

Abstract
We have proposed that maintenance of genomic stability may constitute the basis for the tumor-suppressing activity of the Bre1 (RNF20/RNF40) complex. Revisiting the evidence we presented in our recent publication, we discuss the mechanism by which maintenance of genomic stability by the Bre1 complex is achieved through coordination of events during transcription. Among many functions of Bre1, we focus on the two that, when defective, could lead to the formation of R-loops, the RNA:DNA hybrid structures regarded as a major source of genomic instability. Specifically, we discuss the role of Bre1-mediated H2B ubiquitination in the 3′-end processing of replication-associated histone mRNA and in heterochromatic gene silencing and show how disturbance of these two functions may result in the specific pattern of chromosomal abnormalities we observe in the Bre1-depleted cells.
Keywords: CIN, H2B monoubiquitination, H3K79, R-loop, RNA:DNA hybrids, gene silencing, genomic instability, homologous recombination
Introduction
Once regarded simply as chromatin packaging blocks, nucleosomes are now known as key regulators of gene activity. This regulation is often manifested through post-translational modifications of histones and accounts for coordination of diverse processes, ranging from cell differentiation to DNA damage response. Much attention in the last decade was attracted to a distinctive pathway mediated by the yeast E3 ubiquitin ligase Bre1 and its mammalian homologs RNF20/RNF40 (also known as BRE1A/BRE1B and henceforth in this paper referred to as Bre1), which monoubiquitinate histone H2B at H2BK123 in yeast and H2BK120 in mammals.1-4 H2B ubiquitination is required for the di- and trimethylation of H3K4 and H3K79, respectively.5,6 The main function of H2B ubiquitination is in transcriptional regulation, but it also has been shown to have other functions, including regulation of the cell cycle and in DNA repair (reviewed in ref. 7). Given that Bre1 affects multiple pathways involved in genome maintenance in yeast, it is likely that Bre1 homologs play a major tumor-suppressing role in higher organisms. Consistent with this, it has been suggested that mammalian Bre1 acts as a tumor suppressor by a transcriptional mechanism8 through selective downregulation of specific proto-oncogenes and activation of tumor suppressor genes, including p53, in human cells.3,8 Of special interest, the subset of genes selectively suppressed by Bre1 and including several oncogenes is localized deep in the compact heterochromatin.8 In this model of tumor suppression, a diminished function of Bre1 would be expected to inhibit tumor suppressors and activate oncogenes, leading to uncontrolled cell growth, a central hallmark of cancer. For the most part, this hypothesis is experimentally supported, and loss of Bre1 has been shown to increase anchorage-independent cell proliferation and cell migration.8 However, there is a question about the evolutionary conservation of the mechanism, as expression of p53, known to be regulated by Bre1 in human cells, is not regulated by Bre1 in mice (ref. 8 and our observations). In addition, this hypothesis does not explain one key observation, namely that the early effect of Bre1 loss is manifested as an impairment of growth and increased cell death, rather than accelerated proliferation. Therefore, Bre1 does not act as a classical tumor suppressor and its tumor-suppressing role needs further clarification.
R-loops Contribute to Genomic Instability in Bre1-Deficient Cells
Tumor progression is commonly envisioned as a succession of clonal expansions, each of which is made possible by an acquisition of rare genetic changes that enable the subclones of precancerous cells to grow and disseminate. The core process that facilitates generation of random mutations is genomic instability, which, in many cancers, is manifested in the form of chromosomal instability (CIN). CIN is thought to be an early event in carcinogenesis and unrepaired DNA double-strand breaks (DSBs) to be central to CIN initiation. Loss of Bre1 leads to dramatic increase of CIN in mammalian cells, as we have demonstrated in our recent study.9 We followed the evolution of genomic instability in Bre1-deficient cells from early DSBs and replication stress to specific genomic rearrangements that, in turn, triggered breakage-fusion-bridge cycles, known to accelerate genomic instability (Fig. 1). Intriguingly, a significant proportion of the DSBs and chromosomal abnormalities in Bre1-depleted cells is associated with heterochromatin (Fig. 1C and D).
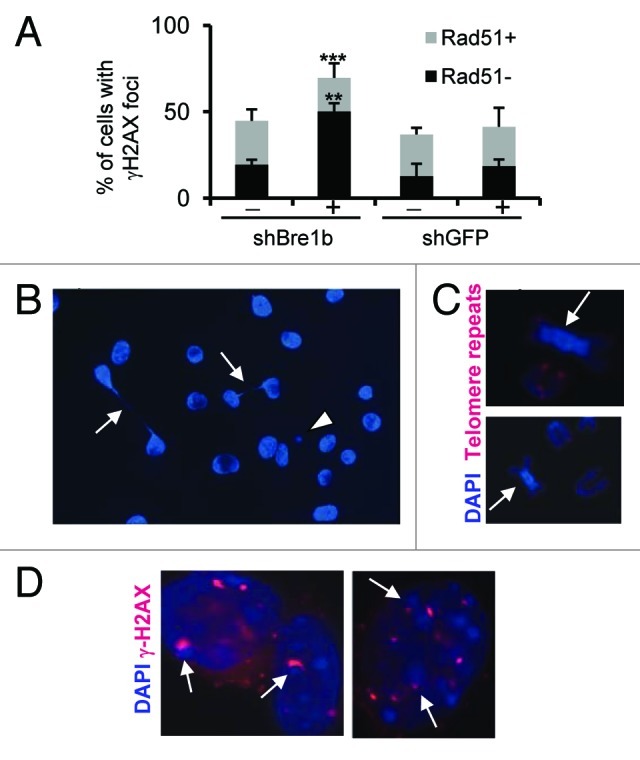
Figure 1. Bre1 depletion leads to persistence of double-strand breaks (DSBs) and chromosomal instability (CIN). (A) Defect in homologous recombination due to depletion of Bre1 increases levels of DSBs, as shown by an increase in number of γH2AX-positive/Rad51-negative cells. CIN in Bre1-depleted cells manifests itself through a bridge-breakage-fusion cycle (B) and chromosomal aberrations, many of which involve heterochromatin (C). In (B) arrows point to anaphase bridges and arrowhead points to a micronucleus formed upon breakage of the bridge. In (C) the arrows point to chromosomes with centromeric heterochromatin amplification. (D) γH2AX co-localizes with heterochromatin of brightly-stained chromocenters (white arrows). Fixation for γH2AX, Rad51 staining, and chromosomal aberration analysis were performed on day 5 after Bre1 knockdown and were performed as described in 9. shBre1b, RNAi against Bre1b (Rnf40), shGFP, RNAi against GFP. (**p < 0.01, and ***p < 0.001, t-test).
A widely accepted mechanism leading to CIN involves excessive DNA damage that often results from DNA repair defects. Consistent with this, we and others have shown that Bre1-mediated H2BK120 ubiquitination is important for the response to DSBs.10-12 This function of Bre1 appears to be in facilitating the timely repair of DSB through induction of H2B ubiquitination at DSB sites and recruitment of repair factors to these sites before the DNA end-resection step.11-13 Thus CIN in Bre1-depleted cells can be explained by the defect in DSB repair, especially in homologous recombination repair (HR), and we demonstrated that chromatid-type aberrations, a signature of an HR defect, were significantly overrepresented among chromosomal rearrangements after loss of Bre1.9 However, this is not the only reason for the increased genomic instability: there is another broad source of genomic instability in the Bre1-deficient cells. By analyzing gene expression after loss of RNF20 and RNF40, we found surprisingly high enrichment of genes involved in RNA biogenesis, with the RNA processing module being the most significantly overrepresented. In fact, a plethora of evidence has been provided recently for a broad category encompassing genes involved in RNA biogenesis and factors involved in its regulation as being a major category of CIN genes.14-20 These studies generalize the concept originally suggested by studies of the Hpr1 mRNA processing factor21 and the splicing factor ASF/SF2.22
Mutations in the RNA biogenesis genes are linked to genomic instability through a common mechanism that involves formation of transcription-associated RNA:DNA hybrids known as R-loops. An R-loop is a three-stranded nucleic acid structure formed by hybridization of a nascent RNA to its template DNA, with exposed single-stranded DNA of the sense strand being more susceptible to DNA damage. The most likely explanation for CIN initiated by R-loops is that it arises from DSBs caused by interference of R-loops with replication and/or transcription machinery (reviewed in refs. 18, 23 and 24). These DSBs are normally subject to repair by recombination, accounting for a transcription-associated hyperrecombination phenotype similar to the one initially reported for yeast THO mutants.21 In the absence of efficient recombination repair, these DSBs persist and induce gross-chromosomal rearrangements (GCRs). In Bre1-depleted cells,9 we observed an increase in numbers of cells with γH2AX foci, which normally form at the sites of DSBs.25 Most of the cells with γH2AX foci are negative for Rad51 foci, suggesting no recombination repair and persistence of DSBs (Fig. 1A). Given the deleterious consequences of R-loops, cells developed multiple ways to get rid of those: RNase H enzymes specifically cleave the RNA within the RNA:DNA hybrid, and a number of helicases are able to unwind the RNA:DNA structures (reviewed in ref. 18). An increased formation of γ-H2AX and GCRs upon perturbation of RNA biogenesis has been shown to be reduced upon overexpression of RNase H1,14-17,19-22 suggesting the R-loop-dependent origin of the DSBs and CIN. Similarly, we attribute most of the genomic instability in the Bre1-deficient cells to the formation of R-loops, as γ-H2AX foci induced in cells upon depletion of Bre1 were fully preventable by overexpressing RNase H1.9
Mechanisms of R-loop Formation in Bre1-Depleted Cells Explain the Occurrence of Chromosomal Rearrangements Involving Heterochromatin
Deleterious interactions between RNA and the antisense DNA are possible at multiple stages during and after transcription, especially when long genes are transcribed;26 therefore, cells have evolved robust mechanisms to prevent R-loop formation. In prokaryotes, re-hybridization of newly synthesized RNA to the DNA is prevented by coupling of transcription and translation, and in eukaryotes, where transcription and translation occur in different cellular compartments, a sophisticated array of factors exists to facilitate proper packaging of nascent mRNA into a ribonucleoprotein particle, thus strongly reducing the ability of mRNA to rehybridize with its template within the transiently opened DNA bubble behind the RNA polymerase.21 Coordination between auxiliary factors during transcription is therefore an important factor preventing the formation of R-loops. Below we discuss possible mechanisms contributing to the R-loop formation in the Bre1-depleted cells.
Analysis of our gene expression array also showed the presence of polyadenylated histone mRNA in the Bre1-depleted cells,9 pointing to a defect in processing of replication-dependent histone mRNA reported by Pirngruber et al.27 Normally, replication-dependent histone transcripts lack poly(A) tails, ending instead in a highly conserved 26 nucleotide sequence that can form a stem-loop, and Bre1 has been shown to be required for the correct recognition of the histone mRNA 3'-end cleavage site.27 Absence of Bre1 results in increased polyadenylation of mRNA due to failure of RNAPII to recognize the stem-loop structure, leading to continuation of transcription until the downstream poly(A) signal is reached. Defects in histone mRNA processing can have two important consequences. First, a defect in processing of mRNA has been shown to increase co-transcriptional formation of R-loops, which could contribute to replication stress in Bre1-deficient cells by blocking progression of replication forks and elevating levels of DSBs.22 The notable upregulation of genes in our microarray that are involved in resolution and avoidance of R-loops would explain the presence of these structures.9 Second, the presense of histone transcripts that have poly(A) tails implies a change in stability of histone mRNA and may lead to inappropriate presence of replication-dependent histones outside of S-phase, thereby interfering with proper incorporation of variant histones and destabilizing centromeric heterochromatin,28 which would explain the prevalence of chromosome aberrations involving centromeric heterochromatin in the Bre1-deficient cells (Fig. 1C). Providing further support for the role of Bre1-mediated H2B ubiquitination in maintaining stability of centromeric heterochromatin, we observed localization of a large fraction (> 60%) of γH2AX foci at the edges of chromocenters (Fig. 1D), the easily visualized bright spots in DAPI-stained cells where pericentric chromatin from different chromosomes cluster together.
Maintenance of heterochromatin outside of the centromere in the mammalian chromosome also requires intact Bre1 function. A defect in the Bre1-Dot1 pathway in S. cerevisiae has been associated with the disruption of gene silencing at telomeres, which contain the main bulk of heterochromatin in budding yeast.1,5 DNA in heterochromatin is relatively inert for transcription, and transcriptionally active genes that relocate in the vicinity of heterochromatin, either as a result of euchromatic gene translocation or reporter gene integration, become silenced. This so called position-effect variegation or chromosomal position effect, is well documented in Drosophila and yeast. We show that Bre1 is important for gene silencing at telomeres in mouse embryonic stem cells (Fig. 2). Two related mechanisms have been proposed to explain how Bre1 maintains the chromosome position effect. In the first, heterochromatic silencing is achieved through uH2BK120-dependent H3K79 methylation of the euchromatin,29 which keeps Sir proteins restricted to heterochromatin. Loss of H2B ubiquitination allows spreading of Sir proteins away from heterochromatin, leading to release of heterochromatic silencing. In another model, gene silencing is achieved through Bre1(RNF20/40)-mediated ubiquitination of H2B and subsequent uH2BK120-dependent methylation of H4K4 at the insulator elements, which serve as a boundary between euchromatin and heterochromatin.30 We propose that under conditions of Bre1 knockdown transcription arising due to the loss of heterochromatic silencing would proceed under suboptimal conditions. This would create more opportunities for the R-loop, and subsequently DSB, formation within the heterochromatic domain, providing an explanation for the involvement of heterochromatin in the GCRs in the Bre1-deficient cells (Fig. 1C and D). The position-effect variegation affects genes within the heterochromatic domain non-specifically, therefore the genes whose transcription is de-repressed as a result of Bre1 deficiency would not necessarily be the ones whose transcription is selectively regulated by Bre1.
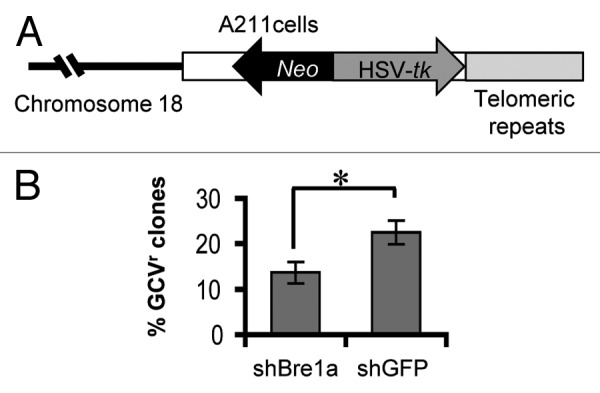
Figure 2. Loss of Bre1 (RNF20) disrupts gene silencing at telomeric chromatin. (A) Clone A211 of mouse embryonic stem cells obtained from Dr. J. P. Murnane 31 contains a single copy of a construct containing the neo gene for positive selection with G418 and a HSV-tk gene for negative selection with ganciclovir incorporated at the telomere. (B) Bre1 loss decreases telomeric gene silencing. A211 cells were infected with lentiviruses containing shRNA against Bre1a and GFP, and grown for 3 d in 300 ug/ml G418 to select for cells expressing the construct. Prolonged culturing without selection results in silencing of the construct and cells become resistant to ganciclovir. Ganciclovir resistant clones were counted upon plating 10,000 cells and selecting in ganciclovir at 2 ug/ml. shBre1, RNAi against Bre1a (Rnf20); shGFP, RNAi against GFP. *p < 0.05, t-test)
In summary, we propose that Bre1(RNF20/40) serves as a tumor suppressor, preventing CIN originating from DSBs associated with R-loops and contributing to timely repair of these DSBs by HR needed for the resolution of these DSBs (Fig. 3). Persistent DSBs resulting from elevated levels of R-loops and inefficient DSB repair account for high levels of CIN and cell death after Bre1 loss. The R-loop-dependent model of CIN in Bre1-deficient cells explains the prevalence of chromosomal rearrangements involving heterochromatin.

Figure 3. Loss of Bre1 (RNF20/40) increases chromosomal instability (CIN), which explains increased cell death and acquisition of the malignant phenotype. Bre1 depletion impairs homologous recombination and the processing of canonical histone mRNAs, both of which act to protect genomic stability during replication. Incorrect processing of replication-dependent histone mRNA 3′-ends, as well as suboptimal transcription of genes in heterochromatin that has become de-silenced as a result of Bre1 deficiency, create conditions that favor co-transcriptional formation of R-loops, blocking replication forks.
Acknowledgments
We are grateful to Dr. John C. Game and Dr. Olga V. Razorenova for helpful discussions and critical reading of the manuscript. The work was supported by grant CA67166 awarded to Prof. J.M Brown by the National Cancer Institute.
Glossary
Abbreviations:
- DSB
double-strand break
- CIN
chromosomal instability
- GCR
gross chromosomal rearrangements
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/21090
References
- 1.Wood A, Krogan NJ, Dover J, Schneider J, Heidt J, Boateng MA, et al. Bre1, an E3 ubiquitin ligase required for recruitment and substrate selection of Rad6 at a promoter. Mol Cell. 2003;11:267–74. doi: 10.1016/S1097-2765(02)00802-X. [DOI] [PubMed] [Google Scholar]
- 2.Hwang WW, Venkatasubrahmanyam S, Ianculescu AG, Tong A, Boone C, Madhani HD. A conserved RING finger protein required for histone H2B monoubiquitination and cell size control. Mol Cell. 2003;11:261–6. doi: 10.1016/S1097-2765(02)00826-2. [DOI] [PubMed] [Google Scholar]
- 3.Kim J, Hake SB, Roeder RG. The human homolog of yeast BRE1 functions as a transcriptional coactivator through direct activator interactions. Mol Cell. 2005;20:759–70. doi: 10.1016/j.molcel.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 4.Zhu B, Zheng Y, Pham AD, Mandal SS, Erdjument-Bromage H, Tempst P, et al. Monoubiquitination of human histone H2B: the factors involved and their roles in HOX gene regulation. Mol Cell. 2005;20:601–11. doi: 10.1016/j.molcel.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 5.Sun ZW, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002;418:104–8. doi: 10.1038/nature00883. [DOI] [PubMed] [Google Scholar]
- 6.Ng HH, Xu RM, Zhang Y, Struhl K. Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. J Biol Chem. 2002;277:34655–7. doi: 10.1074/jbc.C200433200. [DOI] [PubMed] [Google Scholar]
- 7.Game JC, Chernikova SB. The role of RAD6 in recombinational repair, checkpoints and meiosis via histone modification. DNA Repair (Amst) 2009;8:470–82. doi: 10.1016/j.dnarep.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 8.Shema E, Tirosh I, Aylon Y, Huang J, Ye C, Moskovits N, et al. The histone H2B-specific ubiquitin ligase RNF20/hBRE1 acts as a putative tumor suppressor through selective regulation of gene expression. Genes Dev. 2008;22:2664–76. doi: 10.1101/gad.1703008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chernikova SB, Razorenova OV, Higgins JP, Sishc BJ, Nicolau M, Dorth JA, et al. Deficiency in mammalian histone H2B ubiquitin ligase Bre1 (Rnf20/Rnf40) leads to replication stress and chromosomal instability. Cancer Res. 2012;72:2111–9. doi: 10.1158/0008-5472.CAN-11-2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chernikova SB, Dorth JA, Razorenova OV, Game JC, Brown JM. Deficiency in Bre1 impairs homologous recombination repair and cell cycle checkpoint response to radiation damage in mammalian cells. Radiat Res. 2010;174:558–65. doi: 10.1667/RR2184.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakamura K, Kato A, Kobayashi J, Yanagihara H, Sakamoto S, Oliveira DV, et al. Regulation of homologous recombination by RNF20-dependent H2B ubiquitination. Mol Cell. 2011;41:515–28. doi: 10.1016/j.molcel.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 12.Moyal L, Lerenthal Y, Gana-Weisz M, Mass G, So S, Wang SY, et al. Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol Cell. 2011;41:529–42. doi: 10.1016/j.molcel.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chernikova SB, Game JC, Brown JM. Inhibiting homologous recombination for cancer therapy. Cancer Biol Ther. 2012;13:61–8. doi: 10.4161/cbt.13.2.18872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sordet O, Redon CE, Guirouilh-Barbat J, Smith S, Solier S, Douarre C, et al. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO Rep. 2009;10:887–93. doi: 10.1038/embor.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell. 2009;35:228–39. doi: 10.1016/j.molcel.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wahba L, Amon JD, Koshland D, Vuica-Ross M. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol Cell. 2011;44:978–88. doi: 10.1016/j.molcel.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stirling PC, Chan YA, Minaker SW, Aristizabal MJ, Barrett I, Sipahimalani P, et al. R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev. 2012;26:163–75. doi: 10.1101/gad.179721.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aguilera A, García-Muse T. R loops: from transcription byproducts to threats to genome stability. Mol Cell. 2012;46:115–24. doi: 10.1016/j.molcel.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Houlard M, Artus J, Léguillier T, Vandormael-Pournin S, Cohen-Tannoudji M. DNA-RNA hybrids contribute to the replication dependent genomic instability induced by Omcg1 deficiency. Cell Cycle. 2011;10:108–17. doi: 10.4161/cc.10.1.14379. [DOI] [PubMed] [Google Scholar]
- 20.Tuduri S, Crabbé L, Conti C, Tourrière H, Holtgreve-Grez H, Jauch A, et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat Cell Biol. 2009;11:1315–24. doi: 10.1038/ncb1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huertas P, Aguilera A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol Cell. 2003;12:711–21. doi: 10.1016/j.molcel.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 22.Li X, Manley JL. Cotranscriptional processes and their influence on genome stability. Genes Dev. 2006;20:1838–47. doi: 10.1101/gad.1438306. [DOI] [PubMed] [Google Scholar]
- 23.Tuduri S, Crabbe L, Tourrière H, Coquelle A, Pasero P. Does interference between replication and transcription contribute to genomic instability in cancer cells? Cell Cycle. 2010;9:1886–92. doi: 10.4161/cc.9.10.11539. [DOI] [PubMed] [Google Scholar]
- 24.Sordet O, Nakamura AJ, Redon CE, Pommier Y. DNA double-strand breaks and ATM activation by transcription-blocking DNA lesions. Cell Cycle. 2010;9:274–8. doi: 10.4161/cc.9.2.10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Löbrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, et al. gammaH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle. 2010;9:662–9. doi: 10.4161/cc.9.4.10764. [DOI] [PubMed] [Google Scholar]
- 26.Helmrich A, Ballarino M, Tora L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell. 2011;44:966–77. doi: 10.1016/j.molcel.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 27.Pirngruber J, Shchebet A, Schreiber L, Shema E, Minsky N, Chapman RD, et al. CDK9 directs H2B monoubiquitination and controls replication-dependent histone mRNA 3′-end processing. EMBO Rep. 2009;10:894–900. doi: 10.1038/embor.2009.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salzler HR, Davidson JM, Montgomery ND, Duronio RJ. Loss of the histone pre-mRNA processing factor stem-loop binding protein in Drosophila causes genomic instability and impaired cellular proliferation. PLoS One. 2009;4:e8168. doi: 10.1371/journal.pone.0008168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ng HH, Ciccone DN, Morshead KB, Oettinger MA, Struhl K. Lysine-79 of histone H3 is hypomethylated at silenced loci in yeast and mammalian cells: a potential mechanism for position-effect variegation. Proc Natl Acad Sci U S A. 2003;100:1820–5. doi: 10.1073/pnas.0437846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma MK, Heath C, Hair A, West AG. Histone crosstalk directed by H2B ubiquitination is required for chromatin boundary integrity. PLoS Genet. 2011;7:e1002175. doi: 10.1371/journal.pgen.1002175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pedram M, Sprung CN, Gao Q, Lo AW, Reynolds GE, Murnane JP. Telomere position effect and silencing of transgenes near telomeres in the mouse. Mol Cell Biol. 2006;26:1865–78. doi: 10.1128/MCB.26.5.1865-1878.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]