

Abstract
Clinical and epidemiological studies have implicated chronic infections in the development of atherosclerosis. It has been proposed that common mechanisms of signaling via TLRs link stimulation by multiple pathogens to atherosclerosis. However, how pathogen-specific stimulation of TLR4 contributes to atherosclerosis progression remains poorly understood. In this study, atherosclerosis-prone apolipoprotein-E null (ApoE−/−) and TLR4-deficient (ApoE−/−TLR4−/−) mice were orally infected with the periodontal pathogen Porphyromonas gingivalis. ApoE−/−TLR4−/− mice were markedly more susceptible to atherosclerosis after oral infection with P. gingivalis. Using live animal imaging, we demonstrate that enhanced lesion progression occurs progressively and was increasingly evident with advancing age. Immunohistochemical analysis of lesions from ApoE−/−TLR4−/− mice revealed an increased inflammatory cell infiltrate composed primarily of macrophages and IL-17 effector T cells (Th17), a subset linked with chronic inflammation. Furthermore, enhanced atherosclerosis in TLR4-deficient mice was associated with impaired development of Th1 immunity and regulatory T cell infiltration. In vitro studies suggest that the mechanism of TLR4-mediated protective immunity may be orchestrated by dendritic cell IL-12 and IL-10, which are prototypic Th1 and regulatory T cell polarizing cytokines. We demonstrate an atheroprotective role for TLR4 in response to infection with the oral pathogen P. gingivalis. Our results point to a role for pathogen-specific TLR signaling in chronic inflammation and atherosclerosis.
Introduction
The identification of atherosclerosis as a chronic inflammatory disease has emphasized the fundamental role of the immune system in disease pathogenesis (1). Detection of endogenous and microbial ligands by immune competent cells occurs via germline encoded pattern recognition receptors including the innate immune TLRs (2). Engagement via TLRs initiates acute inflammatory responses that are critical in host defense (3). Resident cells in human atherosclerotic plaque express TLRs (4). Animal studies using hyperlipidemic mice have shown that TLR2, TLR4, and the downstream signaling molecule MyD88 play an important role in diet-induced atherosclerosis (5–9). Reduced atherosclerotic development has been observed in TLR4-deficient ApoE−/− mice on high-fat Western diet (5, 6, 8). We have previously shown that TLR2 also plays an important role in atherosclerotic progression independent of dietary lipids in hyperlipidemic ApoE−/− mice (7).
A subset of Gram-negative mucosal pathogens including Chlamydia pneumoniae and Porphyromonas gingivalis induce chronic and systemic inflammatory responses associated with atherosclerosis through TLR signaling (10–12). P. gingivalis induces a local inflammatory response that results in oral bone destruction manifested as periodontal disease, an inflammatory disease that affects 100 million people in the United States (13). In addition to chronic inflammation at the initial site of infection, mounting evidence supports a role for P. gingivalis-mediated periodontal disease as a risk factor for systemic diseases, including atherosclerotic cardiovascular disease as well as diabetes and preterm birth (10, 11, 14–18). P. gingivalis has been detected in human atherosclerotic plaque (19, 20), and animal models of P. gingivalis infection have validated human studies (21–26).
P. gingivalis induces proinflammatory responses primarily through fimbriae-mediated signaling via TLR2 that is MyD88-dependent (27). In support of this, we previously demonstrated that a P. gingivalis fimbriae mutant, fimA, failed to accelerate atherosclerosis in ApoE−/− mice (22). Furthermore, we established that TLR2 signaling contributes in part to P. gingivalis-induced atherosclerosis in mice on a normal chow diet (28). In addition to signaling for proinflammatory responses via TLR2, P. gingivalis has developed mechanisms to evade detection and eradication by the immune system (29, 30). One such mechanism is modification of its lipid A, the biological core of bacterial LPS, universally recognized by the TLR4–MD2 complex (31). In response to environmental stimuli (32) and availability of the essential nutrient heme (33), P. gingivalis uses enzymes to modify the acylation and phosphorylation of its lipid A, resulting in differential recognition by the TLR4 complex (29, 30). In the current study, we demonstrate the unique ability of P. gingivalis to evade TLR4 signaling while inducing TLR2-dependent proinflammatory responses reveals a protective role for TLR4 in chronic inflammatory atherosclerosis.
Materials and Methods
Mice
Male ApoE−/− and C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME). TLR4−/− mice on C57BL/6 background were provided by S. Akira (Osaka University). ApoE−/−TLR4−/− mice were generated in our laboratory. Mouse genotypes were confirmed by PCR, and experimental mice were age-matched. Mice were maintained under specific pathogen-free conditions and cared for in accordance with the Boston University Institutional Animal Care and Use Committee.
Bacteria
P. gingivalis strain 381 was grown anaerobically on blood agar plates (Becton Dickinson) and used to seed-inoculate brain heart infusion broth (pH 7.4; Becton Dickinson) supplemented with yeast extract (Becton Dickinson), hemin (10 μg/ml; Sigma), and menadione (1 μg/ml). CFUs were standardized at an OD at 660 nm of 1 (equivalent to 1 × 109 CFU/ml) by spectrometry (ThermoSpectronic Genesys20). LPS from P. gingivalis 381 was isolated using a modified Tri-Reagent protocol (29).
Oral infection
Three independent experiments were performed with ApoE−/− (total n = 40) and ApoE−/−TLR4−/− (total n = 30) mice, and data were pooled. Mice were fed a normal chow diet (Global 2018; Harlan Teklad, Madison, WI). Six-week-old male mice were given antibiotics (Sulfatrim; Hi-Tech Pharmacal) ad libitum in the drinking water for 10 d, followed by a 2-d antibiotic-free period. One hundred microliters of P. gingivalis 381 (1 × 109 CFU) suspended in vehicle (2% carboxymethylcellulose in PBS) was topically applied to the buccal surface of the maxillary gingiva five times a week for 3 wk (34). Control mice received 100 μl of vehicle. Topical application of P. gingivalis to the buccal surface of the maxillary gingiva five times a week for 3 wk induces alveolar bone loss in ApoE−/−mice (28). Mice were euthanized 13 wk after the final oral challenge (24 wk of age). This time point is consistent with the time frame used in our prior studies (7, 28, 35).
Magnetic resonance angiography
Magnetic resonance angiography (MRA) of the innominate artery was performed with a vertical-bore Bruker 11.7 T Avance spectrometer (Bruker; Billerica, MA) as described (35). Mice were anesthetized with 0.5–2% inhaled isoflurane, placed into a 30-mm vertical probe (Micro 2.5) maintained at 23°C. Respiration was monitored using a monitoring and gating system (SA Instruments, Wahkesha, WI). The ungated 3D gradient echo MRA was acquired with the following parameters: slab thickness = 1.5 cm; flip angle = 45°; repetition time = 20 ms; echo time = 2.2 ms; field of view = 1.5 × 1.5 × 1.5 cm; matrix = 128 × 128 × 128, in-plane; number of average = 4. Total scan time was ∼25 min. Image reconstruction and analysis were performed using Paravision. The 3D reconstruction of the MRA images was achieved by maximum intensity projection. The cross sections were chosen at 0.3- to 0.5-mm distance below the subclavian bifurcation. Lumen area was manually defined and calculated with ImageJ (National Institutes of Health) by two independent observers. Measurement reproducibility had an interclass correlation coefficient of 0.92.
Atherosclerotic plaque assessment
Aortas were harvested and stained with Sudan IV as described (22). Digital micrographs were taken, and total area of atherosclerotic plaque was determined using IPLabs (Scanalytics) by a blinded observer.
Immunohistochemistry
Mice were euthanized (n = 4/group), perfused with 4% paraformaldehyde, and aortic arch with heart tissue was embedded in OCT freezing compound. Five-micrometer serial cryosections were collected every 50 μm in the innominate artery and aortic sinus. In the innominate artery, cryosections were obtained from the region corresponding to the greatest plaque size as revealed by MRA, ∼0.3 mm below the bifurcation of the innominate and subclavian arteries as described (35). Immunohistochemistry was performed using rat anti-mouse F4/80 (no. MCA497R; Serotec, Oxford, U.K.), rat anti-mouse CD4 (BD Biosciences no. 550278 and Caltag Laboratories), rat anti-mouse CD8 (no. 550281; BD Biosciences), rat anti-mouse TLR2 (no. 13-9021-80; eBioscience), anti-mouse Foxp3 (clone MF333F; Enzo), rat anti-mouse IL-17 (no. MAB2276; R&D Systems), or isotype controls (no. MCA1125; Serotec). Biotinylated anti-rat (mouse absorbed) IgG was used as secondary Ab (Vector Laboratories, Burlingame, CA). Digital micrographs were captured. Nuclei were counterstained with hematoxylin, and positive cells were enumerated by microscopy. F4/80+, CD8+ T cells, Foxp3+ T cells, and IL-17+ T cells were counted from three sections from each of four mice/group. For the quantitation of positive staining area, images were acquired using an Olympus BX41 microscope at ×100 magnification, and automated color thresholding was performed using ImageJ.
Flow cytometry
Anti-mouse Abs included CD3 (no. 553062; BD Biosciences), CD4 (no. 553049; BD Biosciences), CD8 (no. 553034; BD Biosciences), Ly6G, CD45 (no. 550994; BD Biosciences) and isotype controls (BD Pharmingen) and IFN-γ (no. 48-7311; eBioscience), IL-17A (no. 51-7177; eBioscience), F4/80 (no. 12-4801; eBioscience), and TLR2 (no. 12-9022; eBioscience). Intracellular cytokine staining was performed using a mouse kit (no. 559311; BD Pharmingen). Samples were acquired on a BD LSR II flow cytometer (Becton Dickinson), and data were analyzed using FlowJo software (Tree Star).
Cell culture
For isolation of bone marrow-derived dendritic cells (DCs), bone marrow cells were cultured in RPMI 1640 containing 10% FBS, 1× nonessential amino acids (MP Biomedicals), 50 μM 2-mercaptoethanol (Life Technologies), 100 μg/ml streptomycin/100 IU penicillin (Cellgro), and 20 ng/ml recombinant mouse GM-CSF (Peprotech) for 11 d. DCs were greater than 95% positive for CD11c. Nonadherent DCs were collected and replated in 24-well dishes at 2 × 105 cells/well in complete media without antibiotics before addition of P. gingivalis at multiplicity of infection of 25 and 50. After 24 h, culture supernatants were collected, and samples were clarified by centrifugation and stored at −80°C for ELISA.
ELISA
Concentrations of IL-6, IL-12p40, and IL-10 in cell culture supernatants were determined by ELISA (BD OptEIA). Plasma was collected from a subset of experimental mice 16 wks postinfection and assayed by ELISA for P. gingivalis-specific Ab isotypes IgG1, IgG2b, IgG2c, and IgG3 as described (36) as follows. Bacteria were washed three times in PBS and fixed overnight at 4°C in 4% paraformaldehyde. Fixed bacteria were washed five times in PBS and protein concentration estimated by bicinchoninic acid protein assay. Immulon 4HXB plates were coated overnight at 4°C with 10 μg/ml P. gingivalis suspension in PBS containing 0.05% sodium azide. Serial dilutions of mouse serum were plated, and determination of IgG isotypes was conducted using the C57BL/6 Clonotyping Kit (SouthernBiotech). Quantitation of IgG was determined using a standard curve. ELISAs were developed using 4-methylumbelliferyl phosphate and read on a spectrofluorometer (BioTek Synergy HT).
Splenocyte restimulation assay
Splenocytes (2 × 106 per milliliter) were collected and stimulated with P. gingivalis soluble Ags (10 μg/ml) in the presence of 1 μg/ml anti-mouse CD28 (eBioscience) for 4 h and 10 μg/ml brefeldin A (eBioscience). Cells were harvested and stained with anti-mouse Abs (BD Pharmingen): CD3, CD4, and CD8. Intracellular cytokine staining for IL-17A (eBioscience) and IFN-γ (eBioscience) was performed using BD Cytofix/Cytoperm kit.
Statistics
Normality of data was determined by visually inspecting for bell-shaped curves. A Mann–Whitney U test was performed to compare two independent samples with Prism 5 software (GraphPad Software, San Diego, CA) with an α equal to 0.05 considered significant. Two-way ANOVA was performed for analysis of percentage plaque between genotypes and infections. A p value < 0.05 was considered significant.
Results
TLR4 deficiency confers enhanced susceptibility to chronic and progressive atherosclerosis after infection with P. gingivalis
ApoE−/− and ApoE−/−TLR4−/− mice were orally infected with 109 CFU P. gingivalis strain 381. The predominant lipid A species expressed by P. gingivalis 381 grown under standard laboratory conditions in the presence of excess heme was tetra-acylated nonphosphorylated (m/z 1380), which is TLR4 inert and immunologically silent (data not shown) (32). The minor lipid A species produced by strain 381 was penta-acylated monophosphorylated (m/z 1690) and has been demonstrated to act as both a weak TLR4 agonist and antagonist (29).
Progression of atherosclerosis in the innominate artery of individual mice was examined in vivo by MRA. The innominate artery exhibits a high degree of lesion progression and expresses features of human disease including vessel narrowing, perivascular inflammation, and plaque disruption (35). The luminal area of the innominate artery of P. gingivalis-infected ApoE−/− and ApoE−/−TLR4−/− mice decreased between baseline and 12 wk compared with uninfected ApoE−/− and ApoE−/−TLR4−/− mice (Fig. 1A) illustrating vessel narrowing and disease progression in infected mice. In P. gingivalis-infected ApoE−/− mice, the luminal area remained unchanged between 12 and 16 wk. However, P. gingivalis-infected ApoE−/−TLR4−/− mice exhibited a progressive decline in luminal area at 0, 12, and 16 wk after the first oral infection, indicative of progressive atherosclerosis. The luminal area was significantly smaller in ApoE−/−TLR4−/− mice compared with ApoE−/− mice at 16 wk (p = 0.03).
FIGURE 1.

TLR4 deficiency confers enhanced susceptibility to atherosclerosis in the innominate artery after infection with P. gingivalis. Innominate arteries were imaged by MRA at baseline (week 0) and at 12 and 16 wk after first oral infection. (A) The temporal change in luminal area (mm2) was calculated for individual mice (n = 5/group). Inset, Representative MRA image indicating the innominate artery (yellow box), where measurements were taken. Uninfected ApoE−/− (blue); P. gingivalis-infected ApoE−/− (red); uninfected ApoE−/−TLR4−/− (green); P. gingivalis-infected ApoE−/−TLR4−/− (purple). (B) Representative hematoxylin staining from each group in innominate artery with F4/80 staining (macrophages stain brown). Scale bar, 20 μm. (C) Visualization of intima, media, and adventitia of representative images. Areas indicated in (B) (blue box). Scale bar, 5 μm. (D) Plaque area within the innominate artery measured from histological images using IPLab software (Becton Dickinson) (n = 5/group). Black bar, Uninfected; gray bar, P. gingivalis infected. **p < 0.01, ***p < 0.001.
Assessed by microscopy in postmortem sections, the innominate artery of uninfected ApoE−/− and ApoE−/−TLR4−/− mice appeared as tightly packed layers of smooth muscle cells with uniform distribution about the circumference of the artery with no apparent lipids and inflammatory cells (Fig. 1B: ApoE−/−, upper left; ApoE−/−TLR4−/−, upper right). Brown staining indicates the presence of macrophages. Plaque formation in the innominate artery of P. gingivalis-infected ApoE−/− mice was modest and superficial, appearing as fatty streaks at the intimal surface (Fig. 1B, lower left). In P. gingivalis-infected ApoE−/−TLR4−/− mice, we observed a significant increase in arterial plaque, which accumulated within subendothelial layers and coincided with the infiltration of inflammatory cells, including macrophages (Fig. 1B, lower right). In contrast to fatty streaks in infected ApoE−/− mice, plaques in infected ApoE−/−TLR4−/− mice protruded into the arterial lumen. Higher resolution reveals the structure of the intima, media, and adventitia in the innominate arteries of each group (Fig. 1C). Corresponding sections from MRA analyses revealed an increase in plaque area within the innominate artery of infected ApoE−/−TLR4−/− mice compared with infected ApoE−/− mice (Fig. 1D, bar graph). No significant differences in plaque area between uninfected ApoE−/− and ApoE−/−TLR4−/− mice were observed.
In the absence of infection, no differences in en face total aortic lesion area, assessed by lipid staining, were observed between uninfected ApoE−/− and ApoE−/−TLR4−/− mice (Fig. 2). Consistent with our previous studies (22), infected ApoE−/− mice developed significantly more plaque than uninfected ApoE−/− controls. Aortas from infected ApoE−/−TLR4−/− mice also demonstrated significantly more plaque than their uninfected, genotype-matched ApoE−/−TLR4−/− controls; however, plaque area was significantly greater in infected ApoE−/−TLR4−/− mice compared with infected ApoE−/− mice. Whereas the increase in lesion area in infected ApoE−/− mice largely localized to the atherosclerosis-prone regions in the aortic arch, lesions in infected ApoE−/−TLR4−/− mice occurred in the proximal as well as the distal aorta.
FIGURE 2.

TLR4 deficiency confers enhanced susceptibility to atherosclerosis in the aorta after infection with P. gingivalis. (A) Sudan IV staining of aorta en face lesions 16 wk after first infection with P. gingivalis. (B) Quantification of lipid content within the total aorta of uninfected (white bars) and P. gingivalis-infected mice (gray bars) (n = 10–13/group). Percentage of aorta occupied by lipids was calculated using IPLab software (Becton Dickinson). *p < 0.05, **p < 0.01, ***p < 0.001.
TLR4 deficiency is associated with increased macrophage infiltration and expression of TLR2 in aortic lesions from P. gingivalis-infected mice
The increased atherosclerotic plaque observed in infected ApoE−/−TLR4−/− mice was accompanied by a significantly increased accumulation of macrophages within the aortic sinus, whereas macrophage accumulation was not significantly increased in infected ApoE−/− mice (Fig. 3A, 3C, left). In agreement with previous findings (22), P. gingivalis infection resulted in increased expression of TLR2 within the aortic sinus of infected ApoE−/− mice, as well as in ApoE−/−TLR4−/− mice, in areas where macrophages were found (Fig. 3B, 3C, right). TLR2 expression was also significantly higher in infected ApoE−/−TLR4−/− mice compared with ApoE−/− mice (Fig. 3C, right).
FIGURE 3.

TLR4 deficiency is associated with increased macrophage influx and TLR2 expression in atherosclerotic plaques after infection with P. gingivalis. Aortic sinus sections from uninfected and P. gingivalis-infected ApoE−/− and ApoE−/−TLR4−/− mice were stained for F4/80+ and TLR2+ cells with hematoxylin counterstaining. Representative aortic sinus sections stained for (A) F4/80 and (B) TLR2. Scale bar, 100 μm. (C) Quantification of positive staining area using ImageJ software. White bar, uninfected; gray bar, P. gingivalis infected. Three sections from n = 4/group were analyzed. Data are mean ± SD positive staining area/slice. Intragenotype comparisons were calculated by Mann–Whitney U test: *p < 0.05, **p < 0.01, ***p < 0.001. Intergenotype comparisons were calculated by two-way ANOVA: p = 0.0001, p = 0.0095.
Greater plaque area and infiltration of macrophages into plaque in ApoE−/−TLR4−/− mice cannot be attributed to differences in plasma cholesterol or triglycerides, as these were similar among all groups (cholesterol, mean ± SE: uninfected ApoE−/−, 476 ± 22; infected ApoE−/−, 449 ± 24; uninfected ApoE−/−TLR4−/−, 500 ± 36; infected ApoE−/−TLR4−/−, 512 ± 24 mg/dl; triglycerides, mean ± SE: uninfected ApoE−/−, 237 ± 21; infected ApoE−/−, 245 ± 18; uninfected ApoE−/−TLR4−/−, 225 ± 24; infected ApoE−/−TLR4−/−, 206 ± 22 mg/dl).
TLR4 deficiency promotes Th17/regulatory T cell imbalance in atherosclerotic lesions after infection with P. gingivalis
In infected ApoE−/− mice, we observed no increase in CD8+ T cells, CD4+ T cells, or IL-17+ cells in the innominate artery compared with uninfected ApoE−/− mice (Fig. 4A, 4C). Accumulation of CD4+ and CD8+ cells within the innominate artery of infected ApoE−/−TLR4−/− mice was dramatically increased compared with infected ApoE−/− mice. The abundance of T cells was accompanied by increased numbers of IL-17–expressing cells and markedly diminished numbers of Foxp3+–expressing regulatory T cells (Tregs) (Fig. 4B, 4C). The marked increase in CD4+, CD8+, and IL-17+ cells and the diminution of FoxP3+ Tregs in infected mice in the absence of TLR4 expression (ApoE−/−TLR4−/− mice) reveal that in the presence of TLR4 expression, TLR4 may be protective after P. gingivalis infection, which serves to prevent the infiltration of IL-17+ T cells and enhance the numbers of Foxp3+ Tregs in the inflammatory lesion.
FIGURE 4.

TLR4 deficiency promotes Th17/Treg imbalance in atherosclerotic lesions after infection with P. gingivalis. Quantitative immunohistochemistry of (A) CD4+, CD8+, IL-17+ staining area in the innominate artery of uninfected and P. gingivalis-infected ApoE−/− and ApoE−/−TLR4−/− mice using ImageJ software or (B) Foxp3+ cell count. Three sections from n = 4 mice/group were analyzed. White bar, uninfected; gray bar, P. gingivalis infected. Data are mean ± SD of positive staining area/slice or cell count/slice. Intragenotype comparisons were calculated by Mann–Whitney U test: ***p < 0.0001. Intergenotype comparisons were calculated by two-way ANOVA (indicated p values). (C) Representative immunohistochemistry of CD4+, CD8+, IL-17+, and Foxp3+ cells in the innominate artery of uninfected and P. gingivalis-infected ApoE−/− and ApoE−/−TLR4−/− mice. Scale bar, 5 μm.
IgG humoral immunity and Th1 responses are altered in the absence of TLR4
Infection with P. gingivalis induced a robust IgG1 response in both ApoE−/− and ApoE−/−TLR4−/− mice, indicating preservation of IgG1-mediated humoral immunity in the absence of TLR4 (Fig. 5A). However, P. gingivalis-infected ApoE−/−TLR4−/− mice produced significantly reduced IgG2b (Fig. 5B) and IgG3 (Fig. 5D) responses compared with ApoE−/− mice—IgG subclasses that are associated with Th1 responses (37). IgG2c levels were increased to a similar level in infected ApoE−/− and ApoE−/−TLR4−/− mice (Fig. 5C).
FIGURE 5.

P. gingivalis-specific Ab isotypes IgG1, IgG2b, IgG2c, and IgG3. P. gingivalis-specific IgG production in uninfected ApoE−/− (n = 15), P. gingivalis-infected ApoE−/− (n = 21), uninfected ApoE−/−TLR4−/− (n = 17), and P. gingivalis-infected ApoE−/−TLR4−/− (n = 14) mice as measured by ELISA. (A) IgG1, (B) IgG2b, (C) IgG2c, and (D) IgG3. White squares, uninfected; gray squares, P. gingivalis infected. Data were analyzed by Student t test. *p < 0.05, **p = 0.0092.
We restimulated splenocytes from experimental mice with P. gingivalis soluble Ags and identified responsive cells that express the effector cytokines IFN-γ and IL-17. T cells from uninfected mice did not exhibit cytokine expression in response to P. gingivalis Ags. We observed a high percentage of IFN-γ–expressing CD4+ (Fig. 6A, 6C) and CD8+ (Fig. 6B, 6C) T cells from P. gingivalis-infected ApoE−/− mice. In contrast, the majority of responsive CD4+ (Fig. 6A, 6D) and CD8+ T cells (Fig. 6B, 6D) from infected ApoE−/−TLR4−/− mice expressed IL-17. A small subset of CD8+ (14%) T cells from infected ApoE−/−TLR4−/− mice also expressed IFN-γ. Although the number of reactive T cells indicates that these responses may not be Ag specific, they were specific to P. gingivalis infection, as T cells from uninfected mice failed to respond to stimulation with Ags. These results suggest that in the absence of TLR4, P. gingivalis infection results in impaired Th1 immunity and IL-17 skewing.
FIGURE 6.

Impaired Th1 immunity and Th17 skewing in P. gingivalis-infected TLR4-deficient mice. Expression of IFN-γ and IL-17 in (A) CD4+ and (B) CD8+ splenic T cells from uninfected and P. gingivalis-infected mice after in vitro stimulation with 10 μg/ml P. gingivalis soluble Ags and 1 μg/ml anti-mouse CD28 for 4 h in the continuous presence of brefeldin A. Data represent mean ± SD, n = 4/group. White bar, uninfected; gray bar, P. gingivalis infected. Intragenotype comparisons were calculated by Mann–Whitney U test. **p < 0.01, ***p < 0.001. Intergenotype comparisons were calculated by two-way ANOVA with Bonferroni’s posttest. (C and D) Representative flow cytometry histogram analysis of cytokine-secreting CD4+ and CD8+ T cells (filled histogram is isotype control).
Th1 and Treg polarizing cytokine production after P. gingivalis infection is impaired in DCs from TLR4-deficient mice
Activation of TLRs on DCs triggers the release of cytokines that play decisive roles in modulating T helper subset differentiation from naive CD4+ cells (38). To investigate the role of P. gingivalis-induced TLR4 activation in DC production of T cell polarizing cytokines, DCs from wild-type and TLR4−/− mice were infected with P. gingivalis, and expression of T cell polarizing cytokines was examined. P. gingivalis induced the production of IL-12 (Fig. 7A), IL-10 (Fig. 7B), and IL-6 (Fig. 7C) in DCs from ApoE−/− mice. Production of these cytokines was markedly reduced in DCs from ApoE−/−TLR4−/− mice. These results suggest that TLR4 is necessary for production of these cytokines after P. gingivalis infection. The abrogated DC production of T cell polarizing cytokines in the absence of TLR4 may be responsible for impaired development of Th1/Treg effector immunity as well as the enhanced IL-17 expression in T cell populations within plaques of ApoE−/−TLR4−/− mice.
FIGURE 7.
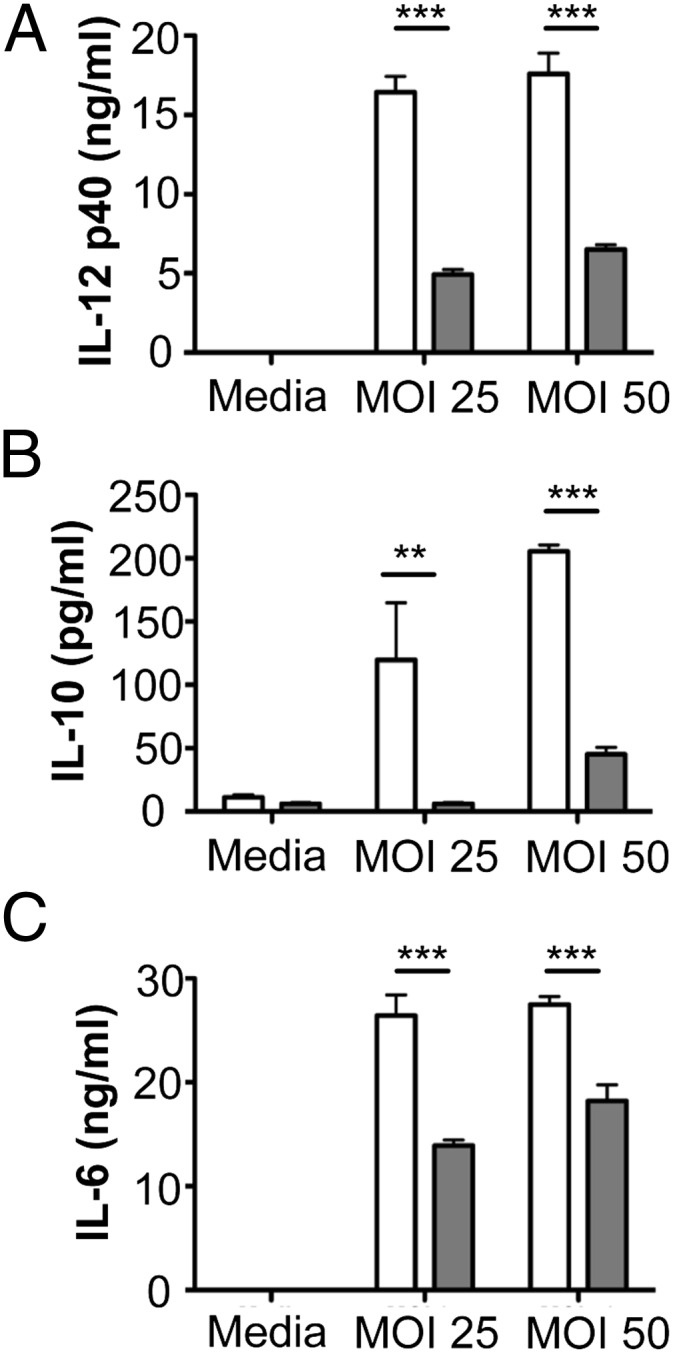
Th1 and Treg polarizing cytokine production after P. gingivalis infection is impaired in DCs from TLR4-deficient mice. Bone marrow-derived DCs from C57BL/6 (white bars) and TLR4−/− (gray bars) mice were infected with P. gingivalis at the indicated multiplicity of infection (MOI). After 24 h, supernatants were analyzed for (A) IL-12, (B) IL-10, and (C) IL-6 by ELISA. Bars represent mean ± SD from triplicate cultures. **p < 0.01, ***p < 0.001 (Student t test).
Discussion
Common chronic infections may contribute to up to 40% of newly developed atherosclerotic cases (39). A role for P. gingivalis-mediated periodontal disease as a risk factor for atherosclerotic cardiovascular disease is well documented (10, 11, 14–18). The observation that innate immune signaling triggered by P. gingivalis is dysregulated within atherosclerotic lesions has sparked interest in the association between oral infection and induction of innate immune cascades in atherosclerosis progression (40). Most experimental studies have focused on the proatherogenic consequence of TLR signaling in mouse models of atherogenesis; many involving the influence of high-fat diet (5, 6, 8, 12). In contrast to studies reporting diminished high-fat diet-induced atherosclerosis in TLR4-deficient mice, we report the unexpected finding that TLR4-deficient mice are markedly more susceptible to atherosclerosis after infection with P. gingivalis. Live animal imaging demonstrated that enhanced disease severity occurred progressively, long after cessation of the infectious stimulus and at two anatomically relevant sites, in large (aortic sinus) and medium (innominate artery) size vessels. Enhanced atherosclerosis progression in ApoE−/−TLR4−/− mice compared with ApoE−/− mice is unlikely to be due to differences in plasma cholesterol or triglycerides, which were similar among all groups. Minimal atherosclerotic lesion area in the innominate artery was observed in uninfected ApoE−/− mice, and this is likely due to the fact that animals were fed a normal chow diet. In our recent study in which atherosclerosis progression was examined using MRA in the innominate artery of uninfected and P. gingivalis-infected ApoE−/− mice, animals were fed a high-fat diet for the duration of the study (35). High-fat diet enhances atherosclerosis progression in ApoE−/− mice. In the absence of high-fat diet and infection, plaque accumulation within the aorta and innominate artery progresses more slowly and is minimal at the time point examined in the current study. Effective control of immune-mediated pathology in P. gingivalis-infected ApoE−/− mice coincided with an increase in Tregs within the innominate artery. In contrast, the exacerbated inflammatory pathology in P. gingivalis-infected ApoE−/−TLR4−/− mice was associated with increased lesion macrophage numbers and T cell infiltration and enhanced expression of IL-17. Tregs play a critical role in maintaining immunological tolerance and controlling the extent of immune-mediated pathology, especially in cases of chronic infection (41, 42). Our studies indicate that in the absence of TLR4, mice fail to develop protective Th1 immunity and are unable to regulate adaptive immune responses mediated by Th17 cells after P. gingivalis infection. We propose that this results in a breakdown of immunological tolerance, owing to impaired Treg function, leading to unrestricted activation of pathogenic T cells that mediate arterial inflammation. The unique TLR4 evasive properties of P. gingivalis lipid A position this organism to disrupt effector T cell mechanisms at the level of DC activation, the interface of innate and adaptive immunity.
TLR2 expression was increased markedly in aortic lesions by P. gingivalis infection in ApoE−/− mice and further increased in ApoE−/−TLR4−/− mice. It is plausible that enhanced TLR2 expression in ApoE−/−TLR4−/− mice may have contributed to increased vascular inflammation and atherosclerosis in ApoE−/−TLR4−/− mice. Thus, the increase in atherosclerosis in ApoE−/−TLR4−/− mice may be a result of not only TLR4 deficiency but also high TLR2 expression. This increased TLR2 expression in activated macrophages and the endothelium may reflect the development and maintenance of a hyperinflammatory state in the absence of TLR4 expression. This observation was an unexpected finding of this study, as was the finding that plaque development was enhanced in the absence of TLR4.
Our results also showed that after in vitro restimulation with Ag, T cells from P. gingivalis-infected ApoE−/−TLR4−/− mice predominantly produced IL-17, whereas IFN-γ was the predominant cytokine produced by T cells from infected ApoE−/− mice. We also demonstrate that TLR4 deficiency was associated with markedly inhibited production of the Th1 polarizing cytokine IL-12 by P. gingivalis-infected DCs. Taken together, our findings suggest that an initially impaired Th1 response in TLR4-deficient mice results in Th17 skewing of the adaptive immune response, which may be the mechanism for exacerbated atherosclerosis. Indeed, research supports a complex relationship between the Th1 and Th17 cell lineages, and many T cells expressing IL-17 coexpress IFN-γ. We previously reported that IFN-γ is significantly upregulated at the protein level in atherosclerotic lesions from P. gingivalis-infected ApoE−/− mice relative to uninfected controls (28). This finding is in agreement with IFN-γ contributing to the development of atherosclerosis as suggested by one study (43). Several studies demonstrate that both IFN-γ and IL-17 may be proatherogenic in mouse models, and circulating levels of both of these cytokines are increased in patients with coronary atherosclerosis (44). A large body of literature also supports a proatherogenic role for IL-17 in mouse models of atherosclerosis, and neutralization of IL-17 has been demonstrated to reduce pathogen- and diet-induced atherosclerosis in ApoE−/− mice (45). These studies demonstrate that the development of atherosclerosis is multifactorial and may be influenced by the inciting stimulus (i.e., high-fat diet versus pathogen-mediated).
Notably, humans are specifically impaired in their ability to recognize penta-acylated lipid A, a phenomenon reflecting bacterial adaptation to the human host (46). Species-specific discrimination of atypical penta-acylated lipid A is mediated by a hypervariable 82-aa sequence in the middle region of TLR4, a region where human polymorphisms are common (31, 46). Our results provide a mechanistic link regarding the conflicting reports on the association of human TLR4 polymorphisms and atherosclerotic diseases. Thus, although we only see a hyperinflammatory phenotype in TLR4-deficient mice, it is plausible that common human TLR4 polymorphisms (47–49) that attenuate receptor signaling may predispose individuals to an increased risk of atherosclerosis associated with bacterial infection, which is in contrast to atherosclerosis risk associated with a Western high-fat diet.
Recent studies reported that ApoE−/−TLR4−/− mice fed a high-fat diet and infected intranasally with C. pneumoniae exhibited diminished atherosclerosis compared with infected ApoE−/− mice (12). A separate report demonstrated a protective role for TLR4 deficiency in diet-induced atherogenesis (8). It is important to note that these results could not be recapitulated under germ-free conditions (50), indicating a potential interaction between hyperlipidemia and indigenous microbes. On the basis of these observations, it was proposed that common mechanisms of signaling via TLR2, TLR4, and MyD88 link stimulation by multiple pathogens and endogenous ligands to atherosclerosis, and that therapeutic TLR4 antagonism could prove beneficial in the treatment of chronic atherosclerosis (8, 51, 52). Our results clearly point to a critical role for specific TLR signaling, in particular, TLR4, in chronic inflammation and atherosclerosis induced by P. gingivalis. Our results raise caution for the safety and efficacy of TLR4 antagonists for the treatment of atherosclerosis, especially in patients with comorbid conditions including periodontal disease and other infectious diseases.
Acknowledgments
We thank Dr. Stephen R. Coates (Department of Periodontics, University of Washington) for analysis of bacterial lipid A. We thank Dr. Xuemei Zhong and Nathalie Bitar (Boston University Immunohistochemistry Core) for technical assistance.
This work was supported by National Institutes of Health grants (National Heart, Lung and Blood Institute Grant R01 HL080387 to C.A.G. and National Institute of Allergy and Infectious Diseases Grant P01 AI078894 to C.A.G.).
- DC
- dendritic cell
- MRA
- magnetic resonance angiography
- Treg
- regulatory T cell.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Cole J. E., Georgiou E., Monaco C. 2010. The expression and functions of toll-like receptors in atherosclerosis. Mediators Inflamm. 2010: 393946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hodgkinson C. P., Ye S. 2011. Toll-like receptors, their ligands, and atherosclerosis. ScientificWorldJournal 11: 437–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akira S., Takeda K. 2004. Toll-like receptor signalling. Nat. Rev. Immunol. 4: 499–511 [DOI] [PubMed] [Google Scholar]
- 4.Edfeldt K., Swedenborg J., Hansson G. K., Yan Z. Q. 2002. Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation 105: 1158–1161 [PubMed] [Google Scholar]
- 5.Bjorkbacka H., Kunjathoor V. V., Moore K. J., Koehn S., Ordija C. M., Lee M. A., Means T., Halmen K., Luster A. D., Golenbock D. T., Freeman M. W. 2004. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat. Med. 10: 416–421 [DOI] [PubMed] [Google Scholar]
- 6.Higashimori M., Tatro J. B., Moore K. J., Mendelsohn M. E., Galper J. B., Beasley D. 2011. Role of toll-like receptor 4 in intimal foam cell accumulation in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 31: 50–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu X., Ukai T., Yumoto H., Davey M., Goswami S., Gibson F. C., III, Genco C. A. 2008. Toll-like receptor 2 plays a critical role in the progression of atherosclerosis that is independent of dietary lipids. Atherosclerosis 196: 146–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michelsen K. S., Wong M. H., Shah P. K., Zhang W., Yano J., Doherty T. M., Akira S., Rajavashisth T. B., Arditi M. 2004. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl. Acad. Sci. USA 101: 10679–10684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mullick A. E., Tobias P. S., Curtiss L. K. 2005. Modulation of atherosclerosis in mice by Toll-like receptor 2. J. Clin. Invest. 115: 3149–3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson F. C., III, Yumoto H., Takahashi Y., Chou H. H., Genco C. A. 2006. Innate immune signaling and Porphyromonas gingivalis-accelerated atherosclerosis. J. Dent. Res. 85: 106–121 [DOI] [PubMed] [Google Scholar]
- 11.Hayashi C., Gudino C. V., Gibson F. C., III, Genco C. A. 2010. Review: Pathogen-induced inflammation at sites distant from oral infection: bacterial persistence and induction of cell-specific innate immune inflammatory pathways. Mol. Oral Microbiol. 25: 305–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naiki Y., Sorrentino R., Wong M. H., Michelsen K. S., Shimada K., Chen S., Yilmaz A., Slepenkin A., Schröder N. W., Crother T. R., et al. 2008. TLR/MyD88 and liver X receptor alpha signaling pathways reciprocally control Chlamydia pneumoniae-induced acceleration of atherosclerosis. J. Immunol. 181: 7176–7185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pihlstrom B. L., Michalowicz B. S., Johnson N. W. 2005. Periodontal diseases. Lancet 366: 1809–1820 [DOI] [PubMed] [Google Scholar]
- 14.Amar S., Gokce N., Morgan S., Loukideli M., Van Dyke T. E., Vita J. A. 2003. Periodontal disease is associated with brachial artery endothelial dysfunction and systemic inflammation. Arterioscler. Thromb. Vasc. Biol. 23: 1245–1249 [DOI] [PubMed] [Google Scholar]
- 15.Dasanayake A. P., Russell S., Boyd D., Madianos P. N., Forster T., Hill E. 2003. Preterm low birth weight and periodontal disease among African Americans. Dent. Clin. North Am. 47: 115–125, x–xi (x-xi.) [DOI] [PubMed] [Google Scholar]
- 16.Genco R. J. 1996. Current view of risk factors for periodontal diseases. J. Periodontol. 67(10, Suppl)1041–1049 [DOI] [PubMed] [Google Scholar]
- 17.Morrison H. I., Ellison L. F., Taylor G. W. 1999. Periodontal disease and risk of fatal coronary heart and cerebrovascular diseases. J. Cardiovasc. Risk 6: 7–11 [DOI] [PubMed] [Google Scholar]
- 18.Tonetti M. S., D’Aiuto F., Nibali L., Donald A., Storry C., Parkar M., Suvan J., Hingorani A. D., Vallance P., Deanfield J. 2007. Treatment of periodontitis and endothelial function. N. Engl. J. Med. 356: 911–920 [DOI] [PubMed] [Google Scholar]
- 19.Haraszthy V. I., Zambon J. J., Trevisan M., Zeid M., Genco R. J. 2000. Identification of periodontal pathogens in atheromatous plaques. J. Periodontol. 71: 1554–1560 [DOI] [PubMed] [Google Scholar]
- 20.Padilla C., Lobos O., Hubert E., González C., Matus S., Pereira M., Hasbun S., Descouvieres C. 2006. Periodontal pathogens in atheromatous plaques isolated from patients with chronic periodontitis. J. Periodontal Res. 41: 350–353 [DOI] [PubMed] [Google Scholar]
- 21.Amar S., Wu S. C., Madan M. 2009. Is Porphyromonas gingivalis cell invasion required for atherogenesis? Pharmacotherapeutic implications. J. Immunol. 182: 1584–1592 [DOI] [PubMed] [Google Scholar]
- 22.Gibson F. C., III, Hong C., Chou H.-H., Yumoto H., Chen J., Lien E., Wong J., Genco C. A. 2004. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation 109: 2801–2806 [DOI] [PubMed] [Google Scholar]
- 23.Lalla E., Lamster I. B., Hofmann M. A., Bucciarelli L., Jerud A. P., Tucker S., Lu Y., Papapanou P. N., Schmidt A. M. 2003. Oral infection with a periodontal pathogen accelerates early atherosclerosis in apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 23: 1405–1411 [DOI] [PubMed] [Google Scholar]
- 24.Li L., Messas E., Batista E. L., Jr., Levine R. A., Amar S. 2002. Porphyromonas gingivalis infection accelerates the progression of atherosclerosis in a heterozygous apolipoprotein E-deficient murine model. Circulation 105: 861–867 [DOI] [PubMed] [Google Scholar]
- 25.Madan M., Amar S. 2008. Toll-like receptor-2 mediates diet and/or pathogen associated atherosclerosis: proteomic findings. PLoS ONE 3: e3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyamoto T., Yumoto H., Takahashi Y., Davey M., Gibson F. C., III, Genco C. A. 2006. Pathogen-accelerated atherosclerosis occurs early after exposure and can be prevented via immunization. Infect. Immun. 74: 1376–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hajishengallis G., Wang M., Liang S. 2009. Induction of distinct TLR2-mediated proinflammatory and proadhesive signaling pathways in response to Porphyromonas gingivalis fimbriae. J. Immunol. 182: 6690–6696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayashi C., Madrigal A. G., Liu X., Ukai T., Goswami S., Gudino C. V., Gibson F. C., III, Genco C. A. 2010. Pathogen-mediated inflammatory atherosclerosis is mediated in part via Toll-like receptor 2-induced inflammatory responses. J. Innate Immun. 2: 334–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coats S. R., Berezow A. B., To T. T., Jain S., Bainbridge B. W., Banani K. P., Darveau R. P. 2011. The lipid A phosphate position determines differential host Toll-like receptor 4 responses to phylogenetically related symbiotic and pathogenic bacteria. Infect. Immun. 79: 203–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coats S. R., Jones J. W., Do C. T., Braham P. H., Bainbridge B. W., To T. T., Goodlett D. R., Ernst R. K., Darveau R. P. 2009. Human Toll-like receptor 4 responses to P. gingivalis are regulated by lipid A 1- and 4′-phosphatase activities. Cell. Microbiol. 11: 1587–1599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller S. I., Ernst R. K., Bader M. W. 2005. LPS, TLR4 and infectious disease diversity. Nat. Rev. Microbiol. 3: 36–46 [DOI] [PubMed] [Google Scholar]
- 32.Curtis M. A., Percival R. S., Devine D., Darveau R. P., Coats S. R., Rangarajan M., Tarelli E., Marsh P. D. 2011. Temperature-dependent modulation of Porphyromonas gingivalis lipid A structure and interaction with the innate host defenses. Infect. Immun. 79: 1187–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Al-Qutub M. N., Braham P. H., Karimi-Naser L. M., Liu X., Genco C. A., Darveau R. P. 2006. Hemin-dependent modulation of the lipid A structure of Porphyromonas gingivalis lipopolysaccharide. Infect. Immun. 74: 4474–4485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baker P. J., Evans R. T., Roopenian D. C. 1994. Oral infection with Porphyromonas gingivalis and induced alveolar bone loss in immunocompetent and severe combined immunodeficient mice. Arch. Oral Biol. 39: 1035–1040 [DOI] [PubMed] [Google Scholar]
- 35.Hayashi C., Viereck J., Hua N., Phinikaridou A., Madrigal A. G., Gibson F. C., III, Hamilton J. A., Genco C. A. 2011. Porphyromonas gingivalis accelerates inflammatory atherosclerosis in the innominate artery of ApoE deficient mice. Atherosclerosis 215: 52–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gibson F. C., III, Gonzalez D. A., Wong J., Genco C. A. 2004. Porphyromonas gingivalis-specific immunoglobulin G prevents P. gingivalis-elicited oral bone loss in a murine model. Infect. Immun. 72: 2408–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Germann T., Bongartz M., Dlugonska H., Hess H., Schmitt E., Kolbe L., Kölsch E., Podlaski F. J., Gately M. K., Rüde E. 1995. Interleukin-12 profoundly up-regulates the synthesis of antigen-specific complement-fixing IgG2a, IgG2b and IgG3 antibody subclasses in vivo. Eur. J. Immunol. 25: 823–829 [DOI] [PubMed] [Google Scholar]
- 38.Peters M., Dudziak K., Stiehm M., Bufe A. 2010. T-cell polarization depends on concentration of the danger signal used to activate dendritic cells. Immunol. Cell Biol. 88: 537–544 [DOI] [PubMed] [Google Scholar]
- 39.Kiechl S., Egger G., Mayr M., Wiedermann C. J., Bonora E., Oberhollenzer F., Muggeo M., Xu Q., Wick G., Poewe W., Willeit J. 2001. Chronic infections and the risk of carotid atherosclerosis: prospective results from a large population study. Circulation 103: 1064–1070 [DOI] [PubMed] [Google Scholar]
- 40.Lundberg A. M., Hansson G. K. 2010. Innate immune signals in atherosclerosis. Clin. Immunol. 134: 5–24 [DOI] [PubMed] [Google Scholar]
- 41.Ait-Oufella H., Salomon B. L., Potteaux S., Robertson A.-K. L., Gourdy P., Zoll J., Merval R., Esposito B., Cohen J. L., Fisson S., et al. 2006. Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 12: 178–180 [DOI] [PubMed] [Google Scholar]
- 42.Zhang S. H., Reddick R. L., Piedrahita J. A., Maeda N. 1992. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 258: 468–471 [DOI] [PubMed] [Google Scholar]
- 43.Gupta S., Pablo A. M., Jiang X., Wang N., Tall A. R., Schindler C. 1997. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J. Clin. Invest. 99: 2752–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eid R. E., Rao D. A., Zhou J., Lo S. F., Ranjbaran H., Gallo A., Sokol S. I., Pfau S., Pober J. S., Tellides G. 2009. Interleukin-17 and interferon-gamma are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation 119: 1424–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen S., Shimada K., Zhang W., Huang G., Crother T. R., Arditi M. 2010. IL-17A is proatherogenic in high-fat diet-induced and Chlamydia pneumoniae infection-accelerated atherosclerosis in mice. J. Immunol. 185: 5619–5627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hajjar A. M., Ernst R. K., Tsai J. H., Wilson C. B., Miller S. I. 2002. Human Toll-like receptor 4 recognizes host-specific LPS modifications. Nat. Immunol. 3: 354–359 [DOI] [PubMed] [Google Scholar]
- 47.Ajdary S., Ghamilouie M.-M., Alimohammadian M.-H., Riazi-Rad F., Pakzad S.-R. 2011. Toll-like receptor 4 polymorphisms predispose to cutaneous leishmaniasis. Microbes Infect. 13: 226–231 [DOI] [PubMed] [Google Scholar]
- 48.Kiechl S., Lorenz E., Reindl M., Wiedermann C. J., Oberhollenzer F., Bonora E., Willeit J., Schwartz D. A. 2002. Toll-like receptor 4 polymorphisms and atherogenesis. N. Engl. J. Med. 347: 185–192 [DOI] [PubMed] [Google Scholar]
- 49.Montes A. H., Asensi V., Alvarez V., Valle E., Ocaña M. G., Meana A., Carton J. A., Paz J., Fierer J., Celada A. 2006. The Toll-like receptor 4 (Asp299Gly) polymorphism is a risk factor for Gram-negative and haematogenous osteomyelitis. Clin. Exp. Immunol. 143: 404–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wright S. D., Burton C., Hernandez M., Hassing H., Montenegro J., Mundt S., Patel S., Card D. J., Hermanowski-Vosatka A., Bergstrom J. D., et al. 2000. Infectious agents are not necessary for murine atherogenesis. J. Exp. Med. 191: 1437–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cole J. E., Mitra A. T., Monaco C. 2010. Treating atherosclerosis: the potential of Toll-like receptors as therapeutic targets. Expert Rev. Cardiovasc. Ther. 8: 1619–1635 [DOI] [PubMed] [Google Scholar]
- 52.Leon C. G., Tory R., Jia J., Sivak O., Wasan K. M. 2008. Discovery and development of toll-like receptor 4 (TLR4) antagonists: a new paradigm for treating sepsis and other diseases. Pharm. Res. 25: 1751–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]