

Abstract
Conditionally replicating adenoviruses (CRAd) are a promising class of gene therapy agents that can overcome already known glioblastoma (GBM) resistance mechanisms but have limited distribution upon direct intratumoral (i.t.) injection. Collagen bundles in the extracellular matrix (ECM) play an important role in inhibiting virus distribution. In fact, ECM pre-treatment with collagenases improves virus distributions to tumor cells. Matrix metalloproteinases (MMPs) are an endogenous class of collagenases secreted by tumor cells whose function can be altered by different drugs including anti-angiogenic agents, such as bevacizumab. In this study we hypothesized that up-regulation of MMP activity during antiangiogenic therapy can improve CRAd-S-pk7 distribution in GBM. We find that MMP-2 activity in human U251 GBM xenografts increases (*p=0.03) and collagen IV content decreases (*p=0.01) during vascular endothelial growth factor (VEGF-A) antibody neutralization. After proving that collagen IV inhibits CRAd-S-pk7 distribution in U251 xenografts (Spearman rho= −0.38; **p=0.003), we show that VEGF blocking antibody treatment followed by CRAd-S-pk7 i.t. injection reduces U251 tumor growth more than each individual agent alone (***p<0.0001). Our data proposes a novel approach to improve virus distribution in tumors by relying on the early effects of anti-angiogenic therapy.
Keywords: anti-VEGF, bevacizumab, oncolytic virus, adenovirus, metalloproteinase, glioma, glioblastoma, brain tumor
Introduction
Glioblastoma (GBM), the most common and aggressive brain tumor,1 is highly resistant to current therapeutic approaches. Surgical debulking, followed by radiation and chemotherapy with temozolomide achieve a mean overall survival of only 15.1 months.2 Therefore, novel agents that can overcome already known glioblastoma resistance mechanisms are needed. A very promising and expanding class of gene therapy vectors includes oncolytic viruses, which comprise mutated viruses (HSV, adenovirus, vaccinia) that can replicate selectively in brain tumors. These agents have proven highly effective in pre-clinical trials in different glioma models. Our group is focused on targeting GBM with conditionally replicative adenoviruses (CRAd). CRAd-S-pk7, a vector that we plan to bring to clinical trials, relies on its poly-lysine (pk7) fiber modifications to attach more effectively to glioma cells and the survivin promoter to selectively induce adenovirus replication in proliferative cells.3 However, a major hurdle to the successful clinical application of gene therapy vectors is their limited distribution upon direct injection.4
Extracellular matrix (ECM) components, especially collagen bundles, form a barrier that inhibits intratumoral distribution of nano-size particles, including oncolytic viruses.5 Therefore, reducing collagen content or inducing its degradation has been proposed as a possible mechanism to increase small particle diffusion in tumors. Moreover, degradation of ECM by using exogenous collagenases has been shown to result in increased oncolytic virus distribution and enhanced therapeutic efficacy.6–8 Relying on exogenous collagenases has its limitations and it would be more beneficial if we could induce such collagenase activity in a tumor selective manner. Matrix metalloproteinases (MMPs) are a class of endogenous collagenases that play an important role in tissue remodeling mainly through collagen degradation. GBM microenvironment is rich in type IV collagen, the substrate of two major MMPs expressed by glioma cells, MMP-2 and 9. Furthermore, over-expression of MMP-9 in glioma cells, through stable transfection, improves oncolytic virus distribution upon direct injection.9 MMP expression is variable among tumors and can also be altered during therapy.10 Therefore, an increase in intratumoral collagenase activity before oncolytic virus injection, should improve its distribution. The ability to alter the tumor microenvironment and increase adenovirus distribution through gene therapy or drugs opens up the possibility for combination therapies. Of all the biological therapies, anti-angiogenic drugs have shown to alter tumor microenvironment the most.
Bevacizumab is a vascular endothelial growth factor A (VEGF-A) neutralization monoclonal antibody approved by the FDA for treatment of progressive/recurrent glioblastomas (GBM) based on data suggesting prolongation of 6 months progression free-survival (PFS-6) in uncontrolled phase II clinical trials.11, 12 Anti-angiogenesis therapy relies on the principle that solid tumors require formation of new vessels to support their growth.13 Pre-clinical studies not only supported this theory but also demonstrated that in the early stages of therapy there was a normalization of the vasculature that could result in improved delivery of chemotherapeutic agents or increased efficacy of radiation therapy.14 This `normalization' window was also found to be associated with significant interstitial pressure reduction,15 presumably due to a decrease in vessel permeability. Since high intra-tumoral (i.t.) interstitial pressure has been shown to inhibit distribution of therapeutic agents within the tumor mass, its reduction creates a window of opportunity to improve their delivery.
In this study we investigated the effects of short term VEGF neutralization via blocking antibody (VEGF-Ab), on the brain tumor microenvironment. Specifically, we show that MMP-2 levels are increased upon treatment with VEGF-Ab in different glioma cell linse as well as in glioma xenograft models. In turn, these high MMP-2 levels degrade collagen IV bundles present in the extracellular matrix of glioblastoma. Moreover, when oncolytic adenovirus injection follows anti-angiogenic therapy it results in increased intra-tumoral distribution and higher adenoviral titers. Finally, we show that combination therapy with VEGF blocking antibody and CRAd-S-pk7 inhibits tumor growth more effectively than each individual agent alone.
Results
Anti-angiogenic therapy has no additive effect to oncolytic adenovirus toxicity in vitro
We first evaluated whether in vitro VEGF antibody blocking (VEGF-Ab) can alter expression of adenovirus receptors in glioma cells. CRAd-S-pk7 attachment and entry into the host cell relies on the interaction between the adenoviral fiber and surface receptors such as coxsackie-adenovirus receptor (CAR), integrins (αvβ3 & αvβ5) and CD138; the latter which interacts with CRAd-S-pk7. We did not detect any significant changes in expression of these receptors on U251 glioma cells after VEGF blockade (Figure 1a). CAR expression varied from 29.2±4.8% in the control group to 29.1±1.4% for the VEGF-Ab treated cells (p=0.98). Similarly, the level of integrins αvβ3 (from 48.7±2.6% to 43±7%; p=0.4), αvβ5 (from 75.2±4% to 75.3±4.8%; p=0.98) and CD138 (from 12.4±1.1% to 12.5±0.1%; p=0.86) did not change significantly after treatment.
Figure 1.

In vitro effects of VEGF neutralization on adenovirus replication. (A) Flow cytometry analysis of U251 glioma cell line for expression of surface receptor CAR, αvβ3, αvβ5 and CD138. The percentages of positive cells for the respective receptors are shown in bar diagrams, below the flow cytometry histograms. (B and C) CRAd-S-pk7 replication in U251 glioma treated with VEGF-Ab was quantified via quantitative real-time PCR for E1A (B) or adenovirus progeny titer (C). (D) Toxicity of CRAd-S-pk7 in combination with VEGF-Ab in U251 glioma cells five days after infection (ns: not significant; *p<0.05; *** p<0.001).
Next we tested whether VEGF-Ab therapy could increase adenovirus replication in vitro. In glioma cells we quantified adenoviral E1A copy numbers (Figure 1b), through real-time quantitative PCR, and the infectious progeny titers produced (Figure 1c). On day 5 we noticed that in cells treated with in VEGF-Ab, adenoviral E1A copies were significantly higher compared to Ig–Control treated cells (9.1*107 vs. 3.6*107 E1A copies/ngDNA; *p=0.01). Also, the infectious progeny produced was 4 fold higher in the VEGF-Ab treated group (4.13*109 vs. 1.02*109 infectious units per ml (IU/ml); ***p<0.0001).
To assess whether increased adenoviral replication in U251 glioma would result in more toxicity, we treated CRAd-S-pk7 infected cells with VEGF-Ab and performed a viability assay 7 days later. VEGf-Ab treatment of infected cells in vitro did not increase CRAd-S-pk7 toxicity to glioma cells (Figure 1d).
VEGF neutralization increases MMP-2 levels in glioma cell lines
To understand how VEGF neutralization can alter the glioma microenvironment we quantified the expression of two major MMPs related to glioma invasion: MMP-2 and MMP-9, following anti-angiogenic therapy. In all four glioma cell lines that were tested, there was a dramatic increase in MMP-2 levels after VEGF-Ab (Figure 2a). No difference was noted in MMP-9 expression after treatment.
Figure 2.

VEGF trapping up-regulates MMP-2 expression in glioma cell lines. (A) Western blot of U87, U118, A172 and U251 glioma cells after treatment with VEGF-Ab for 5 days. Before collection, cell secretion was blocked for 6 hours with Golgi-Plug. Human β-Actin was used as a protein loading control. (B) Quantitative real-time PCR for MMP-2 mRNA levels in glioma cell lines was evaluated using the ΔCT method. Before plotted in the graph, the MMP-2 expression for each cell line was normalized to their Ig-Control treated condition, to which was given an arbitrary value of 1 (dotted, thin black line: Control). The relative expression levels of MMP-2 for all four cell lines upon VEGF-Ab treatment were then presented as fold of their Control treated cells at the indicated time points. (C) Immunocytochemistry (ICC) of glioma cell lines for MMP-2 and collagen IV after treatment with VEGF-Ab. Cells treated in vitro with VEGF-Ab for 5 days were blocked with Golgi-Plug for 6 hours before fixation for ICC. Nuclei are stained blue with DAPI; collagen IV is presented in yellow and MMP-2 in red. Bars equal 50μm.
To understand how VEGF-Ab up-regulates MMP-2, we quantified its mRNA expression at different time-points. We observed that VEGF-Ab up-regulates MMP-2 gene transcription in all four cell lines differently (Figure 2b), as compared to IgG control treated cells. In the case of U87 glioma cell line, we noticed that induction of MMP-2 transcription reached its peak within 24 hours after VEGF-Ab therapy. For U251, RNA MMP-2 peak levels were detected on day 3 of VEGF-Ab treatment; while in U118 and A172 MMP-2 transcription was affected similarly, There was no significant alteration during the first 3 days and the peak was reached on day 5 of therapy. Additionally, the effect of VEGF blocking on the peak levels of MMP-2 varied among the cell lines. U251 glioma cell line achieved the highest MMP-2 transcription, approximately 7-fold higher than control treated cells. Compared to control treated cells, U87 maximal transcription following VEGF-Ab therapy was 4 fold higher. In U118 and A172 the mRNA levels reached an up-regulation of only two fold.
We also looked at the level of collagen IV expression in the same glioma cell lines after VEGF-Ab therapy. Despite up-regulation of its collagenase (MMP-2) levels, there were no detectable changes in collagen IV levels after in vitro therapy (Figure 2c).
High MMP-2 levels reduce collagen IV content in glioma xenografts
To detect the microenvironmental changes induced after short-term (5 days) anti-angiogenic therapy we relied on the highly tumorigenic U251 and U87 glioma cell lines. Flank tumors were left to grow up to 0.5 cm in diameter before starting treatment with VEGF-Ab or Ig-Control. Similar to our in vitro findings, U251 glioma xenografts up-regulated more than 3 fold MMP-2 expression on day 5 after bevacizumab therapy (Figure 3a and b; *p=0.0274). The difference was even more evident in smaller tumors, which in the absence of anti-angiogenic therapy expressed barely detectable levels of MMP-2. Consistent with our in vitro observation, MMP-9 expression was not altered during bevacizumab therapy (data not shown).
Figure 3.

Anti-angiogenic treatment alters the extracellular matrix (ECM) architecture of human glioma xenograft in nude mice. (A) Immunohistochemistry staining for MMP-2, Collagen IV, CD31 and Laminin. (B–E) Quantification of staining intensity was done through a computer based scoring for each of the corresponding IHC slides (n = 5 animals for each group) and mean values ± standard error of measurement (SEM) are presented in bar diagrams. Bars equal 50μm; *p < 0.05; ns, not significant.
Moreover, we tested the possibility that CRAd-S-pk7, by itself, can induce changes in MMP-2 levels in presence or absence of anti-angiogenic therapy. Mice bearing U251 xenografts (n=5) were treated as above, with VEGF-Ab or Ig-Control for 5 days, and then injected 108 IU of CRAd-S-pk7 or PBS intratumorally. Mice were sacrificed 3 days later. We did not find any difference in MMP-2 expression between mice that received CRAd-S-pk7 or PBS; irrespective of whether they were treated VEGF-Ab or Ig-Control before (Figure 4a, b).
Figure 4.
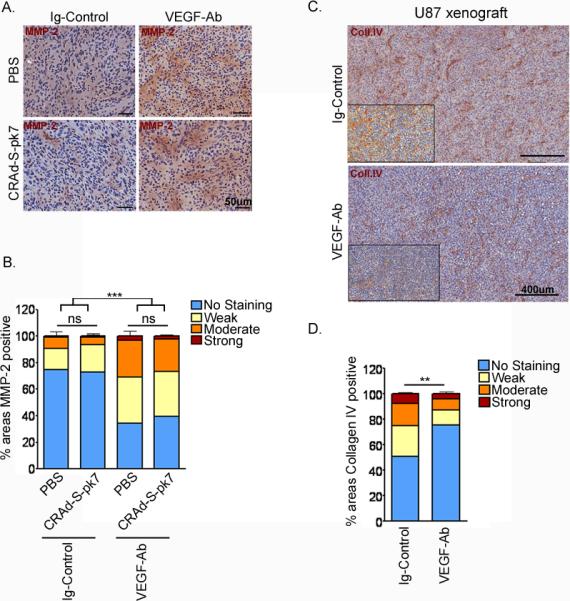
Adenovirus infection does not increase MMP-2 expression. U251 xenografts sections (A) were stained for MMP-2 with HRP and counterstained with hematoxylin; then scanned for intensity of HRP staining, presented as percent of areas with similar intensity (B). Bars represent mean intensity values of all xenografts. ns, not significant; ***p<0.001. (C, D) Sections from U87 xenografts were stained for collagen IV content and scanned for HRP-positivity. (C) Representative images from each group; the insert is the ScanSoft rendered intensity-image of the same slide. (D) Bar graph representation of the staining intensities for each group (n=4). **p=0.008.
Furthermore, VEGF-Ab therapy not only up-regulated MMP-2 but also was associated with significantly reduction of collagen IV content in U251 (Figure 3a and c; *p=0.0133) and U87 xenografts (Figure 4c, d; **p=0.006).
There were several other interesting observations following VEGF-Ab therapy that did not reach statistical significance. First, we observed a trend in reduction of vessel density (Figure 3d and e). Second, CD31, or PECAM, a marker of endothelial cells was found to be non-significantly reduced to 135±68 vessels/mm2 after therapy compared to 345±212 vessels/mm2 in the control group (p=0.54, ns: not significant). Similarly, there was a trend in reduction of laminin expression after bevacizumab therapy that did not reach statistical significance (p=0.197; ns).
High collagen IV content in intracranial glioma correlates negatively with adenovirus distribution
To understand how collagen contents can affect adenovirus distribution in intracranial (i.c.) glioma, we assessed CRAd-S-pk7 localization in relation to collagen bundles. We stained normal mouse brain and mouse brain bearing i.c. U251 glioma for collagen IV and adenoviral hexon protein. In normal mouse brains (Figure 5a, i–iii) collagen IV was found to be organized around vessels. On the other hand, in i.c. U251 glioma xenografts (Figure 5a, iv–vi) we observed extensive disorganized bundles of collagen similar to flank xenografts. Upon VEGF-Ab therapy, there was a substantial reduction in collagen deposits in i.c. glioma (Figure 5a, vii–ix) that remained disorganized.
Figure 5.

Collagen inhibits adenovirus distribution in intracranial glioma xenografts. (A) Collagen IV immunostaining of normal mouse brains (i), mock-treated glioma (iv) and VEGF-Ab treated IC glioma (vii). Representative areas are presented enlarged in (ii), (v) and (viii), respectively; and counterstained with DAPI (iii, vi and ix). (B) Representative area within glioma that depicts the distribution patterns of adenovirus in proximity to collagen bundles. This area was divided in 60 small quadrants and for each of them the percentage of area covered by collagen or adenoviral hexon staining was determined. The threshold was set at 5% and a heat map was constructed for each antigen based on the legend shown in the figure. The quantified distribution patterns were statistically analyzed for possible correlation. Bars equal 150μm.
To show that collagen bundles can indeed block adenovirus distribution we investigated the relationship between collagen content and adenovirus hexon distribution in orthotopic glioma xenografts. An analysis of a representative area (Figure 5b) is shown. Based on immunofluorescent staining intensity we created a heat map graphical representation for each antigen and analyzed their correlation. We found an inverse correlation between collagen content in U251 glioma and CRAd-S-pk7 distribution (Spearman rho= −0.38; **p=0.0028).
VEGF neutralization increased CRAd-S-pk7 distribution and replication in glioma xenografts
We quantified the adenoviral hexon distribution in glioma xenografts after CRAd-S-pk7 i.c. injection. We found that short term anti-angiogenic therapy more than doubled adenovirus distribution (Figure 6a and b; *p=0.044) in the orthotopic glioma. Quantification of the VEGF-Ab effect on adenoviral replication within tumors was performed in U251 flank xenografts. We showed that mice pre-treated with VEGF-Ab before CRAd-S-pk7 administration increased 3 fold intra-tumoral adenoviral E1A copy number, as compared to the control treated group at day seven (3.9*104 vs. 1.5*104 E1Acopies/ngDNA; Figure 6c, *p=0.027). Also, the infectious progeny released by day 7 could be detected only in two out of four animals in the control group; in contrast, we detected progeny in all four animals treated with VEGF-Ab and at higher levels (3*104 vs. 5.4*103 IU/ml; Figure 6d, **p=0.0085).
Figure 6.

VEGF trapping increases adenovirus distribution and replication in glioma xenografts. We assessed CRAd-S-pk7 intracranial distribution and replication in flank tumors with and without concomitant VEGF-Ab therapy. (A) Immunohistochemistry (IHC) staining for detection of adenoviral hexon protein in intracranial xenografts. For each animal we depicted 8 quadrants around the injection tract and quantified hexon distribution (B) within the tumor. Each dot represents a different animal. (C and D) Quantification of CRAd-S-pk7 replication in glioma xenografts. U251 flank tumors were established in nude mice. VEGFAb therapy was started after the tumors reached a diameter of 5mm. Animals received two i.p. injections of VEGF-Ab within 5 days and then injected with 108 IU of CRAd-S-pk7. Adenoviral replication in tumor tissue was quantified at the indicated time points via qRTPCR for E1A (C) and adenoviral progeny titer (D); Bars equal 400μm (H&E) and 200μm (IHC); *p<0.05;.
Combination therapy with VEGF neutralization and CRAd-S-pk7 reduces glioma growth more than each therapy alone
To evaluate whether the higher adenoviral titers and distribution resulted in reduced tumor growth, we treated mice bearing U251 xenografts with VEGF-Ab, CRAd-S-pk7 or combination therapy. Oncolytic adenovirus therapy alone slowed tumor growth at a similar rate to short-term anti-angiogenic therapy. The group that received oncolytic adenovirus after short-term VEGF-Ab therapy had the largest reduction in tumor growth rate (Figure 7; ***p<0.0001). The time needed for the average tumor volume to double was the shortest in the PBS treated animals (2.6 days) and the longest in the combination therapy group (11.9 days). Animals that received CRAd-S-pk7 and VEGF-Ab had a tumor doubling time of 7.6 and 8 days, respectively.
Figure 7.
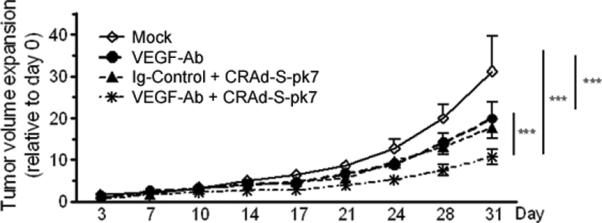
Combining VEGF trapping with CRAd-S-pk7 resulted in more reduced tumor growth rates than each therapy alone. Nude mice with U251 glioma flank xenografts were treated for 5 days with VEGF-Ab; followed by 108 IU of oncolytic adenovirus CRAd-S-pk7. Tumor size was measured twice weekly with caliper. Growth curves represent the mean tumor growth rate, for each group, as compared to the day 0, when the therapy was started. To compare tumor growth curves the log rank test was used; ***p < 0.001
Discussion
This study addresses the effect of anti-angiogenic therapy on oncolytic adenovirus replication and distribution both in vitro and in vivo. We found that VEGF neutralization/blocking increases MMP-2 expression in glioma cell lines. In glioma xenografts, VEGF-Ab up-regulated MMP-2 and reduced the amount of its substrate, collagen IV. When CRAd-S-pk7 was injected intra-tumorally following VEGF-Ab administration, it resulted in increased viral distribution (Figure 6a, b). Short-term VEGF-Ab therapy followed by adenovirus injection resulted in increased intra-tumoral adenoviral titers (Figure 6d). Moreover, this combination therapy was superior in reducing tumor growth rate than each individual therapy (Figure 7).
In vitro VEGF neutralization can disrupt glioma cell metabolism and alter phenotype.16 Cell surface changes can result in a more or less hospitable environment for oncolytic adenovirus replication. Therefore, we first tested whether VEGF neutralization would alter the expression of surface receptors used by adenovirus for attachment and entry into glioma cells. We did not detect any changes in the expression levels of commonly used receptors for adenovirus transduction. But, when glioma cells were treated with VEGF-Ab, we noticed an increase in adenovirus replication on day 5. This difference was not present at earlier time points (Figure 1b and c), attesting to the similar adenovirus transduction efficiency in presence of VEGF-Ab. Higher CRAd-S-pk7 replication, which is driven by a survivin promoter, could result from an increase in the expression of its promoter levels within glioma cells. Bevacizumab induces expression of hypoxia induced factor (HIF-1α) regulated genes and survivin is downstream HIF-1α.17, 18 Of note, adenoviruses lacking survivin promoter did not replicate more in presence of bevacizumab in vitro.19 Nevertheless, higher adenovirus replication did not result in an increase of oncolytic toxicity toward glioma cells. We expected higher adenovirus replication to result in more toxicity, but the same factors that may have increased replication (Survivin up-regulation) have been shown to have anti-apoptotic properties.20
In presence of anti-angiogenic therapy, glioblastomas have demonstrated a propensity to transform their phenotype into a more invasive one.21 This phenotype is characterized by alterations in the microenvironment that results in a lower interstitial pressure. Collagen is one of the main components of the ECM that plays a major role in the generation of interstitial pressure and blocks adenovirus distribution upon direct injection. To better understand how VEGF neutralization can alter collagen composition of gliomas we assessed the expression of collagenases MMP-2 and MMP-9. Over-expression of MMP-9 in neuroblastoma cells resulted in reduced levels of collagen IV and increased oncolytic virus distribution.9 Similarly, oncolytic virus distribution and efficacy was improved in sarcomas expressing MMP-1 and MMP-8.22
In our study we found that short-term anti-angiogenic therapy induced up-regulation of MMP-2 expression in U251 glioma cells both in vitro and in vivo (Figure 2a and 3a). Regulation of MMP-2 expression remains complex and understudied.23 MMP-2 transcription is up-regulated during HIF-1α stabilization, which does not increase MMP-9 levels; similar to what we saw in our study.24, 25 Moreover, multiple downstream targets of HIF-1α are up-regulated during anti-angiogenic therapy.18 Based on these data, we evaluated HIF-1α expression in U251 glioma cells and found it to be up-regulated as early as 8 hours post therapy (Supplementary Figure S1). Furthermore, MMP-2 levels and its activity are regulated post-transcriptionally through proteases, such as tissue inhibitor of metalloproteinases (TIMP2), which are themselves altered during anti-angiogenic therapy.10, 26 Moreover different glioma subtypes (classical, mesenchymal, proneural and neural) can respond to anti-angiogenic therapy and regulate expression of MMPs differently.27 Our study supports previous and recent findings that suggest and active role for VEGF-A on glioma cells through an autocrine loop.28, 29
The importance of MMP-2 in GBM invasion is well established, but its role in tumor growth and ultimately survival remains to be defined. Complete knock-down or reduction of MMP-2 to barely distinguishable levels has a significant impact on tumor growth.30 On the other hand, once a threshold of expression is reached, alterations in its levels do not play a significant role in tumor growth progression.31 To further corroborate the role MMP-2 in GBM progression, we run a gene-based Kaplan-Meier survival analysis at The Cancer Genome Atlas project and found that up-regulation or down-regulation of MMP-2 by a factor of 2 does not have any significant effect on patient survival (Supplementary Figure S2).
During VEGF-A blockade we noted no changes in MMP-9 expression levels, not only in U251 but also in three other cell lines. But higher dose and longer treatment can increase MMP-9 and other metalloproteinases in U87.10 Therefore we have to distinguish between the short and long term effects of VEGF blocking therapy. We focused in the normalization window period (day 5–8) induced by VEGF neutralization. During the early days of bevacizumab therapy there is a reduction in vessel permeability and interstitial pressure that results from ECM alterations. We found that tumors in animals treated with VEGF-Ab had lower content of collagen IV, the substrate of MMP-2. But this early effect of VEGF neutralization seems to rebound during long term therapy, resulting in tumors that express higher levels of collagen.32 Glioblastomas inherently express high levels of collagen that block adenovirus expression upon injection. Therefore any therapy that reduces collagen content, even for a short therapeutic window, can be of benefit to oncolytic virus delivery. VEGF blockade has been shown to increase adenovirus distribution only when given a few days before the intra-tumoral injection.19 When given simultaneously no additive effect was noted.33 It was these observations that argued in favor of VEGF-Ab treatment to precede CRAd-S-pk7 injection.
VEGF neutralization increased adenovirus distribution in intracranial glioma. This was associated with a reduction in the collagen IV content in glioma xenografts (Figure 3b and 5a). Moreover, collagen IV impediment to oncolytic virus distribution has been demonstrated previously in neuroblastoma9 and was also found true in our U251 glioblastoma model (Figure 5b). We discovered an inverse correlation in the distribution of collagen IV and CRAd-S-pk7 in glioma xenografts. Most importantly, with combination therapy (VEGF-Ab followed by CRAd-S-pk7), we found adenovirus replication to be increased (Figure 6c and d) in glioma xenografts and reduced tumor growth compared to each agent alone (Figure 7).
Bevacizumab therapy is currently approved for use in GBM, advanced colon and kidney carcinomas. At the same time, oncolytic virotherapy is an expanding novel field that is targeting the same cancers, but falling short of expectations due to limited intratumoral distribution.4, 34, 35 Therefore, based on our findings, we propose that when combining these agents the bevacizumab therapy should precede intratumoral virus injection. For GBM patients with unresectable tumors that currently receive bevacizumab at their relapse this would mean that oncolytic virotherapy, when introduced in the clinic, should follow bevacizumab during the `normalization window' for best results. Better virus injection techniques, such as convection enhanced delivery (CED), can even increase the benefits we observed with simple injection.
In conclusion, our study shows that anti-angiogenic therapy given prior to adenovirus injection can increase its distribution and replication. Our findings emanate from previous studies on solid tumors, characterizing changes in the microenvironment induced during anti-angiogenic therapy, and therefore are widely applicable outside glioma model. Moreover, these results should also apply to other gene therapy vectors as well.
Material and methods
Cell lines and adenoviral vectors
The human glioma cell lines U251, U87, A172 and U118 were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). U87, A172 and U118 cells were grown in minimal essential medium (MEM) with 10% fetal bovine serum (FBS, Atlanta Biologicals, Lawerenceville, GA, USA), 100 μg/ml penicillin and 100 μg/ml streptomycin. Human glioma cell line U251 human embryonic kidney cell line (HEK-293) and grown in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS, 100 μg/ml penicillin and 100 μg/ml streptomycin (Cellgro, Mediatech Inc., Manassas, VA, USA). All cells were grown in a humidified atmosphere, with 5% CO2 and 37 °C conditions. Cells were subcultured using 1 ml per 106 cells of a 0.25% trypsin/2.21 mmol/l EDTA solution. Trypsin activity was quenched using the appropriate media for each cell type; then washed at 300 relative centrifugal forces and plated at the indicated densities.
The conditionally replicative adenoviral vector CRAd-S-pk7 has been described previously.3 Briefly, the competent vector derives from wild-type adenovirus 5 with human survivin promoter incorporated to drive expression of E1 region. Fiber modification was achieved by insertion of 7 poly-Lysine repeats (pk7) in the C-terminal of knob domain.
Antibodies and reagents
The antibodies anti-human ανβ3 and anti-human ανβ5 were purchased from Chemicon/Millipore (Billerica, MA, USA); the Ig controls and anti-human CD138 from Ebioscience (San Diego, CA, USA); coxsackie-adenovirus receptor (CAR), collagen IV, adenovirus-biotin and rabbit anti-human HIF-1α from Abcam (Cambridge, MA, USA); Laminin from ABR/Thermo-Fisher Scientific (Newington, NH, USA); MMP-9 from Cell Signaling (Danvers, MA, USA) and MMP-2 from Santa Cruz (Santa Cruz, CA, USA). Secondary antibodies or conjugates used were FITC-conjugated anti-mouse IgG from Millipore, Streptavidin-AlexaFluor 555 and Anti-rabbit AlexaFluor 647 from Invitrogen (Carlsbad, CA, USA).
The VEGF trapping antibody (VEGF-Ab) (clone B20-4.1.1),36 a cross species reacting monoclonal antibody to VEGF-A, was provided by Genentech (San Francisco, CA, USA). The concentration of VEGF-Ab for in vitro studies (50μg/ml) was based on the clinically used regimen (10mg/kg) and the extracellular fluid volume where antibodies disperse, to correlate better with physiologically relevant concentrations. For in vivo experiments mice received two intraperitoneal (i.p.) injections, three days apart, of VEGF-Ab (5mg/kg diluted in PBS). As an antibody control we used the same amount of a whole mouse IgG from Jackson ImmunoResearch Laboratories (West Grove, PA, USA).
Flow Cytometry for cell surface receptors
Cells were plated in 6 well plates at 100000 cells per well and treated with VEGF-Ab or Ig-Control for 72 hours. Collection was done by scraping the cells, washing and then staining the live cells with the indicated antibodies. Data were acquired and analyzed in BD FACSCanto™ with CellQuest (Becton Dickinson, San Jose, CA, USA) and FlowJo (TreeStar, Ashland, OR) software. Experiments were performed twice independently, in triplicates.
Western blotting
For western blotting, samples were loaded with an equal amount of total proteins, as measured by Bradford assay read at 630 nm (Bio-Rad, Hercules, CA, USA). To detect levels of MMP-2 and MMP-9, 250000 cells were grown in T25cm2 flasks and treated with VEGF-Ab for 5 days. On the last day cell secretion was blocked with GolgiPlug™ (Ebioscience, San Diego, CA, USA) for 6 hours before collection of cells. Protein lysates were prepared by using a fresh protease inhibitor and RIPA lysis buffer. Fifty μg of proteins were separated in 4–20% ready-to-use gradient gel (Bio-Rad), transferred to the polyvinylidene difluoride (PVDF) membrane following hybridization with either mouse anti human MMP-2 (Santa Cruz), MMP-9 (Cell Signaling) or anti human Actin (Sigma, MO, USA) antibodies. Western blotting assay were done with SuperSignal West Pico chemiluminescent substrate (Bio-Rad). Images were collected in a Bio-Rad Image station.
Glioma viability assay
To test the toxicity of oncolytic adenovirus in combination with VEGF-Ab, cells were plated in a 96-well, 5000 per well, in triplicates. Twenty-four hours later cells were infected with CRAd-S-pk7 in 10 fold dilutions from 10 IU/cell to 0.1 IU/cell and then treated with VEGF-Ab 50μg/ml or control Ig for 3 days before trypan blue exclusion assay. Cells were collected using trypsin-EDTA 0.25% and counted in a cytometer after resuspended in trypan blue. Cells that excluded trypan blue were considered viable. Graphs express percentage of live cell in experimental groups compared to the non-infected control group (Mock).
Quantification of viral titers and E1A copy determinations
To determine the infectious units per ml viral stock (IU/ml), viral progeny in vitro and in vivo we used the Adeno Rapid-X Titer Kit (Clontech, Mountain View, CA, USA). For in vitro study cells were plated in 12-well plates, 2*105 cells per well; incubated 24 hours later with serum-free media containing CRAd-S-pk7, for 1 hour, at the indicated concentrations. Infected cells were collected three and five days later; and freeze-thawed three times to release adenovirus. After spinning down the cells the supernatant was used to infect confluent layers of HEK-293 at different dilutions and 48 hours later viral titers (IU/ml) were determined by counting infected HEK-293 cells. In a similar way, for in vivo experiments, the tissue was collected and homogenized at the time points indicated; the same amount of tissue from each group was taken and freeze-thawed three times.
Since adenovirus replication/packaging is not very effective we also determined E1A copy number in vitro and in vivo. DNA was isolated by using DNeasy Blood and Tissue kit (Qiagen, Valencia, CA, USA). The expression levels of adenoviral DNA were detected by using quantitative real-time PCR with primer sequences recognizing E1A area, which have been described before.37 Samples were run in triplicates and standardized to ng of DNA.
Evaluation of MMP-2 gene expression by qRT-PCR
Relative expression of mRNA transcripts by glioma cell lines was evaluated for the expression of MMP-2. Glioma cells, 105 cells/well in triplicate, were treated with VEGF-Ab or IgG control and cells were collected at different time points. Total cellular RNA was isolated using an RNeasy kit (Qiagen) according to the manufacturer's protocol and in each instance 1 μg of purified mRNA was reverse transcribed to complementary DNA using the iScript cDNA conversion kit (Bio-Rad). Quantitative PCR was conducted using the SYBR Green quantitative PCR kit (Invitrogen) for all experiments. Optimization of annealing temperatures for each transcript was first conducted. Each transcript of interest was amplified in triplicate at its proper annealing temperature and products were analyzed using the Opticon 2 software (Bio Rad). Relative expression was evaluated using the ΔCT method (ΔCT = CT gene of interest– CT GAPDH). Before plotting them in the graph, the MMP-2 expression for each cell line was normalized to their Ig-Control treated condition, to which was given an arbitrary value of 1. The relative expression levels of MMP-2 for all four cell lines upon VEGF-Ab treatment were then presented as fold of their Control treated cells.
Animal experiments
Animals were cared for according to a study-specific animal protocol approved by The University of Chicago Institutional Animal Care and Use Committee.
For immunohistochemistry and adenoviral replication in vivo, seven to eight-week-old male nude mice (Harlan Laboratories, Madison, WI, USA) were injected intracranially (i.c.) or subcutaneously (s.c.). In brief, mice were anaesthetized with an intraperitoneal (i.p.) injection of ketamine hydrochloride (25 mg/ml)/xylazine (2.5 mg/ml) cocktail. For i.c. injection, a midline incision was made, and a 1-mm burr hole centered 2 mm posterior to the coronal suture and 2 mm lateral to the sagittal suture was made. Animals were placed in a stereotactic frame and 25 000 U251 cells, in a 2.5 μl volume, were injected with a Hamilton needle 3 mm deep into the brain. Twenty-one days after tumor implantation, mice received two i.p. injections of 5mg/kg VEGF-Ab three days apart or Ig control (n=3 animals per group); and on day 26 after U251 implantation mice were injected i.c. with 108 IU of CRAd-S-pk7. Mice were sacrificed three days later and viral distribution was quantified.
For flank tumors, 106 U251 or U87 glioma cells in 100 μl were injected in the dorsal chamber (s.c.) bilaterally with a 20 Gauge needle. Three weeks later mice underwent the same therapeutic schedule, two i.p. injections of VEGF-Ab 5mg/kg or Ig control three days apart (n=4 animals per group) and on day 26 mice were sacrificed for IHC or received a dose of 108 IU CRAd-S-pk7 intratumoral (i.t.) injection. The latter were sacrificed after 3 and 7 day for determination of viral titers and E1A expression in xenografts. For determination of the possible role of adenovirus injection in MMP-2 expression, animals were sacrificed three days after receiving i.t.108 IU of CRAd-S-pk7.
For assessment of flank tumor volume progression we injected 106 U251 cells s.c. bilaterally; three weeks later, we separated the animals in four groups of five animals per group with similar mean volume. Mice received either VEGF-Ab (two i.p. injections of 5mg/kg three days apart) or IgG control, followed five days later by direct i.t. injection of 108 IU CRAd-S-pk7. The day of the adenovirus injection was considered day 0. Tumor dimensions were measured twice a week for a month, with caliper, and volume was determined by the formula: a*b*c/2. Growth progression was determined for each tumor by comparing the volume at the indicated time versus day 0.
Immunohistochemistry
Flank s.c. tumor tissues were cut in half and dropped in a 10% formalin solution or frozen in OCT in a dry ice-methylbutane bath. Brain tissues were snap frozen in a mixture of 2-N-methyl-Bromide and Methylbutane; then cut coronally at the injection site in 2 pieces and embedded in OCT in a dry ice-methylbutane bath. Sections of 5 μm for SC tumors, or 10 μm for brain tumor, spanning over 2mm of tissue, were performed and stained with the described antibodies. For quantitative evaluations of IHC staining with HRP-Hematoxylin two different automated image-scanning system were used: Automated Cellular Imaging System (ACIS, Clarient, CA) gives output in brown dots per area (Figure 3); and ScanScope XT (Aperio, Vista, CA) which uses color deconvolution to depict different intensities of staining and gives output in percent of areas that stain at 4 different levels: no staining, weak, moderate and strong staining (Figure 4).
Immunofluorescent staining for adenovirus hexon and collagen was done based on Abcam protocol. Shortly, brain sections were let to dry at room temperature for 10 minutes before fixation and permeabilization with 50/50 mixture of acetone-methanol. After washing with PBS and blocking with 10% BSA, sections were stained for with the described antibodies as suggested in their information sheet. Sections were incubated overnight with the primary antibody and 1 hour with the secondary. After the final wash sections were covered with Prolong® Gold antifade reagent with DAPI (Invitrogen). For capturing fluorescent IHC images we used an inverted Zeiss microscope.
Statistical analysis
The statistical significance difference between means of two groups was evaluated based on unpaired Student T-test. One way ANOVA was used to test for difference between two or more independent groups. The level of significance was set as p<0.05. Calculations were done by using SigmaPlot version 8.02 (SPSS Inc., Chicago, IL, USA).
Supplementary Material
Acknowledgment
We thank Feifei Liu for expertise in statistically analyzing the data and Derek A. Wainwright for optimizing the immunofluorescence staining protocol. A cross species reacting monoclonal antibody to VEGF-A, (clone B20-4.1.1), was provided by Genentech (San Francisco, CA, USA).
Funding and supporting materials This work was supported by the NCI (R01CA122930, R01CA138587), the National Institute of Neurological Disorders and Stroke (U01NS069997), and the American Cancer Society (RSG-07-276-01-MGO).
Footnotes
Supplemental Information Supplementary information is available at Gene Therapy's website.
Conflict of interest The authors declare no conflict of interest.
References
- 1.Bondy ML, Scheurer ME, Malmer B, Barnholtz-Sloan JS, Davis FG, Il'yasova D, et al. Brain tumor epidemiology: consensus from the Brain Tumor Epidemiology Consortium. Cancer. 2008;113(7 Suppl):1953–68. doi: 10.1002/cncr.23741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 3.Ulasov IV, Zhu ZB, Tyler MA, Han Y, Rivera AA, Khramtsov A, et al. Survivin-driven and fiber-modified oncolytic adenovirus exhibits potent antitumor activity in established intracranial glioma. Hum Gene Ther. 2007;18(7):589–602. doi: 10.1089/hum.2007.002. [DOI] [PubMed] [Google Scholar]
- 4.Wojton J, Kaur B. Impact of tumor microenvironment on oncolytic viral therapy. Cytokine Growth Factor Rev. 2010;21(2–3):127–34. doi: 10.1016/j.cytogfr.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pluen A, Boucher Y, Ramanujan S, McKee TD, Gohongi T, di Tomaso E, et al. Role of tumor-host interactions in interstitial diffusion of macromolecules: cranial vs. subcutaneous tumors. Proc Natl Acad Sci U S A. 2001;98(8):4628–33. doi: 10.1073/pnas.081626898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKee TD, Grandi P, Mok W, Alexandrakis G, Insin N, et al. Zimmer JP, et al. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006;66(5):2509–13. doi: 10.1158/0008-5472.CAN-05-2242. [DOI] [PubMed] [Google Scholar]
- 7.Kim JH, Lee YS, Kim H, Huang JH, Yoon AR, Yun CO. Relaxin expression from tumor-targeting adenoviruses and its intratumoral spread, apoptosis induction, and efficacy. J Natl Cancer Inst. 2006;98(20):1482–93. doi: 10.1093/jnci/djj397. [DOI] [PubMed] [Google Scholar]
- 8.Ganesh S, Gonzalez Edick M, Idamakanti N, Abramova M, Vanroey M, Robinson M, et al. Relaxin-expressing, fiber chimeric oncolytic adenovirus prolongs survival of tumor-bearing mice. Cancer Res. 2007;67(9):4399–407. doi: 10.1158/0008-5472.CAN-06-4260. [DOI] [PubMed] [Google Scholar]
- 9.Hong CS, Fellows W, Niranjan A, Alber S, Watkins S, Cohen JB, et al. Ectopic matrix metalloproteinase-9 expression in human brain tumor cells enhances oncolytic HSV vector infection. Gene Ther. 2010;17(10):1200–5. doi: 10.1038/gt.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lucio-Eterovic AK, Piao Y, de Groot JF. Mediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapy. Clin Cancer Res. 2009;15(14):4589–99. doi: 10.1158/1078-0432.CCR-09-0575. [DOI] [PubMed] [Google Scholar]
- 11.Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27(28):4733–40. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 12.Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27(5):740–5. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bergers G, Javaherian K, Lo KM, Folkman J, Hanahan D. Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science. 1999;284(5415):808–12. doi: 10.1126/science.284.5415.808. [DOI] [PubMed] [Google Scholar]
- 14.Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004;6(6):553–63. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 15.Dickson PV, Hamner JB, Sims TL, Fraga CH, Ng CY, Rajasekeran S, et al. Bevacizumab-induced transient remodeling of the vasculature in neuroblastoma xenografts results in improved delivery and efficacy of systemically administered chemotherapy. Clin Cancer Res. 2007;13(13):3942–50. doi: 10.1158/1078-0432.CCR-07-0278. [DOI] [PubMed] [Google Scholar]
- 16.Hu B, Guo P, Bar-Joseph I, Imanishi Y, Jarzynka MJ, Bogler O, et al. Neuropilin-1 promotes human glioma progression through potentiating the activity of the HGF/SF autocrine pathway. Oncogene. 2007;26(38):5577–86. doi: 10.1038/sj.onc.1210348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yun YJ, Li SH, Cho YS, Park JW, Chun YS. Survivin mediates prostate cell protection by HIF-1alpha against zinc toxicity. Prostate. 2010;70(11):1179–88. doi: 10.1002/pros.21152. [DOI] [PubMed] [Google Scholar]
- 18.Rapisarda A, Hollingshead M, Uranchimeg B, Bonomi CA, Borgel SD, Carter JP, et al. Increased antitumor activity of bevacizumab in combination with hypoxia inducible factor-1 inhibition. Mol Cancer Ther. 2009;8(7):1867–77. doi: 10.1158/1535-7163.MCT-09-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Libertini S, Iacuzzo I, Perruolo G, Scala S, Ierano C, Franco R, et al. Bevacizumab increases viral distribution in human anaplastic thyroid carcinoma xenografts and enhances the effects of E1A-defective adenovirus dl922-947. Clin Cancer Res. 2008;14(20):6505–14. doi: 10.1158/1078-0432.CCR-08-0200. [DOI] [PubMed] [Google Scholar]
- 20.Reichert S, Rodel C, Mirsch J, Harter PN, Tomicic MT, Mittelbronn M, et al. Survivin inhibition and DNA double-strand break repair: A molecular mechanism to overcome radioresistance in glioblastoma. Radiother Oncol. 2011;101(1):51–8. doi: 10.1016/j.radonc.2011.06.037. [DOI] [PubMed] [Google Scholar]
- 21.de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, et al. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol. 2010;12(3):233–42. doi: 10.1093/neuonc/nop027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mok W, Boucher Y, Jain RK. Matrix metalloproteinases-1 and -8 improve the distribution and efficacy of an oncolytic virus. Cancer Res. 2007;67(22):10664–8. doi: 10.1158/0008-5472.CAN-07-3107. [DOI] [PubMed] [Google Scholar]
- 23.Chernov AV, Sounni NE, Remacle AG, Strongin AY. Epigenetic control of the invasion-promoting MT1-MMP/MMP-2/TIMP-2 axis in cancer cells. J Biol Chem. 2009;284(19):12727–34. doi: 10.1074/jbc.M900273200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 25.Elstner A, Holtkamp N, von Deimling A. Involvement of Hif-1 in desferrioxamine-induced invasion of glioblastoma cells. Clin Exp Metastasis. 2007;24(1):57–66. doi: 10.1007/s10585-007-9057-y. [DOI] [PubMed] [Google Scholar]
- 26.Lu KV, Jong KA, Rajasekaran AK, Cloughesy TF, Mischel PS. Upregulation of tissue inhibitor of metalloproteinases (TIMP)-2 promotes matrix metalloproteinase (MMP)-2 activation and cell invasion in a human glioblastoma cell line. Lab Invest. 2004;84(1):8–20. doi: 10.1038/sj.labinvest.3700003. [DOI] [PubMed] [Google Scholar]
- 27.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamerlik P, Lathia JD, Rasmussen R, Wu Q, Bartkova J, Lee M, et al. Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J Exp Med. 2012;209(3):507–20. doi: 10.1084/jem.20111424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J, Yu H, Choi K, Choi C. Differential dependency of human cancer cells on vascular endothelial growth factor-mediated autocrine growth and survival. Cancer Lett. 2011;309(2):145–50. doi: 10.1016/j.canlet.2011.05.026. [DOI] [PubMed] [Google Scholar]
- 30.Kargiotis O, Chetty C, Gondi CS, Tsung AJ, Dinh DH, Gujrati M, et al. Adenovirus-mediated transfer of siRNA against MMP-2 mRNA results in impaired invasion and tumor-induced angiogenesis, induces apoptosis in vitro and inhibits tumor growth in vivo in glioblastoma. Oncogene. 2008;27(35):4830–40. doi: 10.1038/onc.2008.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lamfers ML, Gianni D, Tung CH, Idema S, Schagen FH, Carette JE, et al. Tissue inhibitor of metalloproteinase-3 expression from an oncolytic adenovirus inhibits matrix metalloproteinase activity in vivo without affecting antitumor efficacy in malignant glioma. Cancer Res. 2005;65(20):9398–405. doi: 10.1158/0008-5472.CAN-04-4264. [DOI] [PubMed] [Google Scholar]
- 32.Zhang W, Fulci G, Buhrman JS, Stemmer-Rachamimov AO, Chen JW, Wojtkiewicz GR, et al. Bevacizumab With Angiostatin-armed oHSV Increases Antiangiogenesis and Decreases Bevacizumab-induced Invasion in U87 Glioma. Mol Ther. 2011;20(1):37–45. doi: 10.1038/mt.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guse K, Ranki T, Ala-Opas M, Bono P, Sarkioja M, Rajecki M, et al. Treatment of metastatic renal cancer with capsid-modified oncolytic adenoviruses. Mol Cancer Ther. 2007;6(10):2728–36. doi: 10.1158/1535-7163.MCT-07-0176. [DOI] [PubMed] [Google Scholar]
- 34.Gutermann A, Mayer E, von Dehn-Rothfelser K, Breidenstein C, Weber M, Muench M, et al. Efficacy of oncolytic herpesvirus NV1020 can be enhanced by combination with chemotherapeutics in colon carcinoma cells. Hum Gene Ther. 2006;17(12):1241–53. doi: 10.1089/hum.2006.17.1241. [DOI] [PubMed] [Google Scholar]
- 35.Guse K, Sloniecka M, Diaconu I, Ottolino-Perry K, Tang N, Ng C, et al. Antiangiogenic arming of an oncolytic vaccinia virus enhances antitumor efficacy in renal cell cancer models. J Virol. 2010;84(2):856–66. doi: 10.1128/JVI.00692-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuh G, Wu P, Liang WC, Ultsch M, Lee CV, Moffat B, et al. Structure-function studies of two synthetic anti-vascular endothelial growth factor Fabs and comparison with the Avastin Fab. J Biol Chem. 2006;281(10):6625–31. doi: 10.1074/jbc.M507783200. [DOI] [PubMed] [Google Scholar]
- 37.Ulasov IV, Sonabend AM, Nandi S, Khramtsov A, Han Y, Lesniak MS. Combination of adenoviral virotherapy and temozolomide chemotherapy eradicates malignant glioma through autophagic and apoptotic cell death in vivo. Br J Cancer. 2009;100(7):1154–64. doi: 10.1038/sj.bjc.6604969. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.