

Abstract
Background
Neural tube defects (NTDs) are common human birth defects with a complex etiology. To develop a comprehensive knowledge of the genes expressed during normal neurulation, we established transcriptomes from human neural tube fragments during and after neurulation using long Serial Analysis of Gene Expression (long-SAGE).
Methods
Rostral and caudal neural tubes were dissected from normal human embryos aged between 26 and 32 days of gestation. Tissues from the same region and Carnegie stage were pooled (n>=4) and total RNA extracted to construct four long-SAGE libraries. Tags were mapped using the UniGene Homo sapiens 17 bp tag-to-gene best mapping set. Differentially expressed genes were identified by chi-square or Fisher’s exact test and validation was performed for a subset of those transcripts using in situ hybridization. In silico analyses were performed with BinGO and EXPANDER.
Results
We observed most genes to be similarly regulated in rostral and caudal regions, but expression profiles differed during and after closure. In silico analysis found similar enrichments in both regions for biological process terms, transcription factor binding and miRNA target motifs. Twelve genes potentially expressing alternate isoforms by region or developmental stage, and the miRNAs miR-339-5p, miR-141/200a, miR-23ab, and miR-129/129-5p, are among several potential candidates identified here for future research.
Conclusions
Time appears to influence gene expression in the developing central nervous system more than location. These data provide a novel complement to traditional strategies of identifying genes associated with human NTDs, and offer unique insight into the genes associated with normal human neurulation.
Keywords: gene expression, Homo sapiens, long-SAGE, neurulation, neural tube defects
Introduction
Birth defects are the leading cause of death in children under one year of age (Petrini et al. 2002). Neural tube defects (NTDs) are one of the most common birth defects in humans, with an overall incidence of ~1/1000 for all NTD types (Copp et al. 2003). NTDs occur when the neural tube, precursor of the brain and spinal cord, fails to close. Common forms of NTDs include spina bifida and anencephaly in the caudal and rostral neural tube regions respectively. In spina bifida, failed closure leaves part of the spine exposed; this defect can be surgically repaired but the affected individual nonetheless faces an impaired quality of life (McMahon et al. 2011). In anencephaly, the dorsal brain remains exposed; this phenotype is invariably lethal. Dietary supplementation with folate has significantly reduced the frequency of both phenotypes in the US, but in differing magnitudes, suggesting that anencephaly and spina bifida have distinct responses (Williams et al. 2002). Epidemiological differences between NTD phenotypes further suggest differences in their underlying pathogenesis (Garabedian and Fraser 1993). Institution of additional, potentially more specific preventative measures is hindered by our as-yet incomplete understanding of the normal molecular processes of neurulation.
NTDs are known to have a genetic basis; it is estimated that 70% of the variation in human NTD prevalence is determined by heritable factors (Copp and Greene 2010). Over 200 potential candidate genes have been identified in mouse models (Harris and Juriloff 2010), yet none of these genes show consistent and robust association with human NTDs (Copp and Greene 2010). Mouse models are highly valuable for investigating the gene-gene and gene-environment interactions which promote or compromise neurulation, but it is evident that study in the human context is essential to understanding the etiology of human NTDs. However, candidate gene identification for human NTDs is hindered by the dearth of information regarding gene expression patterns during Carnegie stages 7–13, when the neural primordium forms and closes in humans (Detrait et al. 2005). We posit that genes expressed just prior to or during neural tube closure are critical to the etiology of NTDs, and that documenting the normal transcriptome profile and regulatory mechanisms for these genes is a critical requisite for understanding factors which influence human NTD risk.
Serial analysis of gene expression (SAGE) is a powerful technique for generating tissue specific profiles of gene expression (Velculescu et al. 1995). Long-SAGE is an adaptation of SAGE that extracts 17 nt tags from individual transcripts and maps them directly to the human genome (Saha et al. 2002). This quantitative method correlates significantly with array-measured gene expression but is not constrained to a selection of genes, and produces more accurate tag-to-gene assignments than traditional SAGE, making it well-suited to the identification of new and/or novel genes associated with diseases (Lu et al. 2004). SAGE is also capable of detecting small changes in expression levels (Scott and Chrast 2002).
To our knowledge, no previous study has quantitatively profiled global gene expression in caudal and rostral neural tube tissues during the critical periods of normal human neural tube closure. In this study, we generated long-SAGE libraries from total RNA isolated from normal microdissected neural tubes just after neural fold fusion, at Carnegie stage 12 (C12, 26–30 days post fertilization), and after caudal neuropore closure, at Carnegie stage 13 (C13, 30–32 days post fertilization). We also compared the gene expression profiles of rostral (ROS) neural tube, corresponding to the future brain, with caudal (CAU) neural tube, corresponding to future spinal cord, from the same stages, given the gradient of maturation along the rostrocaudal axis that is still relevant at these stages. Our findings suggest that the greatest differences in gene expression of the developing neural tube occur temporally rather than spatially. These data provide a novel complement to traditional strategies of identifying candidate genes associated with developmental disorders, including NTDs, and offer a unique insight into the genes associated with normal neural tube closure in humans.
Methods
Human embryonic neural tube tissues
Normal human embryos were collected after anonymous donations from pregnancies terminated by mifepristone, in agreement with French bioethics law 04–800 and with approval from the Necker hospital ethics committee. The embryos were screened visually for morphological abnormalities and by karyotyping for aneuploidies. Tissues were microdissected from rostral (presomitic) and caudal (post-somite 6) regions of the neural tube. Dissected tissues were pooled from embryos of equivalent Carnegie stages to construct four stage-specific regional libraries: early stage C12 (at the time of neural tube closure) and late stage C13 (neural tube closure completed) for caudal and rostral regions. At least four individuals were incorporated in each pool.
Construction of Long-SAGE libraries
Total RNA was isolated from frozen neural tubes using TRIzol reagent (Invitrogen, Carlsbad, CA) as previously described (Xu et al. 2006). For long-SAGE library construction, we used standard protocols as described by Velculescu et al (1995), with minor modifications. Briefly, long-SAGE was performed with 400–600 ng total RNA. The cDNA was prepared using the SuperscriptII cDNA synthesis kit (Invitrogen, Carlsbad, CA) with gel-purified 5’-biotinylated Oligo(dT)18 (Integrated DNA Technologies, Coralville, IA) according to the manufacturer’s protocol. NlaIII and MmeI restriction enzymes (New England Biolabs, Beverly, MA) were used for tag generation. Purified concatemers were subsequently cloned into the SphI site of pZero-1 (Invitrogen) and transformed in competent ElectroMax DH10B cells (Invitrogen) using a 0.1 cm cuvette and the Gene Pulser II (BioRad, Hercules, CA). Individual long-SAGE library clones were selected and PCR amplified using 96-well format Qiagen Real minipreps. Amplicons were sequenced on an ABI 3700 capillary sequencer using BigDye chemistry (Applied Biosystems, Carlsbad, CA).
SAGE tag extract and statistical analysis
Long-SAGE tags (17 bp) were extracted from sequence files with SAGE 2000 software using a threshold value of PHRED 20 for each base (Margulies and Innis 2000). The long-SAGE tags were extracted from individual sequence files from each SAGE library and sets compared with one another, e.g. contrasting C13 caudal with C12 caudal, to identify tags which were significantly different in expression by region or stage. Differentially expressed genes (p<0.05) were identified by Pearson’s chi-square test as previously described (Man et al. 2000), or by Fisher’s exact test in cases where any expected count value was less than or equal to 10.
Benjamini and Hochberg’s false discovery rate (FDR) method was used to correct for multiple testing (Benjamini and Hochberg 1995). The resulting tag lists were then mapped to the UniGene Homo sapiens 17 bp tag-to-gene best mapping set.
Comparison with established NTD candidate pathways and candidate genes
The list of all differentially expressed genes was checked for membership in KEGG pathways with established importance to neural tube closure using KEGG Mapper (Kanehisa et al. 2011). Pathways were considered NTD-relevant if mentioned in literature reviews (Harris and Juriloff 2010; Copp and Greene 2010). Additionally, differentially expressed genes were compared against a list of 450 previously reported NTD candidate genes compiled from mouse models (Harris and Juriloff 2007, 2010) and manual review of human NTD literature.
Functional analysis
Enrichment of Gene Ontology (GO) Biological Process terms was evaluated for each list of differentially expressed genes using the Cytoscape plugin BinGO (Maere et al. 2005).
Overrepresentation was determined using a hypergeometric test with FDR-based multiple testing correction and all annotated human genes as the background set. Associations with a corrected p-value less than 0.05 were considered to be significant.
Identification of enriched transcription factor motifs
Analysis was performed using the Promoter Integration in Microarray Analysis (PRIMA) algorithm, part of the EXPANDER analysis platform (Shamir et al. 2005). Given one or more lists of genes, PRIMA uses a generalized hypergeometric test to identify transcription factor (TF) binding motifs which are significantly overrepresented in their promoter sequences (Elkon et al. 2003). Motifs used in this analysis come from the TRANSFAC database and are provided with EXPANDER. Human promoter sequences from NCBI build 36.1 are also provided. We used all human genes as the background set, analyzed promoter regions from −1000 to 200 nt around the transcription start site, and retained transcription factors with a Bonferroni-corrected p-value of 0.05 or less.
Identification of enriched miRNA target motifs
Analysis was performed using the Functional Assignment of MiRNAs via Enrichment (FAME) algorithm, part of the EXPANDER analysis platform. This permutation-based algorithm uses predicted binding sites and context scores from TargetScan 5.0 to detect significant enrichment or depletion of miRNA family binding motifs in a group of genes (Ulitsky et al. 2010). We assessed both enrichment and depletion using 200 permutations, with the threshold of significance at p = 0.05. Analyses were performed both with and without FDR-based multiple testing correction.
Validation of differentially expressed genes by in situ hybridization and RT-PCR
Differentially expressed genes were selected for validation based on functional relevance, namely those having prior implication in human NTD pathology or known expression patterns in animal models. Additionally, validated genes were chosen to represent a variety of tag counts and differing magnitudes of expression change. Validation experiments were constrained by sample availability, particularly of rostral tissues.
For in situ hybridizations, intact embryos were fixed in either 4% paraformaldehyde in PBS, pH 7.4, or modified Carnoy’s solution (11% formaldehyde, 60% ethanol, 10% acetic acid). Fixed embryos were embedded in paraffin and sectioned at 5 µm. Genes with differential expression were specifically targeted for hybridization, prioritized by magnitude of expression change. A T7 promoter sequence extension (taatacgactcactatagggaga) was added to the 5’end of each amplification primer. T7F/R and F/T7R primer combinations permitted amplification of sense and antisense templates respectively. PCR products were purified with the QIAquick PCR purification kit (Qiagen). A 10× digoxigenin RNA labelling mix (Roche) and T7 RNA polymerase were used for probe labelling without alkaline lysis as per standard protocols (Wilting et al. 1997), and riboprobes were purified on Sephadex G50 (GE Healthcare) columns. Sections were hybridized and signal developed as previously described (Etchevers et al. 2001).
For RT-PCR reactions, total RNA was extracted from isolated C12 and C13 neural tube regions using the RNeasy Mini kit (Qiagen). Reverse transcription was performed using the GeneAmp PCR Core kit (Applied Biosystems), and qPCR reactions using the LightCycler Fast Start DNA MasterPLUS SYBR Green I system, both according to manufacturer’s protocol. Expression of ACTB was used for normalization in all ΔCt calculations. Primers are available upon request.
Results
We generated and sequenced four long-SAGE libraries using total RNA isolated from the rostral (future brain) and caudal (future spinal cord) portions of neural tubes from normal human embryos at Carnegie stages 12 and 13. A total of 269,043 long-SAGE tags were obtained, representing 137,486 unique transcripts, with an average of 34,371 unique tags per library. We obtained similar tag counts for all four libraries, with no GC content bias in any library. The frequencies of both duplicated ditags and linker contamination were less than 0.5%. By these characteristics, we judged the libraries to be of high quality.
Long-SAGE libraries and the differential expression of genes in space and time around human neurulation
In comparing our long-SAGE libraries for each region and stage, we identified a total of 1359 genes with differential expression in time and/or space (FDR-corrected p <= 0.05). Most of their associated tags had moderate counts (median=11) in their greater condition and little to no expression (median=1) in the other. These ranges are consistent with previously published SAGE data (Waghray et al. 2001). We found several hundred genes to be significantly up- or downregulated at C12 versus C13, many of which were similarly regulated in both regions across the stages (Fig. 1A). The correlation of tags observed between rostral and caudal libraries by stage was accordingly high (at closure R2=0.98, after closure R2=0.89). In contrast, very few genes showed significantly different expression by region at C12, with more differences apparent at C13 (Fig. 1B). The correlation of tags observed during and after neural tube closure within a region was considerably lower (rostral R2=0.42, caudal R2=0.46). Taken together, these results indicate that the greatest differences in gene expression for the developing human neural tube occur over time rather than spatial location in the neural tube.
Figure 1.
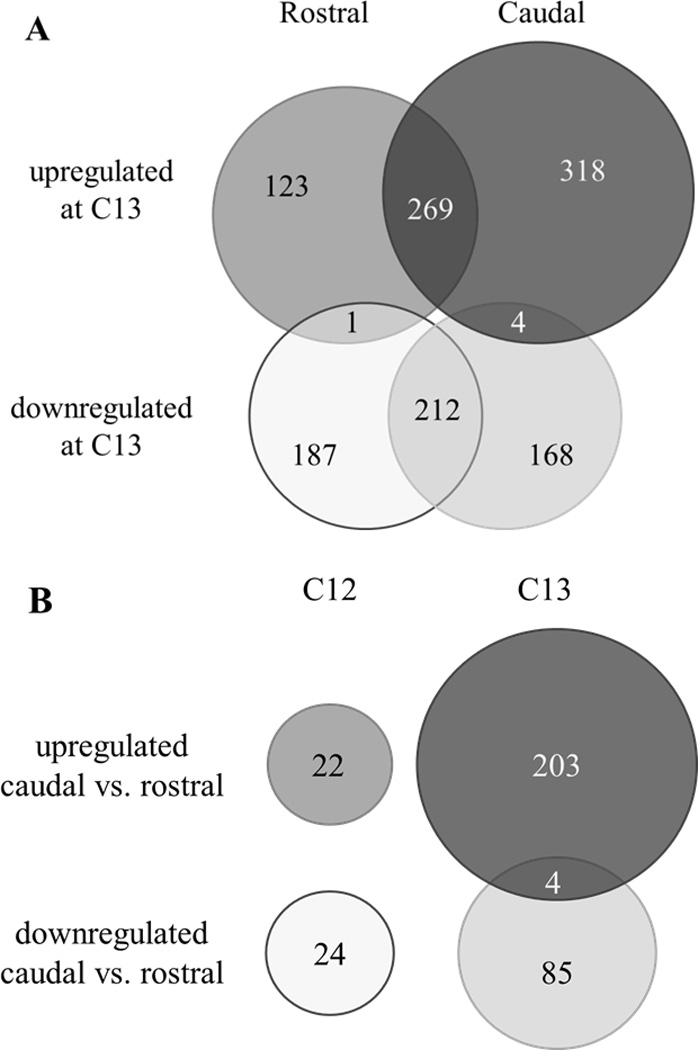
Overview of spatial and temporal gene expression changes in the neural tube at C12 and C13. Many genes are similarly regulated in both rostral and caudal regions (A), while relatively few show significantly different expression between regions either before or after closure (B).
A handful of genes stood out for having opposite directions of differential expression within comparisons (Fig. 1, Suppl. Table 1). Five genes had different tags upregulated and downregulated at C13 relative to C12 in a single region: four caudally (MALAT1, SEC31A, SMARCA4, SRP14) and one rostrally (HNRPA3). Isoforms of these genes may have region-specific expression before and after closure. Another four genes had different tags both upregulated and downregulated in caudal regions at C13 relative to rostral regions (DKFZp547, EEF1A1, HNRPA1, MARCKS). This may represent preferences for different isoforms in caudal and rostral regions after closure.
Long-SAGE data are corroborated by endogenous gene transcription patterns
We used in situ hybridization and RT-PCR of selected genes to validate our long-SAGE differential expression findings. A total of 42 genes were tested by at least one method in at least one region (Suppl. Table 2); in all, 97 validation experiments were performed. In six cases, transcripts were detected by RT-PCR but no SAGE tags were detected for the same region and stage; these discrepancies can most likely be ascribed to the greater sensitivity of RT-PCR. For the genes NOTCH2 and SOX10, expression was detected by SAGE tag counts but either absent from or not confidently detected in the corresponding in situ hybridizations. This may again be a sensitivity issue, as SAGE is more sensitive than in situ hybridization and all involved tag counts were low; in the one such instance also having RT-PCR data, NOTCH2 was detected in agreement with the SAGE results. Only one discrepancy, the presence of MTHFD1 SAGE tags at caudal C12 but no detection of MTHFD transcripts by RT-PCR, cannot be explained by technique sensitivities.
In situ hybridizations for the following genes are presented in Figure 2 as representative examples: SRY-box 11 (SOX11), a transcription factor regulating neural development; Myc-associated zinc finger protein (MAZ), a ubiquitous transcription factor previously implicated in neuronal differentiation; vimentin (VIM), encoding a cytoskeletal filament typical of mesenchymal and neural stem cells; gap junction alpha-1 protein (GJA1), a member of the connexin family and gap junction component; and beta-catenin (CTNNB1), an adherens junction component and member of the canonical Wnt signaling pathway. For the gene SOX11, we extracted three tags, which were all upregulated in both regions post-closure (rostral p<0.02, caudal p<0.0001). This upregulation is corroborated by highly localized caudal staining at C12 (Fig. 2A) and widespread expression in both regions at C13 (Fig. 2B–C). Similarly, we find MAZ upregulated in both regions (rostral p=0.031, caudal p=0.001), and strong C13 MAZ staining both caudally and rostrally (Fig. 2E–F). VIM is also highly upregulated in both regions at C13 (p<2.4E-15, both regions), for which we observed strong staining in the caudal C13 neural tube (Fig. 2H). The gene GJA1 was identified as downregulated postclosure (p<4.5E-15, both regions); we observed localized dorsal and lateral hinge point expression in the rostral but not caudal neural tube at C13 (Fig. 2G). ISL LIM homeobox 1 (ISL1), a TF involved in the development of motor neurons, was discretely expressed in a few spinal cord cells at C13 (Fig. 2I), consistent with postmitotic motoneurons and its low tag count. Caudal neural tube CTNNB1 expression at C13 was more pervasive but of modest intensity (Fig 2K). We additionally stained for genes absent from our long-SAGE libraries as negative controls; shown here is T-box 3 (TBX3), a TF implicated in limb and heart development. TBX3 is transcribed exclusively in the optic vesicle in the rostral CNS at C13, (Fig. 2J), consistent with its absence from the corresponding long-SAGE library.
Figure 2.
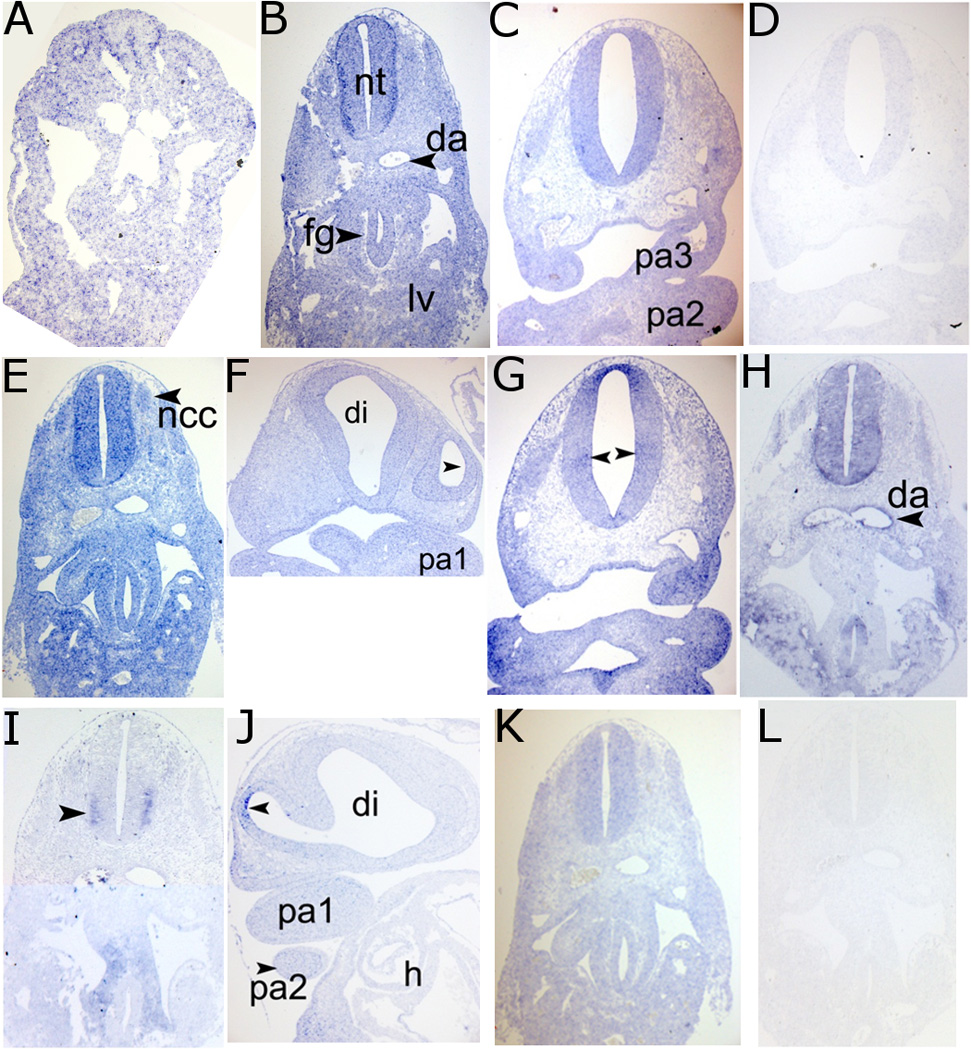
In situ hybridization for selected differentially expressed and control genes, showing expression for: caudal C12 (A), caudal C13 (B), and rostral C13 (C) SOX11; caudal C13 (E) and rostral C13 (F) MAZ; rostral C13 GJA (G), caudal C13 VIM (H) and ISL1 (I); rostral C13 TBX3 (J), and caudal C13 CTNNB1 (K). Representative background staining shown for rostral C13 SOX11 (D) and caudal C13 VIM (L). Labels: da, dorsal aortae; di, diencephalon; fg, foregut; h, heart; lv, liver; nt, neural tube; ncc, neural crest cells; pa1–3, pharyngeal arches 1–3.
Differential expression in NTD candidate pathways and of known candidate genes
The entire pool of differentially expressed genes was examined for membership in various KEGG pathways with established importance to neural tube closure. A total of 125 unique differentially expressed genes were identified in these pathways (Table 1). Differentially expressed genes were uniformly downregulated in four pathways (folate metabolism, retinol metabolism, Notch signaling, and gap junctions), and uniformly upregulated in only one pathway (tight junctions). All other pathways contained both upregulated and downregulated genes, suggesting more complex regulation.
Table 1.
NTD-relevant KEGG pathways containing genes which have either spatial or temporal differential expression around neurulation.
| KEGG ID |
Pathway Name | Genes | Gene List |
|---|---|---|---|
| hsa04810 | Regulation of actin cytoskeleton | 24 | ACTG1, APC, ARHGEF6, ARHGEF7, ARPC2, CFL1, FGFR2, GSN, ITGAV, ITGB1, ITGB8, MYH10, MYL9, NCKAP1, PAK2, PFN1, PIP4K2A, PPP1CB, PPP1CC, PPP1R12A, RAC3, RDX, TMSB4X, WASF1 |
| hsa04310 | Wnt signaling pathway | 17 | APC, CAMK2B, CCND1, CCND2, CSNK2B, CTBP2, DAAM2, DVL3, FZD7, PPP2CB, PPP2R1A, RAC3, RBX1, RUVBL1, SFRP2, SKP1, TP53 |
| hsa04020 | Calcium signaling pathway | 17 | ADCY9, ADRB3, ATP2A2, CALM1, CALM2, CAMK2B, EDNRB, ERBB3, GNAS, GRIN1, ITPKB, ITPR1, P2RX7, PTK2B, RYR3, SLC25A5, VDAC2 |
| hsa04514 | Cell adhesion molecules | 15 | ALCAM, CD99, CLDN11, CNTN1, CNTN2, HLA-B, HLA-DRA, HLA-E, ITGAV, ITGB1, ITGB8, NCAM1, NEO1, SDC3, VCAN |
| hsa00230 | Purine metabolism | 15 | ADCY9, APRT, DGUOK, NME1, NME4, PAICS, PKM2, PNPT1, POLD2, POLR1D, POLR2G, PRPS1, RRM1, RRM2, ZNRD1 |
| hsa04530 | Tight junction | 15 | ACTG1, AMOTL1, ASH1L, CLDN11, CSNK2B, CTNNA2, EPB41L3, EXOC4, GNAI2, MYH10, MYL9, PPP2CB, PPP2R1A, RAB3B, SYMPK |
| hsa04540 | Gap junction | 11 | ADCY9, CDK1, GJA1, GNAI2, GNAS, ITPR1, TUBA1A, TUBA1B, TUBB, TUBB2A, TUBB2B |
| hsa04330 | Notch signaling pathway | 9 | CTBP2, DTX4, DVL3, HDAC1, HDAC2, HES5, JAG1, NCSTN, NOTCH3 |
| hsa04350 | TGF-beta signaling pathway | 8 | BMPR2, ID1, ID3, ID4, PPP2CB, PPP2R1A, RBX1, SKP1, |
| hsa04520 | Adherens junction | 6 | ACTG1, CSNK2B, CTNNA2, RAC3, SORBS1, WASF1, |
| hsa04070 | Phosphatidylinositol signaling system | 6 | CALM1, CALM2, ITPKB, ITPR1, PIK3C2A, PIP4K2A, |
| hsa00270 | Cysteine and methionine metabolism | 5 | AHCYL1, CBS, LDHA, LDHB, MAT2A |
| hsa04210 | Apoptosis | 5 | IRAK1, PRKAR2A, PRKAR2B, TP53, XIAP |
| hsa00670 | One carbon pool by folate | 3 | DHFR, SHMT2, TYMS |
| hsa04340 | Hedgehog signaling pathway | 2 | GAS1, ZIC2 |
| hsa00830 | Retinol metabolism | 2 | CYP2C19, DHRS3 |
Differentially expressed genes were also compared against a list of 450 previously reported NTD candidate genes. In all, we found 61 known candidate genes to be differentially expressed (Suppl Table 3). Of these genes, 24 showed similar temporal regulation in both rostral and caudal regions, while 12 did not exhibit uniform temporal regulation and did show significant spatial differences in expression (Table 2). Finally, differentially expressed genes were compared against all 1660 genes in the Gene Ontology term GO:0007399, “nervous system development” (Suppl. Table 4). A total of 230 genes in this category were identified as differentially expressed in any context, only 31 of which were also established NTD candidate genes.
Table 2.
NTD candidate genes with differential temporal and spatial expression. ROS12, rostral C12; ROS13, rostral C13; CAU12, caudal C13; CAU13, caudal C13.
| Gene | Region | Temporal Expr.Change | p | Spatial Expr. Change | p |
|---|---|---|---|---|---|
| CBS | Rostral | Up ROS13 vs ROS12 | 0.023 | Down CAU13 vs ROS13 | 0.0054 |
| CD99 | Caudal | Up CAU13 vs CAU12 | 0.0001 | Up CAU13 vs ROS13 | 0.0039 |
| FKBP8 | Rostral | Down CAU13 vs ROS13 | 0.323 | ||
| GTF2IRD1 | Caudal | Up CAU13 vs CAU12 | 0.0314 | Up CAU13 vs ROS13 | 0.0321 |
| IGF2 | Caudal | Up CAU13 vs CAU12 | 0.002 | Up CAU13 vs ROS13 | 0.0406 |
| ITGB1 | Caudal | Up CAU13 vs CAU12 | 0.001 | Up CAU13 vs ROS13 | 0.0107 |
| MSX1 | Caudal | Up CAU13 vs CAU12 | 0.0069 | Up CAU13 vs ROS13 | 0.0475 |
| PFN1 | Rostral | Up ROS13 vs ROS12 | 0.0013 | Down CAU13 vs ROS13 | 0.0111 |
| POR | Caudal | Up CAU13 vs ROS13 | 0.0042 | ||
| SALL1 | Caudal | Up CAU13 vs ROS13 | 0.0069 | ||
| SLC2A1 | Caudal | Up CAU13 vs CAU12 | 0 | Up CAU13 vs ROS13 | 0.0002 |
| TPT1* | Caudal | Up CAU13 vs ROS13 | 0.0049 |
Two tags were significantly different between CAU13 and ROS13 for TPT1; the most significant p value is given.
In silico functional classification of genes differentially expressed during neural tube closure
Genes upregulated post-closure in both regions were enriched for terms relating to DNA and protein synthesis, cell cycle regulation, and metabolic processes consistent with cellular and organismal growth (Suppl. Table 5). Genes downregulated post-closure in both regions were enriched for terms relating to nervous system genesis and development and apoptosis (Suppl. Table 6). Rostrally downregulated genes over time were uniquely enriched for additional categories (Suppl. Table 7) all having prior associations with neural tube defects: cytoskeleton reorganization, metal cation metabolism (Mao et al. 2010; Groenen et al. 2003), and response to oxidative stress (Chang et al. 2003).
Identification of enriched transcription factor motifs
Overall, we found significant enrichment for TF motifs in the promoters of upregulated gene groups, but little in downregulated gene groups (Table 3). Any one enriched motif encompassed between 20% and 60% of differentially regulated genes, with proportions typically consistent between gene groups (Figure 3). The majority of enriched motifs are recognized by “ubiquitous” or “conditional” TF families, whose members are widely expressed and choreograph gene expression by combinatorial regulation.
Table 3.
Transcription factor motif enrichment for genes with differential temporal or spatial expression in human neurulation. P-values corrected using Benjamini-Hochberg FDR method. Up, upregulated genes; down, downregulated genes, CAU12, caudal C12; CAU13, caudal C13; ROS12, rostral C12; ROS13, rostral C13.
| Motif | TF Family | Motif Overrepresented In | p-value |
|---|---|---|---|
| M00196 | SP1 | Up CAU13 vs CAU12 | 6.90 × 10−4 |
| Up ROS13 vs ROS12 | 0.024 | ||
| M00287 | NF-Y | Up CAU13 vs CAU12 | 1.25 × 10−10 |
| Up ROS13 vs ROS12 | 2.33 × 10−4 | ||
| Up CAU13 vs ROS13 | 0.029 | ||
| M00649 | MAZ | Up CAU13 vs CAU12 | 0.005 |
| M00652 | NRF-1 | Up CAU13 vs CAU12 | 1.28 × 10−4 |
| Up ROS13 vs ROS12 | 7.96 × 10−5 | ||
| M00716 | ZF5 | Up CAU13 vs CAU12 | 0.001 |
| Up ROS13 vs ROS12 | 0.001 | ||
| M00720 | CAC-binding proteins (KLFs) | Down CAU13 vs CAU12 | 0.027 |
| M00803 | E2F | Up CAU13 vs CAU12 | 2.00 × 10−7 |
| Up ROS13 vs ROS12 | 2.53 × 10−4 | ||
| M00938 | E2F-1 | Up CAU13 vs CAU12 | 0.02 |
| M00940 | E2F-1 | Up ROS13 vs ROS12 | 0.003 |
| M00976 | ARNT | Up ROS13 vs ROS12 | 0.018 |
| M01068 | FOXA1/2 | Up CAU13 vs CAU12 | 0.024 |
| M01104 | OVOL2 | Up CAU13 vs CAU12 | 0.018 |
| M01122 | ZNF219 | Up ROS13 vs ROS12 | 0.027 |
Figure 3.
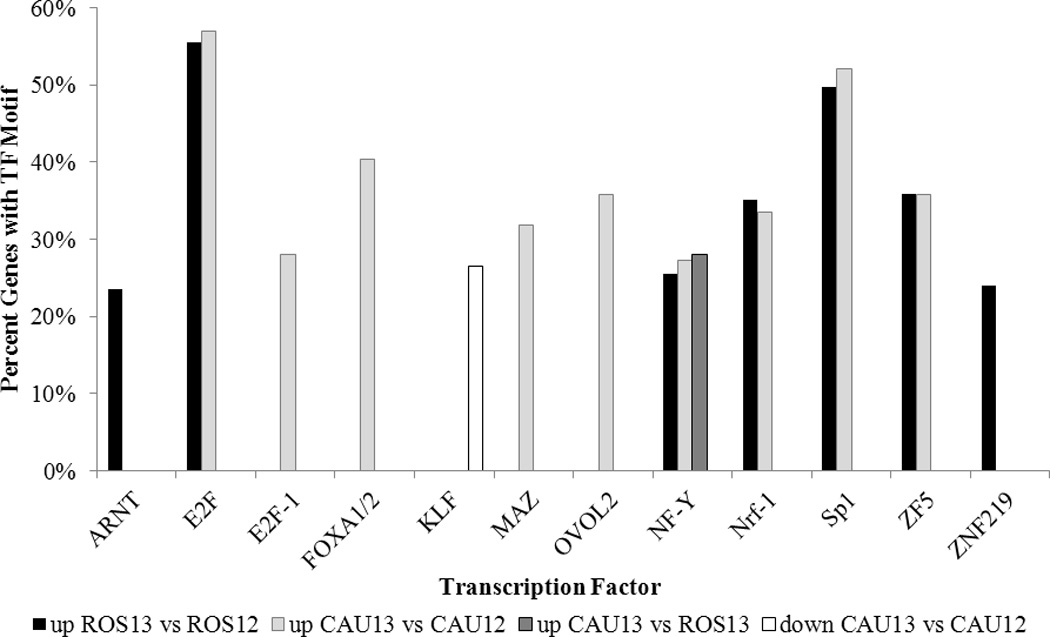
Proportion of differentially expressed genes containing promoter motifs for significant TFs. Up, upregulated genes; down, downregulated genes; CAU12, caudal C12; CAU13, caudal C13; ROS12, rostral C12; ROS13, rostral C13.
Of TFs with enriched motifs, only one, MAZ, was itself significantly upregulated at C13. Indeed, MAZ was upregulated at C13 in both caudal and rostral regions. Two TFs with motifs enriched among differentially regulated genes are previously documented NTD candidate genes: the aryl hydrocarbon receptor nuclear translocator (ARNT), motif enriched in C13 rostrally upregulated genes; and ovo-like 2 (OVOL2), motif enriched in C13 caudally upregulated genes. For downregulated genes, only those downregulated caudally at C13 had any TF motif enrichment; these were enriched for a single motif recognized by Krüppel-like factors (KLF). While tags were extracted for many KLF family members, only KLF9 expression changes achieved significance, downregulated in both regions at C13 (p=0.0143 rostral, p=0 caudal).
miRNA binding site enrichment and depletion
Using FDR-based multiple testing correction, predicted binding sites for only three miRNA families were found overrepresented in the 3’ UTRs of differentially expressed genes (Table 2); none were underrepresented. Enrichment was observed only in groups with the smallest total numbers of differentially expressed genes. Additionally, the absolute numbers of genes with predicted binding sites for these miRNA families were low, suggesting lower signal for miRNA motifs than observed with TF motifs. Considering our small sample sizes, this suggests insufficient power to this study for detecting significant representation of miRNA binding sites. In the interest of obtaining a preliminary perspective, we repeated the analysis without correction. While an analysis without multiple-testing correction returns more false positives, it still captures the overall trends of the system being analyzed.
Without correction, we observed overrepresentation in one or more gene groups for binding sites of a total of 44 distinct miRNA families, including the three significant hits described above (Suppl. Table 8). Overrepresented motifs included the binding sites of several miRNAs known to be specific to non-neural tissues: miR-1/206, expressed in muscle (Lagos-Quintana et al. 2002); miR-150, expressed in mature hematopoietic cells (Zhou et al. 2007); and miR-490/490-3p, expressed in the heart, lung, and GI tract (Ji et al. 2009). These miRNAs may repress transcription of neural genes in non-neural tissues. A total of 16 distinct miRNA families were found to be underrepresented in one or more gene groups (Suppl. Table 9).
Underrepresentation of a binding site sequence indicates genes which are likely to be coexpressed with but not regulated by members of the corresponding miRNA family (Farh et al. 2005). We found depletion of binding sites for known neural-specific miRNAs miR-9 and miR-124, both of which inhibit repressors of neuronal transcripts (Packer et al. 2008; Cao et al. 2007). These observed depletions of neural, and enrichments for non-neural, surveillance miRNA binding sites lend credence to our results.
We found unique or differential enrichment across related gene groups for four miRNA families, indicating interesting targets for follow-up research. Binding sites for miR-339-5p were enriched only within the 3’ UTRs of genes with rostral upregulation after neural tube closure (p=2.71×10−4). Binding sites of the miR-141/200a family were overrepresented only among genes rostrally downregulated after closure (p=0.006). Binding sites for miR-23ab were enriched among genes upregulated post-closure (p=0.021 rostral, p=0.025 caudal), and depleted for genes downregulated post-closure (p=0.003 rostral, p=0.075 caudal). Binding sites for miR-129 were enriched for genes caudally upregulated post-closure (p=0.002) and also among genes rostrally downregulated post-closure (p=0.005).
Discussion
Our long-SAGE data demonstrate that the expression of genes in the rostral neural tube correlates highly with gene expression in the caudal neural tube at the same stage. Greater disparities were found for the genes expressed within a region during and after neural tube closure (Fig. 1A). We therefore conclude that the constellation of genes involved in human neural tube closure is generally similar in both rostral and caudal regions, with expression coordinated by developmental stage rather than spatial location. This regional similarity indicates that candidate genes for one NTD phenotype may also contribute to the etiology of other NTD phenotypes.
We validated the expression of selected genes by in situ hybridization (Fig. 2), obtaining patterns consistent with the trends identified through long-SAGE. Further validation comes from our in silico analyses, which returned enrichments for GO terms, TF motifs, and miRNA binding sites that were all consistent with the progression of embryonic development and established neural tube literature.
A total of 125 unique differentially expressed genes were associated with established NTD-relevant KEGG pathways (Table 1). The majority of these pathways exhibited complex regulation, containing both upregulated and downregulated genes; this underscores the inherent difficulties in understanding NTD causation. We additionally compared our differentially expressed genes with a list of previously reported NTD candidate genes. In all, we identified 61 known candidate genes to be differentially expressed (Suppl Table 3), 12 of which did not have uniform temporal regulation and were differentially expressed between rostral and caudal regions (Table 1). These 12 genes may have region-specific importance in NTDs, as suggested by differential effects of folate (Williams et al. 2002) and differences in frequency between rostral and caudal NTD phenotypes (Garabedian and Fraser 1993).
The 61 established candidate genes we found to have significantly different expression (Suppl. Table 3) constituted only 14% of the entire candidate set. Conversely, of 230 differentially expressed genes in the GO BP term “nervous system development” (Suppl. Table 4), only 31 (13%) were also on our list of candidate genes. These low proportions likely reflect the complexity of NTD etiology and underscore the importance of utilizing several complementary approaches to identify NTD candidate genes. Many established candidate genes are not themselves directly involved in neurulation; any approach focused on neurulation, including ours, may not identify genes of this sort. Furthermore, gene-centric approaches also may not capture interactions with the many environmental factors which have been associated with NTDs. Fully elucidating the mechanisms of NTD causation will require integrating these many different contributors into a more systemic view. In the interest of obtaining insights into this larger picture, we performed several in silico analyses on our groups of differentially expressed genes to identify regulatory factors of possible importance for future studies.
One gene which was itself upregulated after closure, MAZ, encodes a transcription factor whose binding motif was also significantly enriched in the promoters of other differentially expressed genes. MAZ is known to regulate other ubiquitous TFs, such as Specificity Protein 1 (SP1) family members (Song et al. 2001), suggesting a possible cascade wherein increased MAZ expression post-closure leads to subsequent upregulation of additional TFs. MAZ protein can bind to SP1 target motifs, which were also enriched for in C13-upregulated genes; furthermore, MAZ and SP1 have been previously implicated in neuronal differentiation via upregulation of the N-Methyl-D-aspartate receptor 1, and perhaps other neural-specific genes (Okamoto et al. 2002). However, while the mouse homolog of MAZ is highly expressed in mature brain (Song et al. 1999), the gene’s embryonic expression has been undocumented to date.
Two TFs with motifs enriched in the promoters of differentially expressed genes are previously documented NTD candidate genes: ARNT and OVOL2. ARNT knockout mice develop exencephaly (Kozak et al. 1997), and both ARNT and ARNT2 are very strongly expressed in the developing nervous system during and after neural tube closure (Aitola and Pelto-Huikko 2003). We observed enrichment for the ARNT binding motif in rostrally upregulated genes, supporting its importance to cranial development. In contrast, while mice lacking the TF OVOL2 primarily exhibit cranial defects (Mackay et al. 2006), we observed OVOL2 motif enrichment only among genes upregulated caudally. Ovol2 is normally expressed highly in the forebrain region and at low levels throughout the entire embryo; however, one Ovol2 mutant embryo has been documented with spina bifida (Mackay et al. 2006), lending further support to its potential involvement in caudal neurulation.
Genes downregulated in the caudal region after neural tube closure were enriched only for the binding motif recognized by Krüppel-binding factors (Table 3). This could result from either expression of caudal genes being driven by KLFs during neurulation, or these genes being repressed by KLFs after neurulation. Individual KLFs may function as either activators or repressors depending on the context of binding sequence and cofactors (Kaczynski et al. 2003). Expression patterns of KLF3, KLF6, and KLF7 have been previously determined in the developing mouse brain. Specifically, KLF3 expression increases in the midbrain from day E7.5 through E10.5 (Crossley et al. 1996); KLF6 is expressed in surrounding mesenchyme after neural tube closure (E11.5 through E16.5) (Laub et al. 2001b); and KLF7 expression is very low in the brain at E9.0, but increases to extensive expression at E11.5 (Laub et al. 2001a). Mouse neural tube closure completes on day E10 (Greene and Copp 2009), thus E10 corresponds roughly to human C13. Though not achieving significance, our caudal tag counts for KLF3 and KLF6 suggest increasing expression from C12 to C13, consistent with expression patterns in mouse and a potential repressive role for these transcription factors against neurulation genes. Rostral tag counts for these genes were too low to observe any trends.
In testing for the enrichment of miRNA binding sites in 3’ UTRs of differentially expressed genes, we identified four miRNA families which may merit further investigation: miR-339-5p, miR-141/200a, miR-23ab, and miR-129/129-5p. It is thought that some 10% of miRNAs, including miR-200a and miR-23b, are regulated by DNA methylation (Han et al. 2007). Given the increasing evidence which associates aberrant DNA methylation with NTDs (Chen et al. 2010; Chang et al. 2011), misregulation of these miRNAs through hypomethylation may contribute to NTD causation.
Predicted binding sites of miR-339-5p were enriched among genes upregulated rostrally at C13. They were additionally enriched in genes more upregulated at C13 in the caudal region than the rostral, suggesting that this miRNA plays a role in both regions. Its parent stem-loop miR-339 is downregulated in fetal mouse brain upon exposure to ethanol (Wang et al. 2009), a known risk factor for NTDs. miR-339-5p itself has been shown to be downregulated in human anencephaly cases (Zhang et al. 2010). miR-339-5p is known to regulate the histone demethylase KDM6B, and implicated in the susceptibility of Splotch mice to caudal NTDs (Ichi et al. 2010). KDM6B removes H3K27 methyl marks, which have been suggested to influence de novo methylation (Cedar and Bergman 2009) and shown to direct the restoration of perturbed DNA methylation (Wong et al. 2011). These close ties with DNA methylation suggest that miR-339-5p merits further investigation as a potential contributor to NTD causation.
Predicted binding sites of the miR-141/200a family were enriched among genes downregulated rostrally at C13, suggesting that this miRNA may participate in repressing those genes. miR-200a is upregulated in DNMT3b knockout cells (Han et al. 2007), indicating that it is normally repressed by DNA methylation. No member of this family has been explicitly associated with NTDs to our knowledge, but the miR-200 cluster has been implicated in olfactory neurogenesis (Choi et al. 2008) and inhibits the mesenchyme to epithelial transition (Korpal et al. 2008) necessary for both neural crest formation and secondary neurulation. Whether these miRNAs have additional roles in rostral neural tube closure, as our findings suggest, remains to be determined.
Predicted binding sites of the miR-23ab family were enriched for in genes upregulated in both regions post-closure. This suggests that miR-23 family members may play an important role in the temporal regulation of genes around neural tube closure, perhaps by preventing pre-closure translation of genes which are normally expressed post-closure. In adult brains, mir-23 expression is specific to astrocytes, where it is constitutively expressed (Smirnova et al. 2005). Little is known about miR-23 expression during development, but miR-23b has been shown to be upregulated both in DNMT3b knockout cells (Han et al. 2007) and in human anencephaly cases (Zhang et al. 2010), supporting a potential relevance to NTDs.
Predicted binding sites of the miR-129 family were enriched in genes upregulated caudally at C13, but also in genes downregulated rostrally at C13. This suggests that miR-129 may have region-specific roles in rostral and caudal neural tissue. miR-129 is known to be highly expressed in human brain, and present but minimally expressed in other tissues (Lau et al. 2008; Liang et al. 2007). miR-129 has recently been shown to regulate Gsk3b, an inhibitor of Wnt signaling (Liu et al. 2011); as Wnt signaling is integral to neural tube formation, this further indicates that miR-129 may be important to NTD etiology.
To our knowledge, this is the first study to quantitatively compare gene expression before and after neural tube closure in different regions of the human neural tube, providing a uniquely holistic perspective on the normal process of neurulation. Our findings underscore the overall similarity of genetic mechanisms underlying closure throughout the neural tube, suggesting that candidate genes associated with one NTD phenotype (e.g. OVOL2) are likely to contribute to the etiology of other NTD phenotypes as well. From our in silico analyses and corroborating literature, we additionally propose several miRNAs as potential novel regulatory factors of neurulation, though further research and functional validation remains necessary. In conclusion, our transcriptome profiling of genes involved in normal human neural tube closure has highlighted the similarities of closure in rostral and caudal regions, and suggests new candidates for NTD risk that should be further explored.
Supplementary Material
SAGE tag counts for genes having tags with discordant differential expression A) before and after neurulation or B) by region at C13. CAU12, caudal C12; CAU13, caudal C13; ROS13, rostral C13.
Results of validation experiments performed for selected differentially expressed genes. CAU12, caudal C12; CAU13, caudal C13; ROS12, rostral C12; ROS13, rostral C13; ISH, in situ hybridization ; p, present; a, absent; p/a, ambiguous; *in situ image presented in Figure 2; †validation discrepant with SAGE data.
Established NTD candidate genes having significantly different expression surrounding neural tube closure either by region or Carnegie stage. Up, upregulated genes; down, downregulated genes; CAU12, caudal C12; CAU13, caudal C13; ROS12, rostral C12; ROS13, rostral C13. A p-value of 0 indicates significance beyond the ability of the software to report.
Differentially expressed genes which are also members of the GO Biological Process term “nervous system development” (GO:0007399). Up, upregulated genes; down, downregulated genes; CAU12, caudal C12; CAU13, caudal C13; ROS12, rostral C12; ROS13, rostral C13; *genes also in our list of known NTD candidate genes. A p-value of 0 indicates significance beyond the ability of the software to report.
Enriched GO Biological Process terms common to genes upregulated both rostrally and caudally at C13. P-values corrected using Benjamini-Hochberg FDR method.
Enriched GO Biological Process terms common to genes downregulated both rostrally and caudally at C13. P-values corrected using Benjamini-Hochberg FDR method.
GO Biological Process terms solely enriched among genes rostrally upregulated at C13. P-values corrected using Benjamini-Hochberg FDR method.
miRNA families with target motifs overrepresented in the 3’ UTRs of differentially expressed genesets. Values in italic are suggestive. CAU12, caudal C12; CAU13, caudal C13; ROS12, rostral C12; ROS13, rostral C13.
miRNA families with target motifs underrepresented in the 3’ UTRs of differentially expressed genesets. Values in italic are suggestive. CAU12, caudal C12; CAU13, caudal C13; ROS12, rostral C12; ROS13, rostral C13.
Table 4.
miRNA families with significant overrepresentation of their binding sequences in the 3’ UTRs of genes differently regulated around neurulation. Up, upregulated genes; down, downregulated genes; CAU12, caudal C12; CAU13, caudal C13.
| miRNA Family | Group | p | Genes | Genes in Group |
|---|---|---|---|---|
| miR-181 | Up CAU12 vs. CAU13 | 0.004 | 5 | 22 |
| miR-148/158 | Down CAU12 vs. CAU13 | 0.004 | 3 | 24 |
| miR-30a | Down CAU12 vs. CAU13 | 0.019 | 5 | 24 |
Acknowledgments
The authors dedicate this work to the memory of the late Dr. Marcy Speer, who was its major driving force. Samples for this study were collected under National Institutes of Health grant R01-NS039818.
Footnotes
The expression data from this study is in the process of being submitted to the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/).
Literature Cited
- Aitola MH, Pelto-Huikko MT. Expression of Arnt and Arnt2 mRNA in Developing Murine Tissues. Journal of Histochemistry & Cytochemistry. 2003;51:41–54. doi: 10.1177/002215540305100106. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological) 1995;57:289–300. [Google Scholar]
- Cao X, Pfaff SL, Gage FH. A functional study of miR-124 in the developing neural tube. Genes Dev. 2007;21:531–536. doi: 10.1101/gad.1519207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nature Reviews Genetics. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- Chang H, Zhang T, Zhang Z, Bao R, Fu C, Wang Z, Bao Y, Li Y, Wu L, Zheng X, Wu J. Tissue-specific distribution of aberrant DNA methylation associated with maternal low-folate status in human neural tube defects. Journal of Nutritional Biochemistry. 2011 doi: 10.1016/j.jnutbio.2010.10.003. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Chang TI, Horal M, Jain SK, Wang F, Patel R, Loeken MR. Oxidant regulation of gene expression and neural tube development: Insights gained from diabetic pregnancy on molecular causes of neural tube defects. Diabetologia. 2003;46:538–545. doi: 10.1007/s00125-003-1063-2. [DOI] [PubMed] [Google Scholar]
- Chen X, Guo J, Lei Y, Zou J, Lu X, Bao Y, Wu L, Wu J, Zheng X, Shen Y, Wu B-L, Zhang T. Global DNA hypomethylation is associated with NTD-affected pregnancy: a case-control study. Birth Defects Res A. 2010;88:575–581. doi: 10.1002/bdra.20670. [DOI] [PubMed] [Google Scholar]
- Choi PS, Zakhary L, Choi W-Y, Caron S, Alvarez-Saavedra E, Miska EA, McManus M, Harfe B, Giraldez AJ, Horvitz HR, Schier AF, Dulac C. Members of the miRNA-200 family regulate olfactory neurogenesis. Neuron. 2008;57:41–55. doi: 10.1016/j.neuron.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp AJ, Greene NDE. Genetics and Development of Neural Tube Defects. J Pathol. 2010;220:227–230. doi: 10.1002/path.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp AJ, Greene NDE, Murdoch JN. The genetic basis of mammalian neurulation. Nat Rev Genet. 2003;4:784–793. doi: 10.1038/nrg1181. [DOI] [PubMed] [Google Scholar]
- Crossley M, Whitelaw E, Perkins A, Williams G, Fujiwara Y, Orkin S. Isolation and characterization of the cDNA encoding BKLF/TEF-2, a major CACCC-box-binding protein in erythroid cells and selected other cells. Mol. Cell. Biol. 1996;16:1695–1705. doi: 10.1128/mcb.16.4.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detrait ER, George TM, Etchevers HC, Gilbert JR, Vekemans M, Speer MC. Human neural tube defects: developmental biology, epidemiology, and genetics. Neurotoxicol Teratol. 2005;27:515–524. doi: 10.1016/j.ntt.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkon R, Linhart C, Sharan R, Shamir R, Shiloh Y. Genome-Wide In Silico Identification of Transcriptional Regulators Controlling the Cell Cycle in Human Cells. Genome Res. 2003;13:773–780. doi: 10.1101/gr.947203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etchevers HC, Vincent C, Le Douarin NM, Couly GF. The cephalic neural crest provides pericytes and smooth muscle cells to all blood vessels of the face and forebrain. Development. 2001;128:1059–1068. doi: 10.1242/dev.128.7.1059. [DOI] [PubMed] [Google Scholar]
- Farh KK-H, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- Garabedian BH, Fraser FC. Upper and lower neural tube defects: an alternate hypothesis. J. Med. Genet. 1993;30:849–851. doi: 10.1136/jmg.30.10.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene NDE, Copp AJ. Development of the Vertebrate Central Nervous System: Formation of the Neural Tube. Prenat Diagn. 2009;29:303–311. doi: 10.1002/pd.2206. [DOI] [PubMed] [Google Scholar]
- Groenen PM, Peer PG, Wevers RA, Swinkels DW, Franke B, Mariman EC, Steegers-Theunissen RP. Maternal myo-inositol, glucose, and zinc status is associated with the risk of offspring with spina bifida. American Journal of Obstetrics & Gynecology. 2003;189:1713–1719. doi: 10.1016/s0002-9378(03)00807-x. [DOI] [PubMed] [Google Scholar]
- Han L, Witmer PD, Casey E, Valle D, Sukumar S. DNA methylation regulates MicroRNA expression. Cancer Biol. Ther. 2007;6:1284–1288. doi: 10.4161/cbt.6.8.4486. [DOI] [PubMed] [Google Scholar]
- Harris MJ, Juriloff DM. An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Res A. 2010;88:653–669. doi: 10.1002/bdra.20676. [DOI] [PubMed] [Google Scholar]
- Harris MJ, Juriloff DM. Mouse Mutants With Neural Tube Closure Defects and Their Role in Understanding Human Neural Tube Defects. Birth Defects Res A. 2007;79:187–210. doi: 10.1002/bdra.20333. [DOI] [PubMed] [Google Scholar]
- Ichi S, Costa FF, Bischof JM, Nakazaki H, Shen Y-W, Boshnjaku V, Sharma S, Mania-Farnell B, McLone DG, Tomita T, Soares MB, Mayanil CSK. Folic acid remodels chromatin on Hes1 and Neurog2 promoters during caudal neural tube development. J. Biol. Chem. 2010;285:36922–36932. doi: 10.1074/jbc.M110.126714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X, Takahashi R, Hiura Y, Hirokawa G, Fukushima Y, Iwai N. Plasma miR-208 as a Biomarker of Myocardial Injury. Clinical Chemistry. 2009;55:1944–1949. doi: 10.1373/clinchem.2009.125310. [DOI] [PubMed] [Google Scholar]
- Kaczynski J, Cook T, Urrutia R. Sp1- and Krüppel-like transcription factors. Genome Biol. 2003;4:206. doi: 10.1186/gb-2003-4-2-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucl. Acids Res. 2011 doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpal M, Lee ES, Hu G, Kang Y. The miR-200 Family Inhibits Epithelial-Mesenchymal Transition and Cancer Cell Migration by Direct Targeting of E-cadherin Transcriptional Repressors ZEB1 and ZEB2. Journal of Biological Chemistry. 2008;283:14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak KR, Abbott B, Hankinson O. ARNT-deficient mice and placental differentiation. Dev. Biol. 1997;191:297–305. doi: 10.1006/dbio.1997.8758. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of Tissue-Specific MicroRNAs from Mouse. Current Biology. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- Lau P, Verrier JD, Nielsen JA, Johnson KR, Notterpek L, Hudson LD. Identification of dynamically regulated microRNA and mRNA networks in developing oligodendrocytes. J. Neurosci. 2008;28:11720–11730. doi: 10.1523/JNEUROSCI.1932-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laub F, Aldabe R, Friedrich V, Jr, Ohnishi S, Yoshida T, Ramirez F. Developmental Expression of Mouse Krüppel-like Transcription Factor KLF7 Suggests a Potential Role in Neurogenesis. Developmental Biology. 2001a;233:305–318. doi: 10.1006/dbio.2001.0243. [DOI] [PubMed] [Google Scholar]
- Laub F, Aldabe R, Ramirez F, Friedman S. Embryonic expression of Krüppel-like factor 6 in neural and non-neural tissues. Mechanisms of Development. 2001b;106:167–170. doi: 10.1016/s0925-4773(01)00419-1. [DOI] [PubMed] [Google Scholar]
- Liang Y, Ridzon D, Wong L, Chen C. Characterization of microRNA expression profiles in normal human tissues. BMC Genomics. 2007;8:166. doi: 10.1186/1471-2164-8-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Huang T, Zhao X, Cheng L. MicroRNAs modulate the Wnt signaling pathway through targeting its inhibitors. Biochemical and Biophysical Research Communications. 2011;408:259–264. doi: 10.1016/j.bbrc.2011.04.009. [DOI] [PubMed] [Google Scholar]
- Lu J, Lal A, Merriman B, Nelson S, Riggins G. A comparison of gene expression profiles produced by SAGE, long SAGE, and oligonucleotide chips. Genomics. 2004;84:631–636. doi: 10.1016/j.ygeno.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Mackay DR, Hu M, Li B, Rhéaume C, Dai X. The mouse Ovol2 gene is required for cranial neural tube development. Developmental Biology. 2006;291:38–52. doi: 10.1016/j.ydbio.2005.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- Man MZ, Wang X, Wang Y. POWER_SAGE: comparing statistical tests for SAGE experiments. Bioinformatics. 2000;16:953–959. doi: 10.1093/bioinformatics/16.11.953. [DOI] [PubMed] [Google Scholar]
- Mao J, McKean DM, Warrier S, Corbin JG, Niswander L, Zohn IE. The iron exporter ferroportin 1 is essential for development of the mouse embryo, forebrain patterning and neural tube closure. Development. 2010;137:3079–3088. doi: 10.1242/dev.048744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies EH, Innis JW. eSAGE: managing and analysing data generated with serial analysis of gene expression (SAGE) Bioinformatics. 2000;16:650–651. doi: 10.1093/bioinformatics/16.7.650. [DOI] [PubMed] [Google Scholar]
- McMahon K, Cope H, Heise E, Eubanks S, Gregory SG, Ashley-Koch A. Descriptive Life Experiences of Adults with Myelomeningocele. Presented at the 7th International Conference on Neural Tube Defects; Austin, Texas. 2011. [Google Scholar]
- Okamoto S, Sherman K, Bai G, Lipton SA. Effect of the ubiquitous transcription factors, SP1 and MAZ, on NMDA receptor subunit type 1 (NR1) expression during neuronal differentiation. Brain Res. Mol. Brain Res. 2002;107:89–96. doi: 10.1016/s0169-328x(02)00440-0. [DOI] [PubMed] [Google Scholar]
- Packer AN, Xing Y, Harper SQ, Jones L, Davidson BL. The Bifunctional microRNA miR-9/miR-9* Regulates REST and CoREST and Is Downregulated in Huntington’s Disease. J. Neurosci. 2008;28:14341–14346. doi: 10.1523/JNEUROSCI.2390-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrini J, Damus K, Russell R, Poschman K, Davidoff MJ, Mattison D. Contribution of birth defects to infant mortality in the United States. Teratology. 2002;66:S3–S6. doi: 10.1002/tera.90002. [DOI] [PubMed] [Google Scholar]
- Saha S, Sparks AB, Rago C, Akmaev V, Wang CJ, Vogelstein B, Kinzler KW, Velculescu VE. Using the transcriptome to annotate the genome. Nat. Biotechnol. 2002;20:508–512. doi: 10.1038/nbt0502-508. [DOI] [PubMed] [Google Scholar]
- Scott HS, Chrast R. Global Transcript Expression Profiling by Serial Analysis of Gene Expression (SAGE) In: Setlow JK, editor. Genetic Engineering: Principles and Methods. Boston: Kluwer Academic Publishers; 2002. pp. 201–219. [DOI] [PubMed] [Google Scholar]
- Shamir R, Maron-Katz A, Tanay A, Linhart C, Steinfeld I, Sharan R, Shiloh Y, Elkon R. EXPANDER - an integrative program suite for microarray data analysis. BMC Bioinformatics. 2005;6:232. doi: 10.1186/1471-2105-6-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova L, Gräfe A, Seiler A, Schumacher S, Nitsch R, Wulczyn FG. Regulation of miRNA expression during neural cell specification. Eur. J. Neurosci. 2005;21:1469–1477. doi: 10.1111/j.1460-9568.2005.03978.x. [DOI] [PubMed] [Google Scholar]
- Song J, Mangold M, Suske G, Geltinger C, Kanazawa I, Sun K, Yokoyama KK. Characterization and promoter analysis of the mouse gene for transcription factor Sp4. Gene. 2001;264:19–27. doi: 10.1016/s0378-1119(01)00328-6. [DOI] [PubMed] [Google Scholar]
- Song J, Murakami H, Tsutsui H, Ugai H, Geltinger C, Murata T, Matsumura M, Itakura K, Kanazawa I, Sun K, Yokoyama KK. Structural organization and expression of the mouse gene for Pur-1, a highly conserved homolog of the human MAZ gene. Eur. J. Biochem. 1999;259:676–683. doi: 10.1046/j.1432-1327.1999.00081.x. [DOI] [PubMed] [Google Scholar]
- Ulitsky I, Laurent LC, Shamir R. Towards computational prediction of microRNA function and activity. Nucl. Acids Res. 2010;38:e160–e160. doi: 10.1093/nar/gkq570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- Waghray A, Schober M, Feroze F, Yao F, Virgin J, Chen YQ. Identification of Differentially Expressed Genes by Serial Analysis of Gene Expression in Human Prostate Cancer. Cancer Res. 2001;61:4283–4286. [PubMed] [Google Scholar]
- Wang L-L, Zhang Z, Li Q, Yang R, Pei X, Xu Y, Wang J, Zhou S-F, Li Y. Ethanol exposure induces differential microRNA and target gene expression and teratogenic effects which can be suppressed by folic acid supplementation. Hum. Reprod. 2009;24:562–579. doi: 10.1093/humrep/den439. [DOI] [PubMed] [Google Scholar]
- Williams LJ, Mai CT, Edmonds LD, Shaw GM, Kirby RS, Hobbs CA, Sever LE, Miller LA, Meaney FJ, Levitt M. Prevalence of spina bifida and anencephaly during the transition to mandatory folic acid fortification in the United States. Teratology. 2002;66:33–39. doi: 10.1002/tera.10060. [DOI] [PubMed] [Google Scholar]
- Wilting J, Eichmann A, Christ B. Expression of the avian VEGF receptor homologues Quek1 and Quek2 in blood-vascular and lymphatic endothelial and non-endothelial cells during quail embryonic development. Cell Tissue Res. 1997;288:207–223. doi: 10.1007/s004410050807. [DOI] [PubMed] [Google Scholar]
- Wong C-M, Wong CC-L, Ng Y-L, Au SL-K, Ko FC-F, Ng IO-L. Transcriptional Repressive H3K9 and H3K27 Methylations Contribute to DNMT1-Mediated DNA Methylation Recovery. PLoS ONE. 2011;6:e16702. doi: 10.1371/journal.pone.0016702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P-T, Li Y-J, Qin X-J, Scherzer CR, Xu H, Schmechel DE, Hulette CM, Ervin J, Gullans SR, Haines J, Pericak-Vance MA, Gilbert JR. Differences in apolipoprotein E3/3 and E4/4 allele-specific gene expression in hippocampus in Alzheimer disease. Neurobiology of Disease. 2006;21:256–275. doi: 10.1016/j.nbd.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Chang H, Li Y, Zhang T, Zou J, Zheng X, Wu J. MicroRNAs: Potential regulators involved in human anencephaly. The International Journal of Biochemistry & Cell Biology. 2010;42:37–374. doi: 10.1016/j.biocel.2009.11.023. [DOI] [PubMed] [Google Scholar]
- Zhou B, Wang S, Mayr C, Bartel DP, Lodish HF. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. PNAS. 2007;104:7080–7085. doi: 10.1073/pnas.0702409104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SAGE tag counts for genes having tags with discordant differential expression A) before and after neurulation or B) by region at C13. CAU12, caudal C12; CAU13, caudal C13; ROS13, rostral C13.
Results of validation experiments performed for selected differentially expressed genes. CAU12, caudal C12; CAU13, caudal C13; ROS12, rostral C12; ROS13, rostral C13; ISH, in situ hybridization ; p, present; a, absent; p/a, ambiguous; *in situ image presented in Figure 2; †validation discrepant with SAGE data.
Established NTD candidate genes having significantly different expression surrounding neural tube closure either by region or Carnegie stage. Up, upregulated genes; down, downregulated genes; CAU12, caudal C12; CAU13, caudal C13; ROS12, rostral C12; ROS13, rostral C13. A p-value of 0 indicates significance beyond the ability of the software to report.
Differentially expressed genes which are also members of the GO Biological Process term “nervous system development” (GO:0007399). Up, upregulated genes; down, downregulated genes; CAU12, caudal C12; CAU13, caudal C13; ROS12, rostral C12; ROS13, rostral C13; *genes also in our list of known NTD candidate genes. A p-value of 0 indicates significance beyond the ability of the software to report.
Enriched GO Biological Process terms common to genes upregulated both rostrally and caudally at C13. P-values corrected using Benjamini-Hochberg FDR method.
Enriched GO Biological Process terms common to genes downregulated both rostrally and caudally at C13. P-values corrected using Benjamini-Hochberg FDR method.
GO Biological Process terms solely enriched among genes rostrally upregulated at C13. P-values corrected using Benjamini-Hochberg FDR method.
miRNA families with target motifs overrepresented in the 3’ UTRs of differentially expressed genesets. Values in italic are suggestive. CAU12, caudal C12; CAU13, caudal C13; ROS12, rostral C12; ROS13, rostral C13.
miRNA families with target motifs underrepresented in the 3’ UTRs of differentially expressed genesets. Values in italic are suggestive. CAU12, caudal C12; CAU13, caudal C13; ROS12, rostral C12; ROS13, rostral C13.