell. 2003;5:657. doi: 10.1016/s1534-5807(03)00274-0. [DOI] [PubMed] [Google Scholar]
- (252).Tsukahara T, Tsukahara R, Fujiwara Y, Yue J, Cheng Y, Guo H, Bolen A, Zhang C, Balazs L, Re F, Du G, Frohman MA, Baker DL, Parrill AL, Uchiyama A, Kobayashi T, Murakami-Murofushi K, Tigyi G. Mol Cell. 2010;39:421. doi: 10.1016/j.molcel.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Shan L, Li S, Jaffe K, Davis L. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;862:161. doi: 10.1016/j.jchromb.2007.12.003. [DOI] [PubMed] [Google Scholar]
- (254).Reynolds CM, Roche HM. Prostaglandins Leukot Essent Fatty Acids. 2010;82:199. doi: 10.1016/j.plefa.2010.02.021. [DOI] [PubMed] [Google Scholar]
- (255).Jaudszus A, Krokowski M, Mockel P, Darcan Y, Avagyan A, Matricardi P, Jahreis G, Hamelmann E. J Nutr. 2008;138:1336. doi: 10.1093/jn/138.7.1336. [DOI] [PubMed] [Google Scholar]
- (256).Loscher CE, Draper E, Leavy O, Kelleher D, Mills KH, Roche HM. J Immunol. 2005;175:4990. doi: 10.4049/jimmunol.175.8.4990. [DOI] [PubMed] [Google Scholar]
- (257).Yu Y, Correll PH, Vanden Heuvel JP. Biochim Biophys Acta. 2002;1581:89. doi: 10.1016/s1388-1981(02)00126-9. [DOI] [PubMed] [Google Scholar]
- (258).Bassaganya-Riera J, Hontecillas R, Beitz DC. Clin Nutr. 2002;21:451. doi: 10.1054/clnu.2002.0594. [DOI] [PubMed] [Google Scholar]
- (259).Bassaganya-Riera J, Hontecillas R. Clin Nutr. 2006;25:454. doi: 10.1016/j.clnu.2005.12.008. [DOI] [PubMed] [Google Scholar]
- (260).Bassaganya-Riera J, Reynolds K, Martino-Catt S, Cui Y, Hennighausen L, Gonzalez F, Rohrer J, Benninghoff AU, Hontecillas R. Gastroenterology. 2004;127:777. doi: 10.1053/j.gastro.2004.06.049. [DOI] [PubMed] [Google Scholar]
- (261).Moya-Camarena SY, Van den Heuvel JP, Belury MA. Biochim Biophys Acta. 1999;1436:331. doi: 10.1016/s0005-2760(98)00121-0. [DOI] [PubMed] [Google Scholar]
- (262).Houseknecht KL, Vanden Heuvel JP, Moya-Camarena SY, Portocarrero CP, Peck LW, Nickel KP, Belury MA. Biochem Biophys Res Commun. 1998;244:678. doi: 10.1006/bbrc.1998.8303. [DOI] [PubMed] [Google Scholar]
- (263).Brown JM, Boysen MS, Jensen SS, Morrison RF, Storkson J, Lea-Currie R, Pariza M, Mandrup S, McIntosh MK. J Lipid Res. 2003;44:1287. doi: 10.1194/jlr.M300001-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Chung S, Brown JM, Provo JN, Hopkins R, McIntosh MK. J Biol Chem. 2005;280:38445. doi: 10.1074/jbc.M508159200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Tjonneland A, Overvad K, Bergmann MM, Nagel G, Linseisen J, Hallmans G, Palmqvist R, Sjodin H, Hagglund G, Berglund G, Lindgren S, Grip O, Palli D, Day NE, Khaw KT, Bingham S, Riboli E, Kennedy H, Hart A. Gut. 2009;58:1606. doi: 10.1136/gut.2008.169078. [DOI] [PubMed] [Google Scholar]
- (266).Chakravarthy MV, Pan Z, Zhu Y, Tordjman K, Schneider JG, Coleman T, Turk J, Semenkovich CF. Cell Metab. 2005;1:309. doi: 10.1016/j.cmet.2005.04.002. [DOI] [PubMed] [Google Scholar]
- (267).Chakravarthy MV, Lodhi IJ, Yin L, Malapaka RR, Xu HE, Turk J, Semenkovich CF. Cell. 2009;138:476. doi: 10.1016/j.cell.2009.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Jisaka M, Kim RB, Boeglin WE, Brash AR. J Biol Chem. 2000;275:1287. doi: 10.1074/jbc.275.2.1287. [DOI] [PubMed] [Google Scholar]
- (269).Ford-Hutchinson AW. Crit Rev Immunol. 1990;10:1. [PubMed] [Google Scholar]
- (270).Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, Wahli W. Nature. 1996;384:39. doi: 10.1038/384039a0. [DOI] [PubMed] [Google Scholar]
- (271).Lin Q, Ruuska SE, Shaw NS, Dong D, Noy N. Biochemistry. 1999;38:185. doi: 10.1021/bi9816094. [DOI] [PubMed] [Google Scholar]
- (272).Narala VR, Adapala RK, Suresh MV, Brock TG, Peters-Golden M, Reddy RC. J Biol Chem. 2010;285:22067. doi: 10.1074/jbc.M109.085118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Gupta RA, Tan J, Krause WF, Geraci MW, Willson TM, Dey SK, DuBois RN. Proc. Natl. Acad. Sci. U.S.A. 2000;97:13275. doi: 10.1073/pnas.97.24.13275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Hatae T, Wada M, Yokoyama C, Shimonishi M, Tanabe T. J Biol Chem. 2001;276:46260. doi: 10.1074/jbc.M107180200. [DOI] [PubMed] [Google Scholar]
- (275).Schug TT, Berry DC, Shaw NS, Travis SN, Noy N. Cell. 2007;129:723. doi: 10.1016/j.cell.2007.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (276).Fauti T, Muller-Brusselbach S, Kreutzer M, Rieck M, Meissner W, Rapp U, Schweer H, Komhoff M, Muller R. FEBS J. 2006;273:170. doi: 10.1111/j.1742-4658.2005.05055.x. [DOI] [PubMed] [Google Scholar]
- (277).Rieck M, Meissner W, Ries S, Muller-Brusselbach S, Muller R. Mol Pharmacol. 2008;74:1269. doi: 10.1124/mol.108.050625. [DOI] [PubMed] [Google Scholar]
- (278).Borland MG, Foreman JE, Girroir EE, Zolfaghari R, Sharma AK, Amin S, Gonzalez FJ, Ross AC, Peters JM. Mol Pharmacol. 2008;74:1429. doi: 10.1124/mol.108.050609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (279).Naruhn S, Meissner W, Adhikary T, Kaddatz K, Klein T, Watzer B, Muller-Brusselbach S, Muller R. Mol Pharmacol. 2010;77:171. doi: 10.1124/mol.109.060541. [DOI] [PubMed] [Google Scholar]