

Abstract
The ubiquitin-proteasome system (UPS) is involved in the replication of a broad range of viruses. Since replication of the murine hepatitis virus (MHV) is impaired upon proteasomal inhibition, the relevance of the UPS for the replication of the severe acute respiratory syndrome coronavirus (SARS-CoV) was investigated in this study. We demonstrate that the proteasomal inhibitor MG132 strongly inhibits SARS-CoV replication by interfering with early steps of the viral life cycle. Surprisingly, other proteasomal inhibitors (e.g., lactacystin and bortezomib) only marginally affected viral replication, indicating that the effect of MG132 is independent of proteasomal impairment. Induction of autophagy by MG132 treatment was excluded from playing a role, and no changes in SARS-CoV titers were observed during infection of wild-type or autophagy-deficient ATG5−/− mouse embryonic fibroblasts overexpressing the human SARS-CoV receptor, angiotensin-converting enzyme 2 (ACE2). Since MG132 also inhibits the cysteine protease m-calpain, we addressed the role of calpains in the early SARS-CoV life cycle using calpain inhibitors III (MDL28170) and VI (SJA6017). In fact, m-calpain inhibition with MDL28170 resulted in an even more pronounced inhibition of SARS-CoV replication (>7 orders of magnitude) than did MG132. Additional m-calpain knockdown experiments confirmed the dependence of SARS-CoV replication on the activity of the cysteine protease m-calpain. Taken together, we provide strong experimental evidence that SARS-CoV has unique replication requirements which are independent of functional UPS or autophagy pathways compared to other coronaviruses. Additionally, this work highlights an important role for m-calpain during early steps of the SARS-CoV life cycle.
INTRODUCTION
In 2003, a new human coronavirus of zoonotic origin emerged in southern China, causing a worldwide epidemic of an atypical life-threatening pneumonia, the severe acute respiratory syndrome (SARS) (12, 28, 30, 43). The new virus, designated SARS-coronavirus (CoV), exhibited extraordinary pathogenicity with a high mortality rate, in contrast to other known human coronaviruses, which normally cause slight diseases of the upper respiratory or gastrointestinal tract. Coronaviruses, showing extensive genetic diversity and short generation times, are very infectious and are capable of crossing species barriers (20). Since closely related and SARS-CoV-like viruses circulate in bats, their natural animal reservoir (11, 34), SARS or similarly severe diseases might reemerge or emerge. Therefore, the molecular mechanisms of SARS-CoV replication are still important subjects of investigation.
As a member of the family Coronaviridae, SARS-CoV is a positive-stranded, enveloped RNA virus with a genome consisting of around 30 kb (37). Upon binding to its functional receptor, the angiotensin-converting enzyme 2 (ACE2) (33), SARS-CoV is internalized via endocytosis, and the viral RNA is released from the endosome (19, 44, 67, 70). In the cytosol, the SARS-CoV genome is translated into two large polyproteins which are autocatalytically processed to produce the nonstructural proteins (nsps), including all nsps of the viral replicase complex (50, 61, 63). Protected by double membrane vesicles (DMVs) most likely originating from endoplasmic reticulum (ER) membranes (58), the genome is replicated and, by generation of specific sets of subgenomic mRNA, structural and SARS-CoV-specific accessory proteins are produced (61, 63). After assembly and budding processes into the endoplasmic reticulum Golgi intermediate compartment (ERGIC) (25, 59), mature virions are released by exocytosis.
Along with the lysosomal and autophagy pathways by which proteins, protein aggregates, or even whole organelles are degraded, the ubiquitin-proteasome system (UPS) is part of the cellular degradation machinery. Degradation via the proteasome is highly regulated, and for recognition, substrates must be covalently modified with multiple molecules of the small 76-amino-acid protein ubiquitin (6). However, in addition to degradation of misfolded or unfolded proteins or protein aggregates, the UPS also plays major roles in regulating cell cycle progression, apoptosis, antigen processing, signal transduction, and transcriptional regulation (6). For this reason, viruses have evolved mechanisms to exploit the UPS for viral benefit.
To circumvent host innate immune response, human cytomegalovirus blocks antigen presentation by targeting major histocompatibility complex class I (MHC-I) molecules for proteasomal degradation (24, 53). Alternatively, downregulation of CD4 by inducing its proteasomal degradation contributes to the pathogenesis of immunodeficiency in HIV-1 infection (51, 66). In addition to destabilizing antiviral proteins, viruses maintain proviral or viral proteins by manipulating cellular ubiquitination processes by expressing their own deubiquitination proteins (DUBs), which were recently described for SARS-CoV as well (2, 35). On the other hand, the replication of a number of viruses strongly depends on the activity of a functional UPS. For example, the processing of gag proteins of HIV-1 is dependent on proteasomal function (52). If the proteasome is inhibited, the nucleoprotein of influenza A virus is retained in the cytosol (22, 68). UPS facilitates the transport of incoming capsids to the nucleus during herpes simplex virus infection (8), and replication of vaccinia virus, coxsackievirus B3 virus, and orthopoxvirus is impaired if the proteasome is inhibited (49, 54, 62). Of note, replication of another coronavirus, murine hepatitis virus (MHV), is affected upon impairment of proteasomal activity because viral RNA cannot be released from endosomes (72). Furthermore, replication of the recombinant coronaviruses feline infectious peritonitis virus (FIPV)-Δ3abcFL, MHV-EFLM, MHV-nsp2EGFP, and SARS-CoV-GFP was shown to be impaired upon proteasomal inhibition, as demonstrated by reduced reporter gene expression (46). Therefore, a general dependence of coronaviral replication on a functional ubiquitin proteasome pathway was suggested. However, in vivo studies by the same group showed that treatment with the proteasome inhibitor bortezomib of C57BL/6 mice infected with MHV-A59 actually led to increased viral titers and pathology (45).
Due to these conflicting data, we aimed to elucidate the role of the ubiquitin-proteasome system during the SARS-CoV life cycle. By performing infection studies with wild-type (wt) SARS-CoV (strain Frankfurt-1), we confirmed that by treatment with the established proteasomal inhibitor MG132, viral replication is strongly impaired in a dose-dependent manner. This treatment mainly affected early steps in the SARS-CoV life cycle. However, subsequent studies with other proteasomal inhibitors or autophagy-deficient cells revealed that neither inhibition of the proteasome nor induction of autophagy were responsible for the impairment of viral replication observed with MG132. Since treatment of infected cells with MDL28170, an inhibitor of calpain, a protease which is inhibited by MG132 but not by other proteasomal inhibitors, impaired SARS-CoV replication, we assume that inhibition of calpain rather than inhibition of the proteasome accounts for the reduction of viral titers by MG132. Subsequent m-calpain knockdown experiments further confirmed that loss of m-calpain activity strongly reduces production of SARS-CoV particles. These results clearly demonstrate that SARS-CoV, compared to other Coronaviridae, has specific requirements for replication and reveal an important role for m-calpain during early steps of the SARS-CoV life cycle.
MATERIALS AND METHODS
Cell culture and virus.
The Vero E6 cell line (African green monkey kidney) was obtained from the ATCC (CRL-1587D) and cultured in Dulbecco's modified Eagle medium (DMEM) with 10% heat-inactivated fetal calf serum (FCS), 100 U of penicillin, 100 μg of streptomycin per ml, and 25 mM HEPES buffer at 37°C without CO2. The wild-type and ATG5−/− mouse embryonic fibroblast (MEF) cell lines were a kind gift from Noboru Mizushima (Tokyo Medical and Dental University, Japan) and were maintained in DMEM with 10% heat-inactivated FCS, 100 U of penicillin, and 100 μg of streptomycin per ml at 37°C with CO2. SARS-CoV strain Franfurt-1 was a kind gift from Christian Drosten (University of Bonn, Germany). All experiments with SARS-CoV were done in a biosafety level 3 facility. MHV strain JHM was a kind gift from Xuming Zhang (University of Arkansas). The mouse epithelial cell line NCTC clone 1469 (ATCC CCL-9.1), used for propagation of MHV, was cultured in DMEM supplemented with 10% horse serum, 100 U of penicillin, and 100 μg of streptomycin per ml.
Reagents and antibodies.
MG132, lactacystin (Lac), SJA6017/calpain inhibitor VI (CI VI), and calpain inhibitor III (MDL28170; CI III) were purchased from Calbiochem/Merck (Darmstadt, Germany). Bortezomib (BZ; Velcade) and imatinib (Glivec) were obtained from Selleck Chemicals (Houston, TX). All chemical compounds were dissolved in dimethylsulfoxide (DMSO) and stored at −20°C. A ready-made solution of 1 mM staurosporine in DMSO, methyl cellulose (viscosity, 4,000 cP), and crystal violet were purchased from Sigma-Aldrich (Munich, Germany). All cell culture reagents as well as Lipofectamine were obtained from Invitrogen (Darmstadt, Germany). Pefabloc proteinase inhibitor was purchased from Roche Diagnostics GmbH (Mannheim, Germany), and the Coomassie Plus (Bradford) protein assay kit was from Pierce (Rockford, IL). Polyvinylidene difluoride (PVDF) membranes and the chemiluminescent Western blotting detection reagent ECL Plus were obtained from GE Healthcare-Amersham (Freiburg, Germany). The production of the rabbit polyclonal anti-N antibody has been described already (9), and rabbit polyclonal anti-Grp78 (H-129) and mouse monoclonal anti-ubiquitin (P4D1) antibodies were from Santa Cruz Biotechnology Inc. (Heidelberg, Germany). The rabbit polyclonal m-calpain (calpain 2) antibody was obtained from Triple Point Biologics (Forest Grove, OR). Mouse monoclonal anti-ß-actin was from Sigma (Munich, Germany), mouse-monoclonal anti-LC3 antibody was obtained from nanoTools Antikörpertechnik GmbH & Co., KG (Teningen, Germany), and the mouse monoclonal anti-ACE2 antibody was obtained from Enzo Life Science (Lörrach, Germany). Anti-mouse and anti-rabbit horseradish peroxidase-conjugated antibodies were purchased from GE Healthcare-Amersham (Freiburg, Germany).
Infection with SARS-CoV or MHV.
Twenty-four h after seeding, cells were pretreated with the different chemical compounds in media without FCS for 1 h or left untreated. After pretreatment, cells were washed with phosphate-buffered saline (PBS) and infected with SARS-CoV or MHV-JHM at the indicated multiplicities of infection (MOIs) in 1 ml FCS-free medium with or without compounds at 37°C for the indicated incubation times. Inocula were removed, and cells were washed three times with PBS before adding 3 ml complete growth medium with or without compounds as indicated. After incubation for the indicated time periods at 37°C, viral supernatants were harvested and aliquots of 1 ml were stored at −80°C.
Plaque assay.
Confluent Vero E6 (SARS-CoV) or NCTC (MHV) cells in 12-well plates were infected with 6 serial dilutions of viral supernatants in FCS-free medium and incubated for 1 h at 37°C. Afterwards, cells were overlaid with normal growth medium containing 1% (Vero E6) or 2% (NCTC) methylcellulose and incubated at 37°C. Seventy-two hours postinfection (p.i.), the overlay was removed and cells were washed with PBS and fixed in 4% formalin for inactivation of virus for 1 h at room temperature. For staining of fixed-cell monolayers, 2% crystal violet in 3.6% formaldehyde and 20% ethanol was used.
Internalization assay.
After pretreatment for 1 h, Vero E6 cells were infected with SARS-CoV for 15 min at an MOI of 2 as described above. After removal of inocula and completion of PBS washing steps, cells were trypsinized for 10 min on ice. Cells were then resuspended in growth medium and centrifuged at 1,200 rpm for 3 min at 4°C to remove cell debris. After resuspending the cells in 1 ml growth medium, cells were broken up by vigorously passing the homogenate 3 times through a 19-gauge syringe needle. Suspensions were directly diluted for viral titer analysis.
Cell proliferation and viability assay.
For investigation of cell proliferation and viability, a cell proliferation kit (XTT) from Roche Diagnostics GmbH (Mannheim, Germany) was used according to the manufacturer's protocol. Briefly, Vero E6 cells were seeded in 96-well plates and grown to 80% confluence. Compounds at the indicated concentrations and the XTT substrate were added for 16 h. After incubation, absorbance was measured by using an enzyme-linked immunosorbent assay (ELISA) reader.
Transfection of MEF cells.
MEFs were seeded in 25-cm2 flasks and cultured for 24 h. Cells were transfected with 4 μg of DNA for 6 h using Lipofectamine 2000 according to the manufacturer's protocol. The human ACE2 expressing construct (huACE2) (33) was a kind gift from Michael Farzan (Harvard University, Cambridge, MA).
siRNA transfection and m-calpain knockdown experiments.
Vero E6 cells were transfected with 350 pmol of short interfering RNA (siRNA) targeting m-calpain mRNA (HS 188705; CCATCCTGAGAAGGGTTCTAGCAAA) or nonsilencing RNA (negative control; both from Ambion/Invitrogen, Darmstadt, Germany) using Lipofectamine 2000 according to the manufacturer's protocol. Forty-eight hours after transfection, cells were lysed for immunoblot analysis or infected with SARS-CoV (MOI, 0.001). After incubation for 16 h, viral titers were determined by plaque assay.
Cell lysis and immunoblot analysis.
Immunoblot analysis was performed as previously described (15). For cell lysis, cells were washed with PBS and incubated in lysis buffer (10 mM Tris, pH 7.5, 100 mM NaCl, 10 mM EDTA, 0.5% Triton X-100, and 0.5% Na-desoxycholate) and 1 mM Pefabloc (a proteinase inhibitor) for 10 min. Lysates were cleared by centrifugation for 1 min at 14,000 rpm, and supernatants were precipitated with methanol at −20°C overnight. After centrifugation for 30 min at 3,500 rpm at 4°C, pellets were resuspended in TNE buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 5 mM EDTA). Total protein concentrations were determined by using the Coomassie Plus (Bradford) protein assay kit (Pierce) before addition of sample loading buffer (7% SDS, 30% glycerin, 20% 2-mercaptoethanol, 0.01% bromphenol blue in 90 mM Tris-HCl, pH 6.8). After boiling at 95°C, equal protein amounts were analyzed by 12.5% SDS-PAGE. Proteins were electrotransferred to a PVDF membrane, which was blocked with nonfat dry milk (5%) in Tris-buffered saline (TBST) (0.05% Tween 20, 100 mM NaCl, 10 mM Tris-HCl; pH 7.8). After incubation with the appropriate primary and secondary horseradish peroxidase (HRP)-conjugated antibodies, proteins were detected using an enhanced chemiluminescence blotting (ECL plus) kit.
Statistical analysis.
Quantification of immunoblot signals was done by means of the ImageQuant Program (Image Quant Analysis; Molecular Dynamics) and measured relative to ß-actin signals. Statistical analysis was performed with Prism software (Graphpad Software, San Diego, CA) using the unpaired two-tailed t test for pairwise comparisons, always referring band intensities or titers of different conditions to DMSO values. Statistical significance was expressed as the following: *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ns, not significant.
RESULTS
Dose-dependent impairment of SARS-CoV replication by MG132 is independent of proteasome inhibition.
The ubiquitin-proteasome pathway is of superior importance for eukaryotic cells operating as a major protein degradation machinery and being involved in major regulatory processes. Therefore, viruses often exploit the system for its benefits or, on the other hand, strongly depend on its functionality (14, 66). To investigate whether the ubiquitin-proteasome system also plays a role during the SARS-CoV life cycle, initial experiments were done using the proteasome inhibitor MG132. Vero E6 cells were pretreated with different concentrations of MG132 or DMSO (mock) and were subsequently infected with SARS-CoV in the presence or absence of MG132. Supernatants were collected and viral titers were determined by plaque assay. As shown in Fig. 1A, treatment of cells with 2.5 μM MG132 decreased viral titers by ∼6 log equivalents compared to mock-treated cells. Furthermore, a dose-dependent effect of MG132 on SARS-CoV replication was confirmed, in that exposure to 0.5 μM MG132 already had decreased viral titers by approximately 1,000-fold (Fig. 1A).
Fig 1.

MG132 strongly impairs SARS-CoV particle production in a dose-dependent and proteasome-independent manner. (A) Impairment of SARS-CoV replication by MG132 is dose dependent. Vero E6 cells were pretreated with DMSO (mock) or 0.5, 1.5, and 2.5 μM MG132 for 1 h and inoculated with SARS-CoV (MOI, 0.001) in FCS-free medium containing DMSO or MG132. After removal of inocula, cells were treated in media with FCS for 16 h before cell-free viral supernatants were collected. Infectious viral titers were determined by plaque assay. Values represent the means ± standard errors of the means (SEM) of two independent experiments performed in duplicate. Statistical analysis was performed as described in Materials and Methods, and SEM are shown. (B) Effects of different proteasomal inhibitors on SARS-CoV replication. Experiments were performed as described above, using DMSO (mock), 2.5 μM MG132, 5 and 10 μM lactacystin (Lac), or 40 and 80 nM bortezomib (BZ), respectively. (C) Effective inhibition of proteasomal activity by different compounds. Vero E6 cells were treated with MG132, Lac, and BZ at the indicated concentrations for 16 h. Equal amounts of protein extracts were subjected to immunoblot analysis using anti-ubiquitin (upper) and anti-Grp78/BiP (lower) antibody. For each immunoblot, the respective actin loading control is shown below. (D) Fold induction of BiP expression and ubiquitin accumulation. Values represent the means ± SEM of one experiment performed in triplicate. (E) Effects of different proteasomal inhibitors on MHV-JHM replication. NCTC cells were pretreated with DMSO (mock), 2.5 μM MG132, and 5 or 10 μM lactacystin for 1 h and inoculated with MHV-JHM in FCS-free medium containing the different inhibitors. After removal of inocula, cells were treated in media with FCS for 16 h before cell-free viral supernatants were collected. Infectious viral titers were determined by plaque assay as described for SARS-CoV experiments.
To verify that effects of MG132 on viral replication are caused by impairment of the ubiquitin-proteasome pathway, two further proteasomal inhibitors were used under identical conditions, i.e., lactacystin (Lac), an irreversible proteasome inhibitor, and bortezomib (BZ), the first therapeutic proteasome inhibitor for treatment of multiple myeloma and mantle cell lymphoma (48). Although treatment of infected cells with 10 μM Lac and 40 nM BZ slightly, though significantly, reduced viral titers (∼1-log reduction), inhibitory effects were much less pronounced than the effect of MG132 (∼6-log reduction) (Fig. 1B). The concentrations of proteasomal inhibitors and other applied chemical compounds used were not toxic to Vero cells after 16 h of incubation, as verified in XTT viability assays (data not shown).
Proteasomal inhibition leads to accumulation of polyubiquitinated proteins in the cytosol and proteins of the secretory pathway, which are destined for degradation and accumulate in the endoplasmic reticulum (ER) (26), making ER stress and the subsequent unfolded protein response (UPR) unavoidable. BiP/Grp78 is the major ER chaperone induced upon activation of UPR and serves as an indicator of proteasomal dysfunction (4, 32). To ensure effective and comparable impairment of proteasomal activity by the inhibitors used, we analyzed the accumulation of polyubiquitinated proteins and the induction of BiP/Grp78 expression. Therefore, cells were treated with proteasomal inhibitors as described above, and cell lysates were analyzed by immunoblotting using anti-ubiquitin and anti-BiP antibodies, respectively. Treatment with the different proteasomal inhibitors significantly induced BiP expression compared to mock-treated cells, as well as showing dose-dependent effects for Lac and BZ (Fig. 1C; data are quantified in Fig. 1D). Similarly, polyubiquitinated proteins were induced with a 2.5- to 3.5-fold increase (Fig. 1C; data are quantified in Fig. 1D).
It has been demonstrated in a previous study that replication of MHV (strain JHM) is dependent on proteasome function (72). In this study, all of the proteasome inhibitors used were able to significantly reduce viral replication. Since our data are in sharp contrast to those of earlier work, we aimed at directly comparing the effects of proteasomal inhibition on MHV and SARS-CoV replication under our experimental conditions. Therefore, murine NCTC cells were pretreated with inhibitors as described for SARS-CoV experiments and infected for 16 h with MHV. As demonstrated in Fig. 1E and in line with the previous publication (72), treatment with 2.5 μM MG132 as well as with lactacystin in two different concentrations strongly decreased MHV titers in NCTC cells.
Whereas effective and comparable impairment of the proteasome by all three inhibitors was confirmed, MG132 has a unique potency to inhibit SARS-CoV replication in a dose-dependent and, most likely, proteasome-independent way.
MG132 treatment inhibits early steps during the SARS-CoV life cycle without affecting virus internalization.
Having seen that MG132 strongly interferes with SARS-CoV replication, we next wanted to determine which steps of SARS-CoV replication are affected. Therefore, viral particle production was determined in time course experiments, and viral replication kinetics of mock- or MG132-treated cells were compared. Vero E6 cells were pretreated with or without MG132 before infection with SARS-CoV. At 0, 8, or 16 h p.i. with continuous MG132 treatment, viral supernatants were analyzed by plaque assay or infected cells were lysed to determine intracellular levels of the viral nucleocapsid protein (N) by immunoblot analysis. N associates with the viral RNA to form the helical nucleocapsid (60) and is most abundantly expressed during infection (27). Therefore, it can be considered a marker for the production of viral proteins in total. Viral loads increased exponentially under mock infection conditions, reaching the highest levels at 16 h p.i. (Fig. 2A), as did the production of N protein (Fig. 2B). In contrast, production of infectious viral particles in MG132-treated cells was severely inhibited, and very low viral titers were achieved only at 16 h p.i. (Fig. 2A), with N protein being almost undetectable by immunoblot analysis (Fig. 2B). This let us assume that treatment with MG132 also affects production of viral proteins. To confirm effective proteasomal inhibition during this infection experiment, we analyzed lysates of infected cells for BiP/Grp78 expression. As expected, MG132 treatment strongly induced BiP expression during infection (Fig. 2B, upper). Interestingly and contrary to our expectation, infection of mock-treated cells did not induce Grp78/BiP expression; moreover, a reduction of BiP was observed.
Fig 2.

MG132 inhibits early steps in SARS-CoV life cycle without affecting virus internalization. (A) Replication kinetics of SARS-CoV upon mock or MG132 treatment. Vero E6 cells were pretreated with DMSO (mock) or 2.5 μM MG132 for 1 h and then inoculated with SARS-CoV (MOI, 0.001) in FCS-free media containing DMSO or MG132, respectively. After removal of inocula, cells were grown in normal growth medium and viral supernatants were collected at 0, 8, and 16 h p.i. Viral titers were determined by plaque assay (in PFU). Values represent the means ± SEM of two independent experiments performed in duplicate. (B) Vero E6 cells were infected as described for panel A, cell lysates were collected at the indicated time points p.i., and they were subjected to immunoblot analysis with anti-N and anti-Grp/BiP78 antibodies, respectively. Actin was used as a loading control (lower panel). (C) Experimental setup for time frame experiments. (D) Time frame experiment. Vero E6 cells were infected with SARS-CoV at an MOI of 0.001 for 10 min in media without FCS. After removal of inocula, growth medium was added and cells were treated for 2-h time frames with DMSO or 2.5 μM MG132. After each time frame, cells were washed with PBS and new growth medium was added. After an overall incubation time of 8 h, viral supernatants were collected. Titers were determined by plaque assay and are shown as PFU/ml. Values represent the means ± SEM of two independent experiments performed in duplicate. (E) Internalization assay. Vero E6 cells were pretreated with DMSO (mock) or 2.5 μM MG132 for 1 h, followed by infection with SARS-CoV (MOI, 2), and treatment with DMSO or MG132 was continued for 15 min. After removal of inocula, cells were washed with PBS, trypsinated, and disrupted. Viral titers of the suspension were determined by plaque assay. Values represent the means ± SEM of one experiment performed in triplicate.
We next addressed the question of whether MG132 interferes with initial steps during infection, e.g., by inhibiting uptake of virus or inhibition of later steps, such as viral protein production. Since effects of MG132 are reversible, we treated Vero E6 cells for 2-h time frames to identify sensitive steps during infection. Previous experiments had revealed that virus is internalized within 15 min and that a SARS-CoV replication cycle from adsorption to release of infectious progeny takes about 7 to 8 h (data not shown). Based on these findings, we decided to shorten inoculation times from 1 h to 15 min to record very early effects of MG132. In addition, since MG132 effects on viral titer production in a first replication cycle could be compensated by a highly productive second round of infection, it was necessary not to exceed an 8-h interval, the time of one replication cycle. We therefore infected Vero E6 cells with SARS-CoV for 15 min, removed viral inocula by extensive washing, and incubated cells with medium containing DMSO (mock treatment) or MG132 for 2-h time frames. After each time frame, cells were intensively washed and incubated with growth medium without MG132. Cell-free viral supernatants were collected after an overall incubation time of 8 h. A schematic overview of the experimental design is shown in Fig. 2C. While treatment of infected cells with MG132 during the first 2 h p.i. strongly inhibited viral titer production, treatment during later steps of the viral life cycle (2 to 4 h) had a much weaker or even no effect during the latest time frame of 4 to 6 h (Fig. 2D). To exclude the possibility that the compound MG132 directly affects the infectivity of viral particles, we incubated virus with cell-free medium alone or with MG132. Subsequent titer analysis revealed no differences (data not shown).
Since time frame experiments had shown that a very early step during the viral life cycle must be affected, we wanted to elucidate whether MG132 treatment interferes with virus internalization. We pretreated Vero cells with MG132 or DMSO as described above, infected them at an MOI of 2 for 15 min in the presence of MG132, removed inocula and virus bound to its receptor by extensive washing and trypsination, respectively, disrupted the cells, and subsequently determined infectious titers corresponding to viable virus. However, no titer differences in the presence or absence of MG132 were detected (Fig. 2E). Of note, treatment of infected cells with chlorpromazine, an inhibitor of clathrin-mediated endocytosis which was shown to inhibit SARS-CoV entry (19), significantly inhibited SARS-CoV uptake in our control studies, verifying the experimental set-up (data not shown).
Altogether, we demonstrate that MG132 treatment impairs initial steps of viral replication in cells without harming viral particles before adsorption. Notably, MG132 does not interfere with virus internalization.
MG132 does not inhibit SARS-CoV replication by induction of autophagy, and SARS-CoV replication is independent of autophagic mechanisms.
Since we excluded that viral internalization is affected by MG132 treatment, we assumed that the viral life cycle is impaired at a postinternalization step. It is known that the autophagolysosomal pathway can be induced by proteasomal inhibitors (10, 69), and SARS-CoV is taken up by endocytosis (19, 67, 70). Therefore, one scenario is that reduction of viral titers is caused by rapid degradation of virus upon fusion of endosomes with autophagosomes due to an increased autophagic flux induced by MG132 treatment. To investigate this hypothesis, we first confirmed that MG132 treatment induces autophagy in Vero E6 cells. Vero E6 cells were treated for 16 h with DMSO, MG132, and imatinib, a known inducer of autophagy, as a positive control (13). The conversion of LC3-I to LC3-II, which serves as a reliable marker for autophagosome formation (21), was analyzed by immunoblotting. MG132 induced the formation of LC3-II nearly to the same extent as the positive-control imatinib (∼8-fold) (Fig. 3A and B). If MG132 reduced viral titers by induction of autophagy, imatinib, as a known strong inducer of autophagy, is supposed to have similar effects on SARS-CoV replication. To test this, we pretreated Vero E6 cells with 10 μM imatinib, infected them with SARS-CoV, and incubated cells for 16 h with imatinib before collecting viral supernatants for titer analysis. However, compared to MG132 treatment, there was no impact of imatinib on SARS-CoV replication (Fig. 3C).
Fig 3.
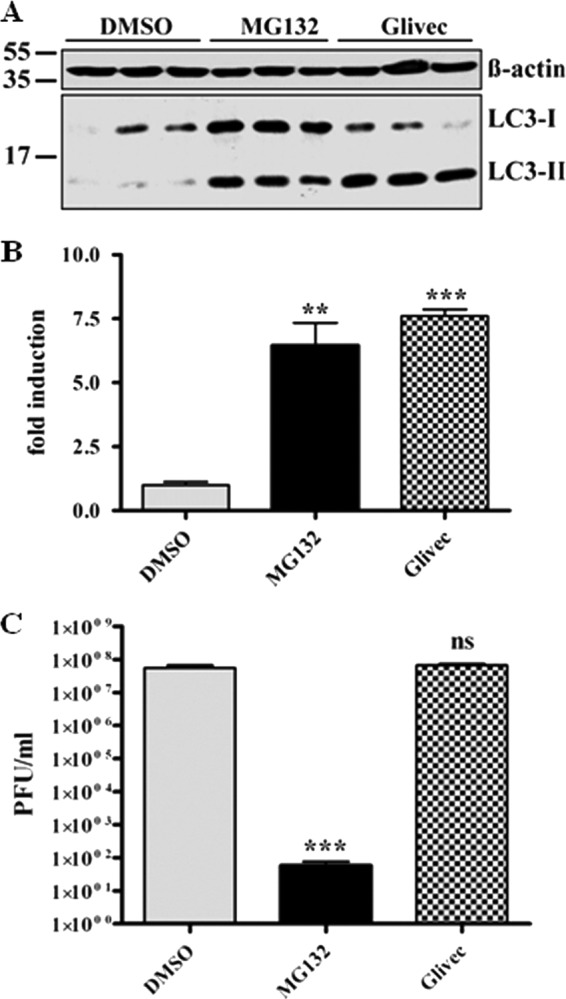
SARS-CoV replication is not affected by induction of autophagy in Vero E6 cells. (A) LC3-II signal in Vero E6 cells upon MG132 treatment. Vero E6 cells were treated with DMSO, 2.5 μM MG132, or 10 μM imatinib for 16 h. Equal amounts of total protein were subjected to SDS-PAGE, and LC3-I and -II signals were determined using an anti-LC3 antibody. Actin was used as a loading control (upper panel). (B) Fold induction of LC3-II upon treatment. Values represent the means ± SEM of one experiment performed in triplicate. (C) SARS-CoV replication upon MG132 and imatinib treatment. Vero E6 cells were pretreated with DMSO (mock), 2.5 μM MG132, or 10 μM imatinib for 1 h and were inoculated with SARS-CoV (MOI, 0.001) with FCS-free media containing the compounds. After removal of inocula, cells were further cultivated in growth medium containing FCS and compounds for 16 h before viral supernatants were collected. Viral titers were determined by plaque assay and are shown as PFU/ml. Values represent the means ± SEM of two independent experiments performed in duplicate.
To further verify that the MG132 effect is independent of autophagic pathways, autophagy-deficient mouse embryonic fibroblasts (MEFs) were employed (31). ATG5−/− MEFs are derived from ATG5 knockout mice and therefore do not express the Atg5 protein, which is part of the Atg5/Atg12 conjugation complex and is indispensable for formation of autophagolysosomes (39). Induction of autophagy by MG132 and imatinib in wild-type MEFs was confirmed in our experimental setup by immunoblotting (Fig. 4A and B). As expected, induction of autophagy either by MG132 or imatinib was not observed in ATG5−/− MEFs (Fig. 4A). As MEFs, being of murine origin, are not susceptible to SARS-CoV infection, we rendered them susceptible by transient transfection with huACE2, the functional SARS-CoV receptor. Effective transfection and expression was confirmed by immunoblot analysis 24 and 48 h after transfection (Fig. 4C). Ultimately, 24 h after transfection (or mock transfection, i.e., w/o ACE2), wild-type (wt) and ATG5−/− huACE2 MEFs were left untreated or were treated with DMSO (mock), MG132, or imatinib. After 1 h, cells were infected with SARS-CoV and incubated for 16 h with the different compounds before supernatants were collected and viral titers determined. As expected, wt huACE2 MEFs could be successfully infected with SARS-CoV (Fig. 4D, column 2). This was also the case for transfected Atg5−/− huACE2 MEFs, indicating that the autophagy gene ATG5 is not required for SARS-CoV replication (Fig. 4D, column 6). Of note, treatment with MG132 of ATG5−/− huACE2 MEFs resulted in a strong reduction (i.e., 3 orders of magnitude) of viral titers, comparable to those of cells lacking the viral receptor (Fig. 4D, compare column 6 to 7 and 5). As these cells are entirely deficient in induction of autophagy, this demonstrates that the MG132 effects in our scenario are not based on alterations of autophagy pathways.
Fig 4.

SARS-CoV replication does not require the autophagy gene ATG5, and replication inhibition by MG132 is independent of induction of autophagy. (A) LC3-I/II signal in wild-type (wt; right side) and ATG5−/− (left side) MEFs upon MG132 treatment. Cells were treated with DMSO, 2.5 μM MG132, or 10 μM imatinib for 16 h. Equal amounts of protein were subjected to SDS-PAGE, and LC3-I/II signals were determined by immunoblotting using an anti-LC3 antibody. Actin was used as a loading control (upper panel). (B) Fold induction of LC3-II in wt MEF upon treatment. Values represent the means ± SEM of one experiment performed in triplicate. (C) Transient expression of huACE2 in MEFs. Wild-type (left) and ATG5−/− (right) MEFs were transiently transfected with 4 μg of pcDNA3.1-ACE2. Equal protein amounts collected at 24 and 48 h posttransfection were subjected to immunoblot analysis using an anti-ACE2 antibody. Actin was used as a loading control (lower panel). (D) wt and ATG5−/− huACE2 MEFs were pretreated with DMSO (mock), 2.5 μM MG132, or 10 μM imatinib for 1 h and were inoculated with SARS-CoV (MOI, 0.2) in FCS-free medium containing compounds. After removal of inocula, treatment was continued in growth medium with FCS for 16 h before viral supernatants were collected. Viral titers were determined by plaque assay and are shown as PFU/ml. Values represent the means ± SEM of two independent experiments.
Taken together, our data convincingly show that the ATG5 gene is not required for SARS-CoV replication and that autophagy pathways are not involved in the mechanism by which MG132 blocks SARS-CoV replication.
Reduction of SARS-CoV replication by MG132 is caused by inhibition of m-calpain.
The preceding studies have shown that despite effective impairment of proteasomal activity of all used proteasomal inhibitors, only MG132 had a strong inhibitory effect on viral titer. Peptide aldehydes are known inhibitors of serine and cysteine proteases. Since MG132, a tripeptide aldehyde (benzyloxycarbonyl-leucyl-leucyl-leucinal, or ZLLLal), comprised an unparalleled capability of inhibiting SARS-CoV replication, we hypothesized that inhibition of another proteolytic enzyme is responsible for inhibition of viral titer production. Besides high potency to inhibit the proteasome, MG132 has been described to impair the cysteine protease m-calpain (64). Therefore, our next studies focused on enlightening possible functions of calpains during the SARS-CoV life cycle. To achieve this, Vero E6 cells were pretreated for 1 h with the calpain inhibitors MDL28170/calpain inhibitor III and SJA6017/calpain inhibitor VI, inoculated with SARS-CoV, and incubated for 16 h with continued treatment. Viral supernatants were collected and titer analysis was performed. Interestingly, we found that treatment of infected cells with 10 μM MDL28170 led to an even more pronounced decrease of viral particle production (i.e., >7 orders of magnitude) than did MG132 (Fig. 5A). Calpain inhibitor VI had no significant effect on viral replication. As CI VI primarily targets μ-calpain (18), whereas MDL28170 inhibits both μ- and m-calpain, we assume that SARS-CoV replication was strongly inhibited by impairment of m-calpain activity. This assumption is affirmed by the fact that MG132 also affects m-calpain (64).
Fig 5.
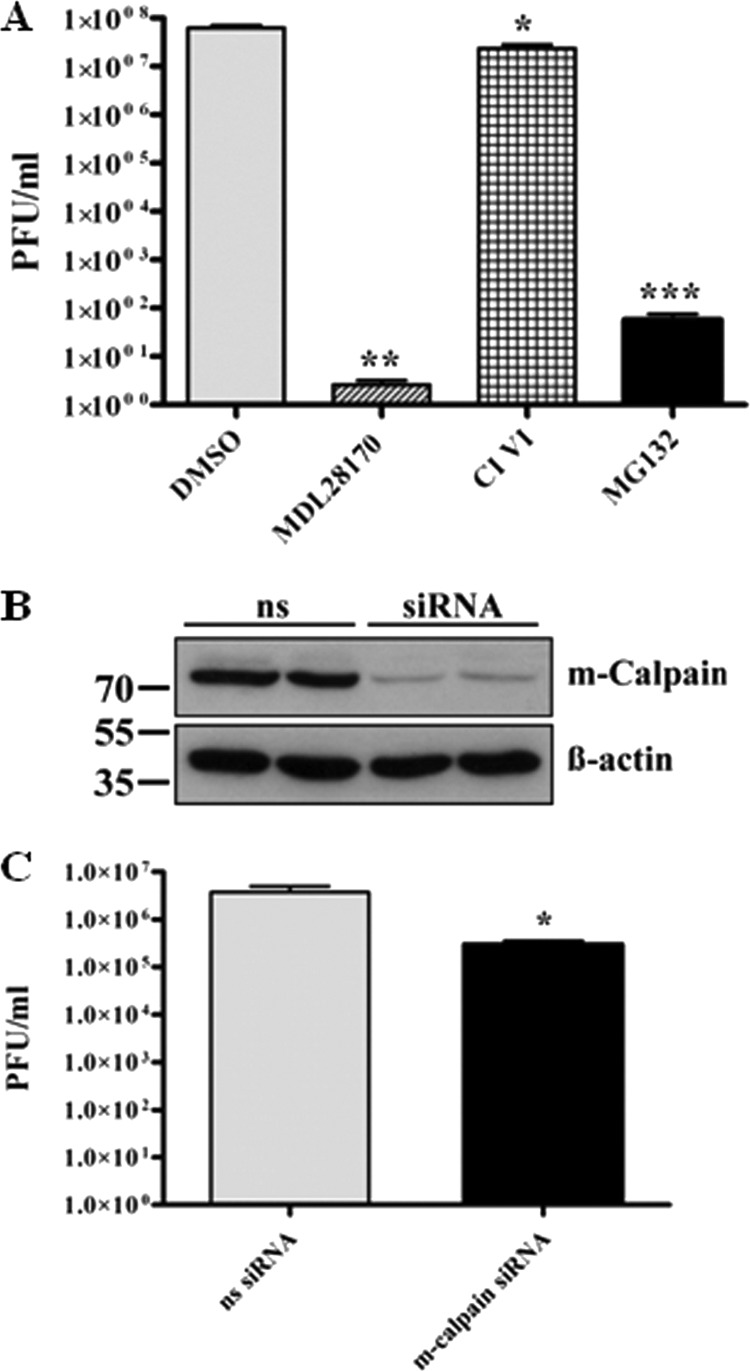
M-calpain activity is necessary for efficient SARS-CoV replication. (A) Vero E6 cells were pretreated with DMSO (mock), 2.5 μM MG132, 5 μM CI VI, or 10 μM MDL28170 for 1 h and then inoculated with SARS-CoV (MOI, 0.001) in FCS-free media containing the respective compounds. After removal of inocula, cells were incubated in growth medium supplemented with FCS and the respective compounds for 16 h, and then viral supernatants were collected. Viral titers were determined by plaque assay and are shown as PFU/ml. Values represent the means ± SEM of two independent experiments performed in duplicate. (B) Vero E6 cells in 25-cm2 flasks were transfected with nonsilencing (ns) siRNA or 350 pmol siRNA against m-calpain. Forty-eight h after transfection, cells were lysed and equal protein amounts were subjected to immunoblot analysis using an m-calpain (Calpain 2) antibody (upper panel). Actin was used as a loading control (lower panel). (C) Vero E6 cells were transfected as described for panel B and infected with SARS-CoV (MOI, 0.001) 48 h posttransfection. Inocula were removed after 1 h, and cells were incubated with normal growth medium for 16 h. Viral titers were determined as described for panel A.
To verify that inhibition of m-calpain specifically reduces SARS-CoV titer production, siRNA knockdown experiments were performed. Vero E6 cells were transiently transfected with siRNA targeting m-calpain or nonsilencing siRNA. Forty-eight hours posttransfection, reduction of m-calpain protein levels was confirmed by Western blot analysis (Fig. 5B). In parallel, transfected Vero E6 cells were infected with SARS-CoV as previously described. Sixteen h p.i., viral titers were determined by plaque assay. As shown in Fig. 5C, siRNA knockdown of the cysteine protease m-calpain reduced SARS-CoV titers more than 90%.
These data further support that MG132 does not impair viral particle production by affecting the ubiquitin-proteasome system but by inhibition of m-calpain.
DISCUSSION
In this study, we investigated the effect of several known proteasomal inhibitors on SARS-CoV replication. Surprisingly, only MG132 strongly inhibited SARS-CoV replication by affecting early steps during the viral life cycle. In sharp contrast to previous studies and our own control experiments with MHV, the inhibitory effect of MG132 on SARS-CoV seemed to be independent of proteasomal inhibition. Since a postfusion step was supposed to be affected, we investigated the impact of autophagy during the viral life cycle, demonstrating that an induction of autophagy did not affect SARS-CoV replication. Infection studies with wild-type and ATG5−/− huACE2 mouse embryonic fibroblasts showed that SARS-CoV replication does not require Atg5 or a functional autophagy system. In line with this, the antiviral effect of MG132 also was independent of induction of autophagy. Infection experiments with calpain inhibitors and knockdown experiments then confirmed that inhibition of m-calpain rather than inhibition of the ubiquitin-proteasome pathway is responsible for inhibition of SARS-CoV replication by MG132.
MG132 strongly inhibits early steps of SARS-CoV replication independent of proteasomal impairment.
The ubiquitin-proteasome system, with its degradative and regulatory responsibilities, is essential for cell function and is also relevant for replication of a broad range of viruses (14). Previous studies and our own control experiments have shown that the activity of a functional proteasome is important for the replication of murine hepatitis coronavirus (MHV) (46, 72). Here, several proteasome inhibitors, including Lac, had similar effects on viral replication (36, 46, 72), contrary to our finding that only MG132 can inhibit SARS-CoV replication. This indicates that effects of MG132 on SARS-CoV replication are independent of proteasomal inhibition and that UPS is differently involved in the replication of coronaviruses. Among all used proteasome inhibitors, MG132 showed maximal potency to reduce MHV replication in vitro (36, 72), and both MG132 and BZ reduced viral titers and inflammatory responses in MHV-1-infected A/J mice, leading to improved pathology and survival (36). In contrast to this, treatment with higher BZ concentrations of C57B1/6 mice infected with the MHV-A59 strain led to an increase of viral titers and even enhanced disease, which was discussed to be due to a reduced inflammatory response and protective immune response (45). Furthermore, certain strain differences (strain JHM versus A59) in the effects of proteasomal inhibition on MHV replication in vitro have been discussed (72). Therefore, it appears plausible that replication of the more distant SARS-CoV is independent of a functional proteasome. Raaben and colleagues recently reported that treatment with BZ reduced not only replication of recombinant MHV and FIPV but also GFP expression in Vero cells infected with a recombinant GFP-tagged SARS-CoV (46). Of note, effects on this recombinant SARS-CoV were less prominent than effects on MHV or FIPV, with a reduction of GFP expression to approximately 40% of the control level when 80 nM BZ was used. At similar BZ concentrations, we found a reduction of SARS-CoV titers of ∼0.5 log, which correlates with a 50% reduction of PFU/ml. However, in terms of viral load, this reduction is not significant. Therefore, the discrepancy between the former study (46) and our results might be explained by the different viruses used (SARS-CoV-GFP with deleted ORF7a/b [56] versus wt SARS-CoV), the lower bortezomib concentrations used in our study, a result of toxicity at higher concentrations, and different read-out systems. Although titers of SARS-CoV-GFP are similar to those of wt SARS-CoV in cell culture (56), it may replicate differently upon proteasome inhibition due to the deletion of ORF7a/b. In contrast, SARS-CoV strain Frankfurt-1 only has a short deletion (45 nucleotides) in ORF7b (57, 63).
In summary, we demonstrate here that MG132 exerts an unparalleled capability among tested proteasomal inhibitors in impairing early steps of SARS-CoV replication, pointing to a proteasome-independent mechanism.
MG132 does not inhibit SARS-CoV replication by induction of ER stress, the unfolded protein response, or autophagy.
Induction of ER stress, due to the accumulation of proteins destined for ER-associated degradation (ERAD), and activation of the unfolded protein response (UPR) are well-known effects of treatment with proteasomal inhibitors. However, the prominent effect of MG132 on SARS-CoV replication most likely is independent of ER stress induction and UPR, since all of the proteasomal inhibitors used caused ER stress and UPR induction at similar levels.
Interestingly, Grp78/BiP expression in mock-treated cells infected with SARS-CoV was not upregulated, although overexpression of several SARS-CoV proteins, e.g., the spike glycoprotein or the ORF3a and ORF6 proteins, was described to induce ER stress (5, 38, 71). Therefore, SARS-CoV might have evolved mechanisms to reduce ER stress or UPR during infection of Vero cells. However, an increase in Grp78 levels is only one marker of ER stress, and further studies are needed to elucidate the ability of SARS-CoV to prevent ER stress or to interfere with certain branches of UPR in Vero cells.
Autophagy is a process shared by all eukaryotic cells by which long-lived proteins, aggresomes, or whole organelles are engulfed by double membrane vesicles–the so-called autophagosomes–and degraded by their delivery to lysosomes (29, 39). Besides its participation in multiple cellular processes, autophagy also serves as an intrinsic immune defense against certain viruses (29) by facilitating antigen presentation, type I interferon production, or xenophagy (29), a process where cytoplasmic virions, viral components, or membrane-protected viral replication factories are disposed by autophagic engulfment and delivery to degrading lysosomes. Work by Lai's group had shown that upon treatment with MG132, MHV virions were retained in endosomes and possibly degraded by fusion of these endosomes with lysosomal vesicles (72). However, induction of autophagy with imatinib (13) did not affect SARS-CoV replication, and both wild-type and autophagy-deficient (ATG5−/−) huACE2 MEFs (31) were equally susceptible to SARS-CoV infection. This set of experiments also indicated that Atg5 expression, and probably also autophagy, are not required for SARS-CoV replication. This is in line with studies demonstrating that Atg5 and Atg7 proteins are not necessary for MHV replication (47, 73). On the other hand, a lack of autophagy is not beneficial for viral replication, as similar viral titers were produced in wt and Atg5−/− cells. Whereas components of the autophagic machinery like the nonlipidated microtubule-associated protein 1 light chain 3 (LC3-I) are essential for DMV biogenesis and thus for MHV replication (47), and possibly also for SARS-CoV replication, an intact autophagic pathway obviously is not of relevance for SARS-CoV replication.
MG132 inhibits SARS-CoV replication by impairing m-calpain activities.
As a tripeptide aldehyde (ZLLLal), MG132 was found to inhibit m-calpain (64), a cysteine protease. Calpains have been reported previously to play a role in SARS-CoV replication (1). Our data involving chemical inhibition of m-calpain and siRNA knockdown strongly suggest that MG132 blocks SARS-CoV replication, because it inhibits m-calpain rather than interfering with proteasomal activities.
Calpains are a class of ubiquitously expressed cysteine proteases whose functions in the cell are still not fully elucidated. Among other things, they are involved in the regulation of cytoskeleton-membrane interaction, cytoskeleton remodeling, and vesicular trafficking (3, 16, 17, 41). Whereas μ-calpain represents the cytosolic fraction of calpains, m-calpain is mostly associated with cellular membranes, vesicles, subcellular organelles, or lipid rafts (23, 40, 65). Picornaviridae utilize cellular membranes as a site for viral replication and for establishment of the replication complex (RC), as do Coronaviridae. Since formation of the echovirus 1 RC strongly depends on m-calpain activity (65), formation of the DMVs by SARS-CoV might also depend on m-calpain activity. Besides effects on the cellular m-calpain, MG132 and MDL28170 might impair the activity of viral proteases 3CLpro/Mpro (main protease) and PLpro (papain-like protease), which also belong to the family of cysteine proteases. Likewise, the adenovirus protease is inhibited by the calpain inhibitor MDL28170 (7). Both MG132 and MDL28170 also have been demonstrated to inhibit cathepsin L-mediated processing of viral glycoproteins, including SARS-CoV S (42, 55), thus SARS-CoV S-mediated viral entry could be prevented (55). Although inhibition of cathepsin L by the chemical inhibitors, in addition to inhibition of m-calpain, might increase their significant effect on SARS-CoV replication, results obtained upon m-calpain knockdown indicate an important role of m-calpain for SARS-CoV replication.
In summary, our data demonstrate that SARS-CoV replication does not fundamentally rely on the ubiquitin-proteosome pathway, conferring an exceptional status to SARS-CoV among other coronaviruses. Inhibitory effects of MG132 during early steps of SARS-CoV replication are most likely due to inhibition of cellular m-calpain. Further studies are needed to elucidate whether inhibition of the cellular m-calpain or of a possible m-calpain-like activity of a viral enzyme is responsible alone or in combination for the massive loss of SARS-CoV infectious titers. Given that binding specificities can be optimized and bioavailability improved, calpain inhibitors represent a promising tool to fight SARS-CoV infection.
ACKNOWLEDGMENTS
We kindly thank Noboru Mizushima (Tokyo Medical and Dental University, Tokyo, Japan) for providing wild-type and ATG5−/− mouse embryonic fibroblast cell lines and Christian Drosten (Medical Centre Bonn, University of Bonn, Germany) for providing infectious SARS-CoV. We are grateful to Michael Farzan (Harvard Medical School, Cambridge, MA) for providing the huACE2-expressing pcDNA3.1 construct and to Xuming Zhang (University of Arkansas for Medical Science) for providing MHV. We especially thank Max Nunziante for proofreading.
Footnotes
Published ahead of print 11 July 2012
REFERENCES
- 1. Barnard DL, et al. 2004. Inhibition of severe acute respiratory syndrome-associated coronavirus (SARSCoV) by calpain inhibitors and beta-d-N4-hydroxycytidine. Antivir. Chem. Chemother. 15:15–22 [DOI] [PubMed] [Google Scholar]
- 2. Barretto N, et al. 2005. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 79:15189–15198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beckerle MC, Burridge K, Demartino GN, Croall DE. 1987. Colocalization of calcium-dependent protease-II and one of its substrates at sites of cell-adhesion. Cell 51:569–577 [DOI] [PubMed] [Google Scholar]
- 4. Bush KT, Goldberg AL, Nigam SK. 1997. Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J. Biol. Chem. 272:9086–9092 [DOI] [PubMed] [Google Scholar]
- 5. Chan CP, et al. 2006. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 80:9279–9287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ciechanover A. 1994. The ubiquitin-mediated proteolytic pathway–mechanisms of action and cellular physiology. Biol. Chem. Hoppe-Seyler 375:565–581 [DOI] [PubMed] [Google Scholar]
- 7. Cotten M, Weber JM. 1995. The adenovirus protease is required for virus entry into host-cells. Virology 213:494–502 [DOI] [PubMed] [Google Scholar]
- 8. Delboy MG, Roller DG, Nicola AV. 2008. Cellular proteasome activity facilitates herpes simplex virus entry at a postpenetration step. J. Virol. 82:3381–3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Diemer C, et al. 2008. Cell type-specific cleavage of nucleocapsid protein by effector caspases during SARS coronavirus infection. J. Mol. Biol. 376:23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ding WX, et al. 2007. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 171:513–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Drexler JF, et al. 2010. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J. Virol. 84:11336–11349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Drosten C, et al. 2003. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 348:1967–1976 [DOI] [PubMed] [Google Scholar]
- 13. Ertmer A, et al. 2007. The anticancer drug imatinib induces cellular autophagy. Leukemia 21:936–942 [DOI] [PubMed] [Google Scholar]
- 14. Gao G, Luo HL. 2006. The ubiquitin-proteasome pathway in viral infections. Can. J. Physiol. Pharmacol. 84:5–14 [DOI] [PubMed] [Google Scholar]
- 15. Gilch S, et al. 2001. Intracellular re-routing of prion protein prevents propagation of PrPSc and delays onset of prion disease. EMBO J. 20:3957–3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harris AS, Morrow JS. 1990. Calmodulin and calcium-dependent protease-I coordinately regulate the interaction of fodrin with actin. Proc. Natl. Acad. Sci. U. S. A. 87:3009–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huttenlocher A, et al. 1997. Regulation of cell migration by the calcium-dependent protease calpain. J. Biol. Chem. 272:32719–32722 [DOI] [PubMed] [Google Scholar]
- 18. Inoue J, et al. 2003. Structure-activity relationship study and drug profile of N-(4-fluorophenylsulfonyl)-l-valyl-l-leucinal (SJA6017) as a potent calpain inhibitor. J. Med. Chem. 46:868–871 [DOI] [PubMed] [Google Scholar]
- 19. Inoue Y, et al. 2007. Clathrin-dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ACE2 with the cytoplasmic tail deleted. J. Virol. 81:8722–8729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jackwood MW. 2006. The relationship of severe acute respiratory syndrome coronavirus with avian and other coronaviruses. Avian Dis. 50:315–320 [DOI] [PubMed] [Google Scholar]
- 21. Kabeya Y, et al. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khor R, McElroy LJ, Whittaker GR. 2003. The ubiquitin-vacuolar protein sorting system is selectively required during entry of influenza virus into host cells. Traffic 4:857–868 [DOI] [PubMed] [Google Scholar]
- 23. Kifor O, Kifor I, Moore FD, Butters RR, Brown EM. 2003. M-calpain colocalizes with the calcium-sensing receptor (CaR) in caveolae in parathyroid cells and participates in degradation of the CaR. J. Biol. Chem. 278:31167–31176 [DOI] [PubMed] [Google Scholar]
- 24. Kikkert M, et al. 2001. Ubiquitination is essential for human cytomegalovirus US11-mediated dislocation of MHC class I molecules from the endoplasmic reticulum to the cytosol. Biochem. J. 358:369–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Klumperman J, et al. 1994. Coronavirus M-proteins accumulate in the Golgi complex beyond the site of virion budding. J. Virol. 68:6523–6534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kostova Z, Wolf DH. 2003. For whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connection. EMBO J. 22:2309–2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krokhin O, et al. 2003. Mass spectrometric characterization of proteins from the SARS virus: a preliminary report. Mol. Cell. Proteomics 2:346–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ksiazek TG, et al. 2003. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348:1953–1966 [DOI] [PubMed] [Google Scholar]
- 29. Kudchodkar SB, Levine B. 2009. Viruses and autophagy. Rev. Med. Virol. 19:359–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuiken T, et al. 2003. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 362:263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kuma A, et al. 2004. The role of autophagy during the early neonatal starvation period. Nature 432:1032–1036 [DOI] [PubMed] [Google Scholar]
- 32. Li JZ, Lee AS. 2006. Stress induction of GRP78/BiP and its role in cancer. Curr. Mol. Med. 6:45–54 [DOI] [PubMed] [Google Scholar]
- 33. Li W, et al. 2003. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li W, et al. 2005. Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676–679 [DOI] [PubMed] [Google Scholar]
- 35. Lindner HA, et al. 2005. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 79:15199–15208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ma XZ, et al. 2010. Proteasome inhibition in vivo promotes survival in a lethal murine model of severe acute respiratory syndrome. J. Virol. 84:12419–12428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marra MA, et al. 2003. The genome sequence of the SARS-associated coronavirus. Science 300:1399–1404 [DOI] [PubMed] [Google Scholar]
- 38. Minakshi R, et al. 2009. The SARS coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS One 4:e8342 doi:10.1371/journal.pone.0008342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mizushima N, Ohsumi Y, Yoshimori T. 2002. Autophagosome formation in mammalian cells. Cell Structure Function 27:421–429 [DOI] [PubMed] [Google Scholar]
- 40. Morford LA, et al. 2002. Calpain II colocalizes with detergent-insoluble rafts on human and Jurkat T-cells. Biochem. Biophys. Res. Commun. 295:540–546 [DOI] [PubMed] [Google Scholar]
- 41. Nixon RA. 2003. The calpains in aging and aging-related diseases. Ageing Res. Rev. 2:407–418 [DOI] [PubMed] [Google Scholar]
- 42. Pager CT, Dutch RE. 2005. Cathepsin L is involved in proteolytic processing of the Hendra virus fusion protein. J. Virol. 79:12714–12720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peiris JS, et al. 2003. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 7:439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Raaben M, Grinwis GCM, Rottier PJM, de Haan CAM. 2010. The proteasome inhibitor velcade enhances rather than reduces disease in mouse hepatitis coronavirus-infected mice. J. Virol. 84:7880–7885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Raaben M, et al. 2010. The ubiquitin-proteasome system plays an important role during various stages of the coronavirus infection cycle. J. Virol. 84:7869–7879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reggiori F, et al. 2010. Coronaviruses hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host. Microbe 7:500–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Richardson PG, Mitsiades C, Hideshima T, Anderson KC. 2006. Bortezomib: proteasome inhibition as an effective anticancer therapy. Annu. Rev. Med. 57:33–47 [DOI] [PubMed] [Google Scholar]
- 49. Satheshkumar PS, Anton LC, Sanz P, Moss B. 2009. Inhibition of the ubiquitin-proteasome system prevents vaccinia virus DNA replication and expression of intermediate and late genes. J. Virol. 83:2469–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Satija N, Lal SK. 2007. The molecular biology of SARS coronavirus. Ann. N. Y. Acad. Sci. 1102:26–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schubert U, et al. 1998. CD4 glycoprotein degradation induced by human immunodeficiency virus type 1 Vpu protein requires the function of proteasomes and the ubiquitin-conjugating pathway. J. Virol. 72:2280–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schubert U, et al. 2000. Proteasome inhibition interferes with Gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc. Natl. Acad. Sci. U. S. A. 97:13057–13062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shamu CE, Flierman D, Ploegh HL, Rapoport TA, Chau V. 2001. Polyubiquitination is required for US11-dependent movement of MHC class I heavy chain from endoplasmic reticulum into cytosol. Mol. Biol. Cell 12:2546–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Si XN, et al. 2005. Pyrrolidine dithiocarbamate reduces coxsackievirus B3 replication through inhibition of the ubiquitin-proteasome pathway. J. Virol. 79:8014–8023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Simmons G, et al. 2005. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U. S. A. 102:11876–11881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sims AC, Burkett SE, Yount B, Pickles RJ. 2008. SARS-CoV replication and pathogenesis in an in vitro model of the human conducting airway epithelium. Virus Res. 133:33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Snijder EJ, et al. 2003. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 331:991–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Snijder EJ, et al. 2006. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 80:5927–5940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stertz S, et al. 2007. The intracellular sites of early replication and budding of SARS-coronavirus. Virology 361:304–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Surjit M, Lal SK. 2008. The SARS-CoV nucleocapsid protein: a protein with multifarious activities. Infect. Genet. Evol. 8:397–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tan YJ, Lim SG, Hong W. 2006. Understanding the accessory viral proteins unique to the severe acute respiratory syndrome (SARS) coronavirus. Antiviral Res. 72:78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Teale A, et al. 2009. Orthopoxviruses require a functional ubiquitin-proteasome system for productive replication. J. Virol. 83:2099–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Thiel V, et al. 2003. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 84:2305–2315 [DOI] [PubMed] [Google Scholar]
- 64. Tsubuki S, Saito Y, Tomioka M, Ito H, Kawashima S. 1996. Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. J. Biochem. 119:572–576 [DOI] [PubMed] [Google Scholar]
- 65. Upla P, et al. 2008. Calpain 1 and 2 are required for RNA replication of echovirus 1. J. Virol. 82:1581–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Viswanathan K, Fruh K, DeFilippis V. 2010. Viral hijacking of the host ubiquitin system to evade interferon responses. Curr. Opin. Microbiol. 13:517–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang S, et al. 2008. Endocytosis of the receptor-binding domain of SARS-CoV spike protein together with virus receptor ACE2. Virus Res. 136:8–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Widjaja I, et al. 2010. Inhibition of the ubiquitin-proteasome system affects influenza A virus infection at a postfusion step. J. Virol. 84:9625–9631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wu WKK, et al. 2008. Induction of autophagy by proteasome inhibitor is associated with proliferative arrest in colon cancer cells. Biochem. Biophys. Res. Commun. 374:258–263 [DOI] [PubMed] [Google Scholar]
- 70. Yang ZY, et al. 2004. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J. Virol. 78:5642–5650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ye Z, Wong CK, Li P, Xie Y. 2008. A SARS-CoV protein, ORF-6, induces caspase-3 mediated, ER stress and JNK-dependent apoptosis. Biochim. Biophys. Acta 1780:1383–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yu GY, Lai MAC. 2005. The ubiquitin-proteasome system facilitates the transfer of murine coronavirus from endosome to cytoplasm during virus entry. J. Virol. 79:644–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhao Z, et al. 2007. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 3:581–585 [DOI] [PubMed] [Google Scholar]