

Abstract
We present the first genomic-scale analysis addressing the phylogenetic position of turtles, using over 1000 loci from representatives of all major reptile lineages including tuatara. Previously, studies of morphological traits positioned turtles either at the base of the reptile tree or with lizards, snakes and tuatara (lepidosaurs), whereas molecular analyses typically allied turtles with crocodiles and birds (archosaurs). A recent analysis of shared microRNA families found that turtles are more closely related to lepidosaurs. To test this hypothesis with data from many single-copy nuclear loci dispersed throughout the genome, we used sequence capture, high-throughput sequencing and published genomes to obtain sequences from 1145 ultraconserved elements (UCEs) and their variable flanking DNA. The resulting phylogeny provides overwhelming support for the hypothesis that turtles evolved from a common ancestor of birds and crocodilians, rejecting the hypothesized relationship between turtles and lepidosaurs.
Keywords: turtles, ultraconserved elements, phylogenomics, evolution, archosaurs
1. Introduction
The evolutionary origin of turtles has confounded the understanding of vertebrate evolution [1] (figure 1). Historically, turtles were thought to be early-diverging reptiles, called anapsids, based on their skull morphology and traits such as dermal armour [2]. Recent morphological studies that included soft tissue and developmental characters [3] allied turtles with lepidosaurs, a group including squamates (lizards and snakes) and tuataras. However, homoplasy stemming from the derived skeletal specializations of turtles limits the utility of phylogenetic inference based on morphological data to resolve turtle placement [4,5].
Figure 1.
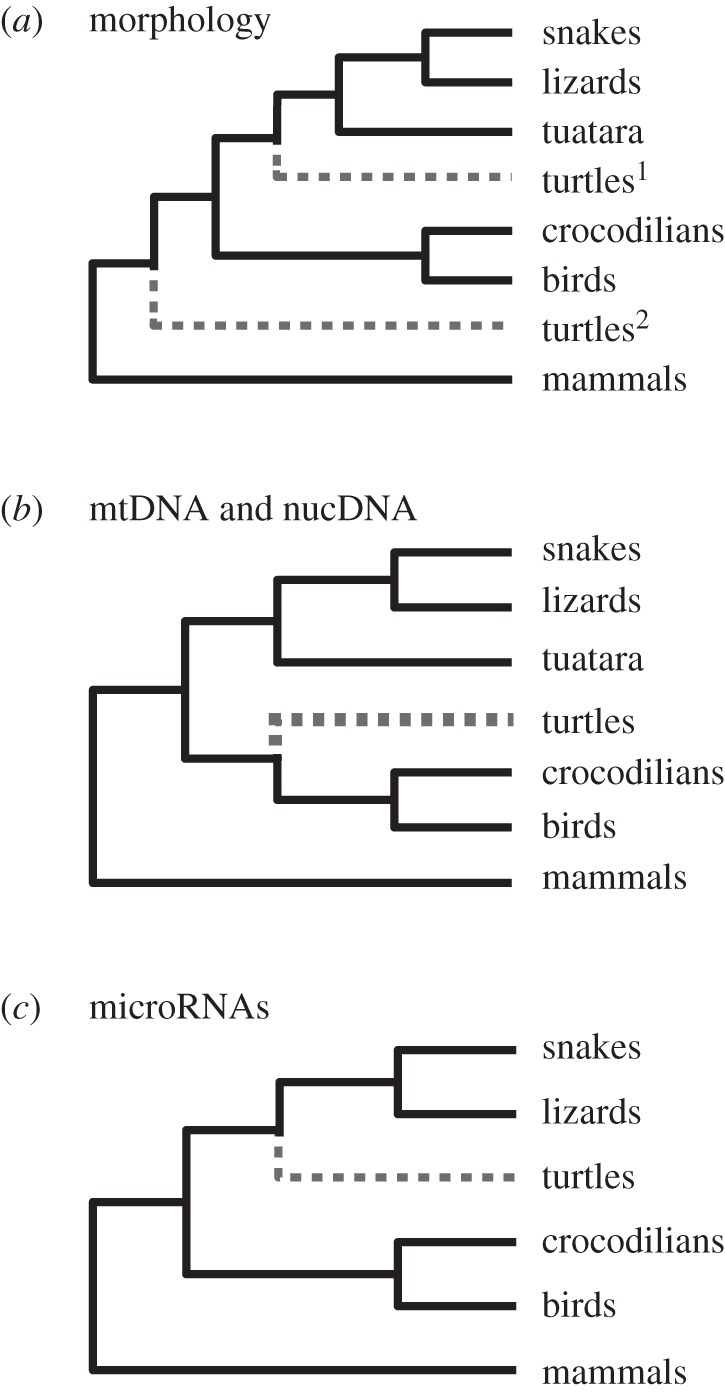
(a) Depicts the primary morphological hypotheses: turtles most basally branching reptilian lineage [2]2 or turtles related to lepidosaurs [3].1 (b) Depicts the primary molecular hypothesis of a turtle–archosaur alliance [4–14]. (c) Depicts the tree derived from miRNA loci [15].
Molecular studies using mitochondrial [4,6–8,16] and nuclear DNA [5,9–14,17] typically place turtles sister to archosaurs (crocodilians and birds; figure 1). This molecular hypothesis was recently contradicted by a phylogeny reconstructed from microRNAs [15] that allied turtles with lepidosaurs. Lyson et al. [15] suggested that prior molecular evidence for a turtle–archosaur relationship may be the result of analytical artefacts. If true, the hypothetical relationship between turtles and lepidosaurs (Ankylpoda) should appear throughout the genomes of these organisms.
Here, we test the Ankylopoda hypothesis and address the evolutionary origin of turtles. We reconstruct a reptile phylogeny using ultraconserved elements (UCEs) [18] and their flanking sequence that we obtained using sequence capture of DNA from a tuatara and two species each of crocodilians, squamates and turtles (table 1). We used UCEs because they are easily aligned portions of extremely divergent genomes [19], allowing many loci to be interrogated across evolutionary timescales, and because sequence variability within UCEs increases with distance from the core of the targeted UCE [20], suggesting that phylogenetically informative content in flanking regions can inform hypotheses spanning different evolutionary timescales. To break up long branches and mitigate potential problems with long-branch attraction, we selected species representing the span of diversity within major reptilian lineages (i.e. the most divergent crocodilians, lepidosaurs and turtles).
Table 1.
University of California Santa Cruz (UCSC) genome build or specimen ID for each sample, the number of ∼100 bp sequence reads, and the total number of UCEs assembled.
| common name | binomial | specimen ID/genome build | reads | assembled UCEs |
|---|---|---|---|---|
| African helmeted turtle | Pelomedusa subrufa | H20145a | 11 200 032 | 1972 |
| American alligator | Alligator mississippiensis | HCD-2620a | 3 528 983 | 2320 |
| Carolina anole | Anolis carolinensis | H16061a | 3 100 147 | 2111d |
| corn snake | Pantherophis guttata | H15909a | 3 362 738 | 2168 |
| human | Homo sapiens | UCSC hg19 | NA | 1748 |
| painted turtle | Chrysemys picta | H2662a | 4 467 644 | 2261 |
| red junglefowl | Gallus gallus | UCSC galGal3 | NA | 2360d |
| saltwater crocodile | Crocodylus porosus | LM-67b | 3 261 088 | 2218 |
| tuatara | Sphenodon tuatara | UMFS-10956c | 5 651 932 | 2199 |
| zebra finch | Taeniopygia guttata | UCSC taeGut1 | NA | 2345d |
aFrom the LSU Museum of Natural Science.
bFrom the Darwin Crocodile Farm courtesy of L. Miles, S. Isberg and C. Moran.
cFrom the University of Michigan Museum of Zoology courtesy of R. Nussbaum and G. Schneider.
dAlthough we identified 2386 UCEs in these organisms, from which we designed capture probes, owing to slight adjustments to matching and filtering algorithms, we only recover ca 98% of these UCEs when re-screening these genomic sequences.
2. Material and methods
We enriched DNA libraries prepared with Nextera kits (Epicentre, Inc., Madison, WI, USA) using a synthesis (Mycroarray, Inc., Ann Arbor, MI, USA or Agilent, Inc., Santa Clara, CA, USA) of RNA probes [20] targeting 2386 UCEs and their flanking sequence. We generated sequences for each enriched library using single-end, 100-base sequencing on an Illumina GAIIx. After quality filtering, we assembled reads into contigs using Velvet [21], and we matched contigs to the UCE loci, removing duplicate hits. We generated alignments using MUSCLE [22], and we excluded loci having missing data in any taxon. Following alignment, we estimated the appropriate finite-sites substitution model for each locus using MrAIC.
We prepared a concatenated dataset by partitioning loci by substitution model prior to analysis using two runs of MrBayes [23] for 5 000 000 iterations (four chains per run; burn-in: 50%; thinning: 100). We also used each alignment to estimate gene trees incorporating 1000 multi-locus bootstrap replicates, which we integrated into STEAC and STAR [24] species trees. Additional details concerning UCE sequence capture methods and phylogenetic methods are available in Faircloth et al. [20].
3. Results
We enriched genomic DNA for UCEs in corn snake (Pantherophis guttata), African helmeted turtle (Pelomedusa subrufa), painted turtle (Chrysemys picta), American alligator (Alligator mississippiensis), saltwater crocodile (Crocodylus porosus) and tuatara (Sphenodon tuatara) (table 1). We sequenced a mean of 4.9 million reads from each library, and from these reads, we assembled an average of 2648 (±314 s.d.) contigs.
We supplemented these taxa with UCEs extracted from the chicken (Gallus gallus), zebra finch (Taeniopygia guttata), Carolina anole lizard (Anolis carolinensis) and human (Homo sapiens) genome sequences. We combined the in silico and in vitro data and generated alignments across all taxa and excluded all loci having missing data from any taxon. This resulted in 1145 individual alignments with a mean length of 406 bp (±100 bp s.d.) per alignment, totalling 465 Kbp of sequence. Tracer showed that both Bayesian analyses converged quickly, having effective sample size (ESS) scores for log likelihood of 170 and 220. Because posterior probabilities for all nodes were 1.0, AWTY (http://ceb.csit.fsu.edu/awty) showed zero variance in the tree topology throughout either run. Bayesian analysis of concatenated alignments and species-tree analysis of 1145 independent gene histories showed turtles to be the sister lineage of extant archosaurs with complete support (figure 2). Removing the snake, which had a very long branch, and re-running all analyses did not change the results.
Figure 2.
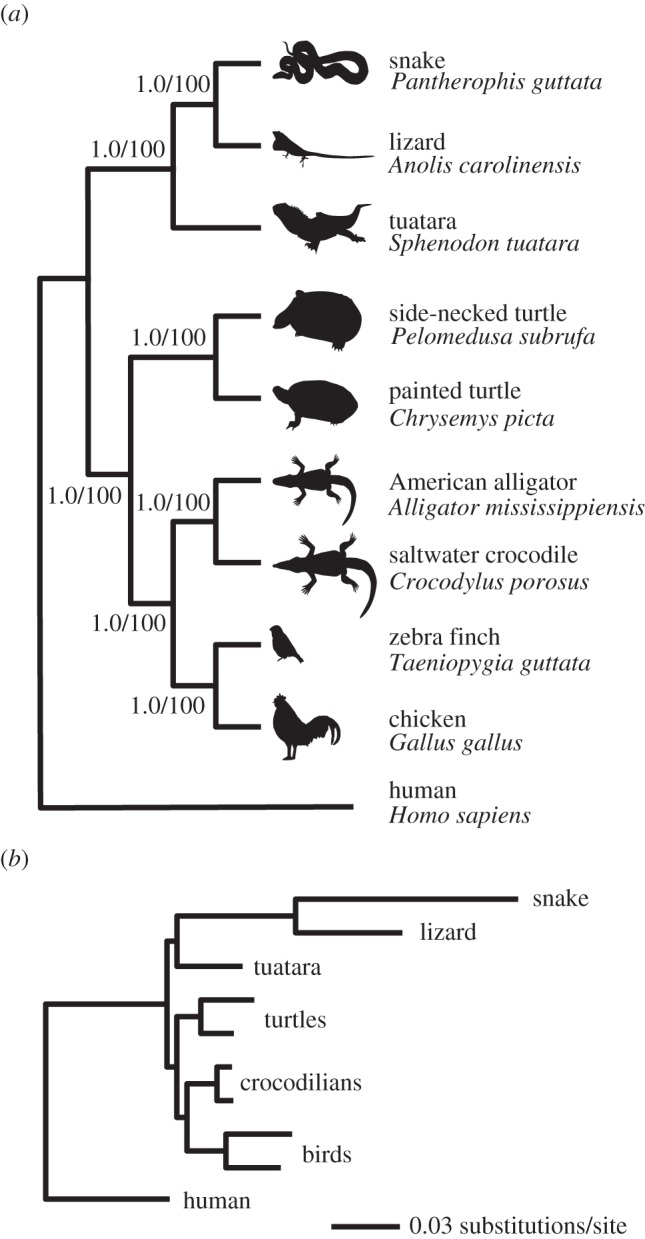
(a) Reptilian phylogeny estimated from 1145 ultra-conserved loci using Bayesian analysis of concatenated data and species-tree methods, yielding identical topologies. Node labels indicate posterior probability/bootstrap support. (b) Phylogram of the UCE phylogeny generated with STEAC.
4. Discussion
Genomic-scale phylogenetic analysis of 1145 nuclear UCE loci agreed with most other molecular studies [4–14], supporting a sister relationship between turtles and archosaurs. We found no support for the turtles–lepidosaur relationship predicted by the Ankylopoda hypothesis [15] (figure 2). The combination of taxonomic sampling, the genome-wide scale of the sampling and the robust results obtained, regardless of analytical method, indicates that the turtle–archosaur relationship is unlikely to be caused by long-branch attraction or other analytical artefacts.
Although our results corroborate earlier studies, many of these studies did not include tuatara. Because tuatara is an early-diverging lepidosaur, it is important to include this taxon in studies of turtle evolution as it breaks up the long-branch leading to squamates (figure 2b). Of the studies including tuatara, two [6,11] found results similar to this study, but both were based on a single locus. The third study [5] was unable to produce a well-resolved tree from four nuclear genes when the authors included tuatara in the dataset. Our study is the first to produce a well-resolved reptile tree that includes the tuatara and multiple loci.
The discrepancy between our results showing a strong turtle–archosaur relationship and microRNA (miRNA) results, which showed a strong turtle–lepidosaur relationship, may be due to several factors. Lyson et al. [15] used the presence of four miRNA gene families, detected among turtles and lepidosaurs and undetected in the other taxa analysed, to support the turtle–lepidosaur relationship. Because complete genomes are unavailable for turtles, tuatara and crocodilians, and because expressed miRNA data are lacking for most reptiles, the authors collected miRNA sequences from small RNA expression libraries. miRNAs have tissue and developmental-stage-specific expression profiles [25,26], which could make the detection of certain miRNAs challenging. Because preparing and sequencing libraries is a biased sampling process, the detection probability for specific targets is variable, and some miRNAs are likely to be more easily detected than others. Thus, failures to detect miRNA families are not equivalent to the absence of miRNA families [27]. We suggest that at least some of the four miRNA families currently thought to be unique to lizards and turtles may be present but as yet undiscovered in other reptiles.
This work is the first to investigate the placement of turtles within reptiles using a genomic-scale analysis of single-copy DNA sequences and a complete sampling of the major relevant evolutionary lineages. Because UCEs are conserved across most vertebrate groups [20] and found in groups including yeast and insects [19], our framework is generalizable beyond this study and relevant to resolving ancient phylogenetic enigmas throughout the tree of life [28]. This approach to high-throughput phylogenomics—based on thousands of loci—is likely to fundamentally change the way that systematists gather and analyse data.
(a). Additional information
We provide all data and links to software via Dryad repository (doi:10.5061/dryad.75nv22qj) and GenBank (JQ868813–JQ885411).
Acknowledgements
We thank R. Nilsen, K. Jones, M. Harvey, R. Nussbaum, G. Schneider, D. Ray, D. Peterson, C. Moran, L. Miles, S. Isberg, C. Mancuso, S. Herke, two anonymous reviewers and the LSU Genomic Facility. National Science Foundation grants DEB-1119734, DEB-0841729 and DEB-0956069, and an Amazon Web Services Education Grant supported this study. N.G.C., B.C.F., J.E.M. and T.C.G. designed the study; N.G.C. and B.C.F. performed phylogenetic analysis; B.C.F. created datasets; J.E.M. performed laboratory work; all authors helped write the manuscript.
References
- 1.Lee M. S. Y., Reeder T. W., Slowinski J. B., Lawson R. 2004. Resolving reptile relationships. In Assembling the tree of life (eds Cracraft J., Donoghue M. J.), pp. 451–467 Oxford, UK: Oxford University Press [Google Scholar]
- 2.Lee M. 1997. Reptile relationships turn turtle. Nature 389, 245–246 10.1038/38422 (doi:10.1038/38422) [DOI] [Google Scholar]
- 3.Rieppel O. 1999. Turtle origins. Science 283, 945–946 10.1126/science.283.5404.945 (doi:10.1126/science.283.5404.945) [DOI] [PubMed] [Google Scholar]
- 4.Janke A., Erpenbeck D., Nilsson M., Aranason U. 2001. The mitochondrial genomes of the iguana (Iguana iguana) and the caiman (Caiman crocodylus): implications for amniote phylogeny. Proc. R. Soc. Lond. B 268, 623–631 10.1098/rspb.2000.1402 (doi:10.1098/rspb.2000.1402) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hedges S., Poling L. 1999. A molecular phylogeny of reptiles. Science 283, 998–1001 10.1126/science.283.5404.998 (doi:10.1126/science.283.5404.998) [DOI] [PubMed] [Google Scholar]
- 6.Rest J. S., Ast J. C., Austin C. C., Waddell P. J., Tibbetts E. A., Hay J. M., Mindell D. P. 2003. Molecular systematics of primary reptilian lineages and the tuatara mitochondrial genome. Mol. Phylogenet. Evol. 29, 289–297 10.1016/S1055-7903(03)00108-8 (doi:10.1016/S1055-7903(03)00108-8) [DOI] [PubMed] [Google Scholar]
- 7.Kumazawa Y., Nishida M. 1999. Complete mitochondrial DNA sequences of the green turtle and blue-tailed mole skink: statistical evidence for archosaurian affinity of turtles. Mol. Biol. Evol. 16, 784–792 10.1093/oxfordjournals.molbev.a026163 (doi:10.1093/oxfordjournals.molbev.a026163) [DOI] [PubMed] [Google Scholar]
- 8.Zardoya R., Meyer A. 1998. Complete mitochondrial genome suggests diapsid affinities of turtles. Proc. Natl Acad. Sci. USA 95, 14 226–14 231 10.1073/pnas.95.24.14226 (doi:10.1073/pnas.95.24.14226) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katsu Y., Braun E. L., Guillette L. J., Jr, Iguchi T. 2009. From reptilian phylogenomics to reptilian genomes: analyses of c-Jun and DJ-1 proto-oncogenes. Cytogenet. Genome Res. 127, 79–93 10.1159/000297715 (doi:10.1159/000297715) [DOI] [PubMed] [Google Scholar]
- 10.Shedlock A. M., Botka C. W., Zhao S., Shetty J., Zhang T., Liu J. S., Deschavanne P. J., Edwards S. V. 2007. Phylogenomics of nonavian reptiles and the structure of the ancestral amniote genome. Proc. Natl Acad. Sci. USA 104, 2767–2772 10.1073/pnas.0606204104 (doi:10.1073/pnas.0606204104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hugall A. F., Foster R., Lee M. S. Y. 2007. Calibration choice, rate smoothing, and the pattern of tetrapod diversification according to the long nuclear gene RAG-1. Syst. Biol. 56, 543–563 10.1080/10635150701477825 (doi:10.1080/10635150701477825) [DOI] [PubMed] [Google Scholar]
- 12.Shen X.-X., Liang D., Wen J.-Z., Zhang P. 2011. Multiple genome alignments facilitate development of NPCL markers: a case study of tetrapod phylogeny focusing on the position of turtles. Mol. Biol. Evol. 61, 346–359 10.1093/molbev/msr148 (doi:10.1093/molbev/msr148) [DOI] [PubMed] [Google Scholar]
- 13.Tzika A. C., Helaers R., Schramm G., Milinkovitch M. C. 2011. Reptilian-transcriptome v. 1.0, a glimpse in the brain transcriptome of five divergent Sauropsida lineages and the phylogenetic position of turtles. EvoDevo 2, 19. 10.1186/2041-9139-2-19 (doi:10.1186/2041-9139-2-19) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iwabe N., Hara Y., Kumazawa Y., Shibamoto K., Saito Y., Miyata T. 2005. Sister group relationship of turtles to the bird–crocodilian clade revealed by nuclear DNA-coded proteins. Mol. Biol. Evol. 22, 810–813 10.1093/molbev/msi075 (doi:10.1093/molbev/msi075) [DOI] [PubMed] [Google Scholar]
- 15.Lyson T. R., Sperling E. A., Heimberg A. M., Gauthier J. A., King B. L., Peterson K. J. 2011. MicroRNAs support a turtle+lizard clade. Biol. Lett. 8, 104–107 10.1098/rsbl.2011.0477 (doi:10.1098/rsbl.2011.0477) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hedges S. B., Moberg K. D., Maxson L. R. 1990. Tetrapod phylogeny inferred from 18S and 28S ribosomal RNA sequences and a review of the evidence for amniote relationships. Mol. Biol. Evol. 7, 607–633 [DOI] [PubMed] [Google Scholar]
- 17.Cao Y., Sorenson M. D., Kumazawa Y., Mindell D. P., Hasegawa M. 2000. Phylogenetic position of turtles among amniotes: evidence from mitochondrial and nuclear genes. Gene 259, 139–148 10.1016/S0378-1119(00)00425-X (doi:10.1016/S0378-1119(00)00425-X) [DOI] [PubMed] [Google Scholar]
- 18.Bejerano G., Pheasant M., Makunin I., Stephen S., Kent W. J., Mattick J. S., Haussler D. 2004. Ultraconserved elements in the human genome. Science 304, 1321–1325 10.1126/science.1098119 (doi:10.1126/science.1098119) [DOI] [PubMed] [Google Scholar]
- 19.Siepel A., et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050 10.1101/gr.3715005 (doi:10.1101/gr.3715005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faircloth B. C., et al. In press Ultraconserved elements anchor thousands of genetic markers for target enrichment spanning. 10.1093/sysbio/SYS004 (doi:10.1093/sysbio/SYS004) [DOI] [PubMed] [Google Scholar]
- 21.Zerbino D. R., Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 10.1101/gr.074492.107 (doi:10.1101/gr.074492.107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edgar R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 10.1093/nar/gkh340 (doi:10.1093/nar/gkh340) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ronquist F., et al. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542(doi:10.1093/sysbio/sys029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu L., Yu L., Kubatko L., Pearl D. K., Edwards S. V. 2009. Coalescent methods for estimating phylogenetic trees. Mol. Phylogenet. Evol. 53, 320–328 10.1016/j.ympev.2009.05.033 (doi:10.1016/j.ympev.2009.05.033) [DOI] [PubMed] [Google Scholar]
- 25.Lagos-Quintana M., Rauhut R., Yalcin A., Meyer J., Lendeckel W., Tuschl T. 2002. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 12, 735–739 10.1016/S0960-9822(02)00809-6 (doi:10.1016/S0960-9822(02)00809-6) [DOI] [PubMed] [Google Scholar]
- 26.Landgraf P., et al. 2007. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129, 1401–1414 10.1016/j.cell.2007.04.040 (doi:10.1016/j.cell.2007.04.040) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.MacKenzie D., Nichols J., Lachman G., Droege S., Royle J., Langtimm C. 2002. Estimating site occupancy rates when detection probabilities are less than one. Ecology 83, 2248–2255 10.1890/0012-9658(2002)083[2248:ESORWD]2.0.CO;2 (doi:10.1890/0012-9658(2002)083[2248:ESORWD]2.0.CO;2) [DOI] [Google Scholar]
- 28.McCormack J. E., Faircloth B. C., Crawford N. G., Gowaty P. A., Brumfield R. T., Glenn T. C. 2012. Ultraconserved elements are novel phylogenomic markers that resolve placental mammal phylogeny when combined with species tree analysis. Genome Research 22, 746–754 10.1101/gr.125864.111 (doi:10.1101/gr.125864.111) [DOI] [PMC free article] [PubMed] [Google Scholar]