

Abstract
Most HIV replication occurs in solid lymphoid tissue, which has prominent architecture at the histological level, which separates groups of productively infected CD4+ cells. Nevertheless, current population models of HIV assume panmixis within lymphoid tissue. We present a simple “metapopulation” model of HIV replication, where the population of infected cells is comprised of a large number of small populations, each of which is established by a few founder viruses and undergoes turnover. To test this model, we analyzed viral genetic variation of infected cell subpopulations within the spleen and demonstrated the action of founder effects as well as significant variation in the extent of genetic differentiation between subpopulations among patients. The combination of founder effects and subpopulation turnover can result in an effective population size much lower than the actual population size and may contribute to the importance of genetic drift in HIV evolution despite a large number of infected cells.
HIV has an enormous evolutionary potential because of the combination of a high mutation rate (≈3 × 10−5 substitutions/site/generation; ref. 1), a short generation time (≈2.6 days; refs. 2 and 3), and a large number of infected cells within the host (107–108 infected cells; ref. 4). Many analyses of the pattern of nucleotide substitution in the viral env gene have detected evolution in response to positive selection (5–16), although relatively few studies have linked these patterns of viral genetic variation to selection by defined immune responses or for changes in viral phenotype associated with coreceptor usage. In the presence of antiretroviral agents that target viral protease and reverse transcriptase, positive selection has been described in numerous studies, which demonstrate convergent evolution in different individuals of mutations that confer drug resistance (17–20). Although specific mutations have been identified with escape from immune responses and escape from antiretrovirals, viral genetic variation reflects more than just these focused positive selection pressures and may reflect more subtle processes of within-host competition between viruses for higher replicative potential.
Despite an enormous potential to respond to selection, there can be substantial differences between infected individuals in the evolution of drug resistance. For example, the frequency of single drug-resistant mutants in viral protease and reverse transcriptase before therapy can vary dramatically between hosts (21–23), apparently by random. Indirect observations of the apparently stochastic way in which multiply resistant mutants emerge during therapy are also consistent with a role of genetic drift (18, 24, 25). In addition, three studies of the evolution of env gene sequences have found substantially lower genetic variation than expected for a population of the order of 107 (24, 26, 27). These studies estimated the “effective” population size to be of the order of 103, reflecting the high relatedness between env sequences sampled at a given time. On the basis of the level of variation in the frequency of M184V mutation in reverse transcriptase, Frost et al. (23) estimated an effective population size of 105–106, similar to with estimates of 104–105 estimated from patterns of variation in protease (28). Hence, stochastic models of drug resistance evolution that assume an effective population size close to the actual population size (29) may dramatically underestimate the role of genetic drift in generating variation between individuals in the emergence of drug-resistant strains.
Despite empirical evidence for an effective population size low enough to generate considerable variation in the frequency of rare drug-resistant mutants, the means by which a large actual population size is translated into an effective population size at least 10- to 100-fold smaller are not understood and reveal a significant gap in our understanding of viral dynamics. Although many models of HIV-1 evolution assume that the population of infected cells within the host is well-mixed (28, 30), there is direct evidence that this is an unrealistic assumption. On a macroscopic spatial scale, it has been recognized for some time that HIV genetic variability is spatially heterogeneous (31–36). Studies of proviral clones isolated from individual splenic white pulps have revealed that such differentiation can also occur on a microscopic scale (37). Despite many potential target cells for HIV infection in splenic tissue, the average number of infected cells is low, with a high frequency of putative recombinants, suggesting high levels of mixing at a local scale but poor mixing of virus at larger spatial scales (38).
To take into account the evidence for population structuring, we have applied a simple “metapopulation” model (39) of HIV replication, in which small subpopulations of infected cells are established by a small number of founding viruses and turn over at a high rate. To test this model, we analyzed the pattern of genetic variation in the V1/V2 region of the env gene within and between infected white pulps in the spleens of six HIV-1-infected individuals. Founder effects are shown to play an important role in determining the effective rate of recombination, and that subpopulation turnover can result in an effective population size that is much lower than the number of infected cells in the body, thus contributing to the action of genetic drift in the evolution of HIV populations.
Methods
Sequence Data.
White pulps were microdissected from spleens and ≈20 proviral clones of the V1/V2 region of env ≈300 bp long were sequenced from two to five pulps per spleen, as described elsewhere (ref. 37; M.-J.D. and S.W.-H., unpublished work). Sequences were aligned with clustalw (40) and checked by eye. Although in peripheral blood, proviral DNA from peripheral blood monoclear cells (PBMCs) can differ from viral RNA in plasma, presumably because of the “archiving” of genetic variation in PBMCs over time (3, 5), this is unlikely to be an issue here, as (M.-J.D. and S.W.-H., unpublished work) have shown that viral RNA and proviral DNA obtained from the same pulp are virtually indistinguishable.
Population Genetic Analysis.
Genetic differentiation between subpopulations was quantified by using estimates of FST, the fraction of the total genetic variation found between subpopulations (41) obtained from an analysis of molecular variance (AMOVA) in arlequin ver. 1.1 (42) assuming a Jukes–Cantor (43) model to correct for multiple hits. The significance of departures from the null hypothesis of a random distribution of genetic variation was determined by using 10,000 randomizations of sequences between populations.
Estimates of the number of pairwise differences and the number of mutations for each sample were obtained by using dnasp ver. 3.0 (44). Tajima's D statistic was calculated by using a β distribution with mean 0 and variance 1 to estimate the P value against the values of D expected under a coalescent model of a population of constant size (45). Values of D ≤ 1.84 give a P value of approximately less than 0.05 when the sample size is ≈20 (46).
FST and Tajima's D were calculated, correcting for heterogeneity in the substitution rate across sites (47). Rate variation was modeled as a γ distribution with shape parameter 0.3, similar to the values of the shape parameter estimated from the V3 region of env (48).
We tested for recombination within each individual by testing for long regions of sequence that are sufficiently long and/or similar to suggest recombination either between sequences in the sample or their ancestors (“inner” fragments) or between a sequence in the sample and another sequence that was unknown (“outer sequence” fragments), either because the other sequence was not present in the sample or because further mutation had obscured the relationship between the two sequences. Global P values, corrected for multiple comparisons, were obtained by simulation using the program geneconv ver. 1.81 (49). To compare the extent of recombination between sequences within each pulp, the minimum number of recombination events was estimated by analyzing pairs of variable sites by using the method of Hudson and Kaplan (50). Sites at which three or four nucleotides were segregating were not included.
Results
To develop a biologically plausible model of HIV replication within solid tissue, a simple model of molecular evolution in a metapopulation was considered as originally studied by M. Slatkin (51) and recently revisited in the context of nucleotide diversity by Pannell and Charlesworth (52). In this model, the total number of infected cells in an individual, NT, remains constant and consists of n subpopulations (where n >>1), each made up of NS infected cells, NT = nNS. A proportion of subpopulations, denoted x, goes extinct every generation and is replaced by new subpopulations, each of which are founded by k infected cells drawn randomly from the whole population. It is assumed that the probability of founders being drawn from the same subpopulation is zero. This assumption is intended to reflect long-range migration from a well-mixed pool of latently infected cells to pockets of uninfected cells, as hypothesized by Cheynier et al. (37). For simplicity in dealing with subpopulation age structure, it was assumed that the k founders immediately grow into a subpopulation of NS infected cells and that generations are discrete. A schematic of the model is given in Fig. 1.
Figure 1.

Schematic of the metapopulation model. The total pool of HIV-infected cells, NT, is assumed to consist of a large number, n (for illustrative purposes, n = 16 × 5), of subpopulations, each consisting of NS cells (here, NS = 9), as shown on the Right. A proportion x of subpopulations go extinct every generation, and new subpopulations are established in their place by k founders (here, k = 1), which for mathematical simplicity are assumed to increase to NS infected cells instantly, as shown on the Left.
At equilibrium, FST, defined as the amount of genetic variation between subpopulations expressed as a fraction of the total variation (41), is given by the expression.
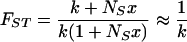 |
1 |
The approximation above is for moderate amounts of subpopulation turnover, x ≫ 1/NS. For example, for NS = 100, the approximation is valid if the average lifetime of a subpopulation is less than 100 generations (≈200 days). Given the low average frequency of infected cells within splenic tissues, with <1% of CD4+ T cells harboring proviral DNA (37), subpopulation turnover is likely to be much higher than this.
To test whether the metapopulation model provides a better fit to the pattern of viral genetic variation in solid tissues than a panmictic model, sequence data of the V1/V2 hypervariable region of the envelope gene obtained from two or more white pulps each from the spleens of six HIV-1-infected individuals were analyzed (ref. 37; M.-J.D. and S.W.-H., unpublished work). FST was calculated for each spleen, both using all of the pulps, to get an overall measure of between host variation in genetic differentiation (Table 1), as well as for each pair of pulps, to indicate potential within-host variation in genetic differentiation (Fig. 2). Estimates of FST allowing for heterogeneity in the substitution rate along the sequence were slightly higher but were very similar (results not shown).
Table 1.
Results of analysis of molecular variance (AMOVA) for each patient
| Patient | Number of pulps analyzed | Between-pulp variation Va | Total variation Vb | FST (⩵Va/Vb) |
|---|---|---|---|---|
| B | 3 | 1.453 | 2.446 | 0.594 |
| L | 4 | 0.836 | 2.266 | 0.369 |
| M | 2 | −0.041 | 2.380 | −0.017ns |
| N | 2 | 0.055 | 0.706 | 0.078 |
| P | 5 | 0.463 | 2.154 | 0.215 |
| S | 4 | 0.146 | 1.629 | 0.090 |
ns, not significant at the 5% level.
Figure 2.

Plots of genetic variation (as measured by the variance components in an analysis of molecular variance) between pairs of infected pulps against total genetic variation. Lines indicate contours of FST; 0.1 (dotted line), 0.5 (dashed line), and 1 (solid line). Data from different individuals are represented by different symbols (B = ο, L = Δ, M = +, n = ×, P = ◊, S = ----). With the exception of the pair of pulps from patient M, all values of FST are significantly greater than zero (P < 0.05), indicating deviation from panmixis, although there is great variation in FST between pulps.
With the exception of patient M, where the estimate of between-pulp variation was negative, reflecting sampling error coupled with homogeneity of viral genetic variation across pulps, FST was significantly higher than expected, based on a random distribution of variation ranging from 0.08 for patient N to 0.59 for patient B (Table 1). FST for pairs of infected pulps was also higher than expected from a random distribution of variation (with the exception of a single comparison in patients P and S) and also varied dramatically between individuals, ranging from very low (<0.1) to very high (0.76) levels of divergence between pulps (Fig. 2). Levels of genetic differentiation were rather similar between pulps within a patient, with the exception of patient P, which shows two clusters of points because of a single pulp with a very high (≈6%) diversity (Fig. 2). Hence the homogenous model was rejected. In addition, the metapopulation model can easily account for the high observed variation in FST between individuals, by subtle differences in the number of founders, k, as with moderate amounts of subpopulation turnover, such that NSx ≫ 1, FST is approximately equal to 1/k.
The metapopulation model predicts the occurrence of founder effects, random changes in frequency of genetic variants because of sampling when the number of founders are low. These effects can be detected by using Tajima's D statistic, which compares pairwise diversity with the number of variable sites in a sample of sequences (45). Pairwise diversity is an estimator of the evolutionary distance to the most recent common ancestor (MRCA), whereas the number of variable sites is an estimator of the overall length of the phylogeny. As founder effects result in phylogenetic trees with short distances to the MRCA (hence low pairwise distances) and with a star-like shape (hence many variable sites), founder effects can give rise to values of D less than 0.
Fig. 3 shows that there is a positive correlation between within-pulp diversity and Tajima's D (Spearman's rank correlation = 0.70, P < 0.001), with a significant bottleneck effect when within-pulp diversity is low, as expected if low within-pulp diversity arises because of a low number of founders. Estimates of Tajima's D, corrected for heterogeneity in the substitution rate along sites (47), were more strongly negative (not shown). No significant bottleneck effect was found when within-pulp diversity is high, consistent with the metapopulation model, where high levels of diversity are generated by multiple, genetically divergent founders seeding pockets of uninfected cells.
Figure 3.

Estimates of Tajima's D against pairwise diversity for viruses sequenced from each pulp. The dashed line indicates significantly (P, approximately less than 0.05) negative values of D (<1.84), whereas the dotted line indicates D = 0 (the expected value under a constant population evolving neutrally). Data from different individuals are represented by different symbols (see Fig. 2 legend). There is a significant (P < 0.001) correlation between Tajima's D and pairwise diversity.
Metapopulation dynamics may also play a role in setting the effective recombination rate, as recombinant viruses are produced only when multiple viruses infect the same cell (which occurs at a local spatial scale) and can be detected only when these viruses are genetically different, which under our metapopulation model is affected by the processes of birth and death of subpopulations. We first tested for the presence of recombinant viruses within each individual as a whole by looking for long regions in which pairs of sequences were unusually similar (“inner” fragments), suggesting recombination between them or their ancestors, or unusually different (“outer” fragments), suggesting recombination with a sequence that is either not represented in the sample, or where further mutation or recombination has obscured the relationship. Fragments could be identified in four of six patients at the P = 0.05 level (B, L, M, and P), and in one additional patient (S) at the P = 0.1 level. We hypothesized that within each individual, more recombination events would be observed in white pulps with higher diversity, as multiple genetically divergent founders increase the opportunities for new genetic variants to be produced locally by recombination. To test this hypothesis, we first estimated the minimum number of recombination events within each pulp from the number of pairs of variable sites where all four combinations of mutations were observed (“four-types”). Four-types were identified in 17/20 pulps. Although these combinations can also be produced by independent substitutions at each site, rather than by recombination between the sites, the number of four-types was usually much higher than the minimum number of recombination events (median 8.1-fold higher), with 13/17 pulps where four-types were at least 4-fold higher (Fig. 4). The large difference in the number of four-types and the minimum number of recombination events in the majority of pulps analyzed is a consequence of the clustered distribution of the “four-types” along the sequence, which is more likely to arise under recombination than under convergent evolution. There was a significant positive correlation between pairwise diversity and the minimum number of recombination events estimated from the pattern of four-types (Spearman's rank correlation = 0.48, P < 0.05; Fig. 5), consistent with the hypothesis that the effective recombination rate may be affected by the number of founding viruses and subpopulation turnover via levels of within pulp diversity.
Figure 4.

Estimates of the number of “four-types,” i.e., the number of pairs of sites where all four combinations of two mutants are present, against the minimum number of recombination events required to produce them, plotted by pulp for each patient (symbols as in Fig. 2 legend). The number of recombination events that need to be invoked to explain the data is much lower than the number of four-types (as shown by the large number of points above the dotted line, which has an intercept 0 and slope 1).
Figure 5.

Estimates of minimum number of recombination events against pairwise diversity for viruses sequenced from each pulp. Data from different individuals are represented by different symbols (see Fig. 2 legend). There is a significant (P < 0.05) positive correlation between the minimum number of recombination events and pairwise diversity.
Discussion
The assumption that HIV populations are panmictic is inconsistent with the pattern of genetic variation in solid tissues. Rather, they support a “metapopulation” model, as originally defined in terms of population turnover as a “population of populations,” with an age structure established by the birth of new populations through colonization and their death through extinction (39). Large differences between individuals in the level of viral genetic structuring within the spleen can arise because of relatively small differences in the number of founders, with high levels of population structure, low levels of diversity within subpopulations, and a lesser role of recombination arising when the number of founders is low. The metapopulation concept, in a more general sense where a population is made up of discrete subunits that exchange genes, has been invoked in a large number of studies in ecology, conservation biology, and population genetics (53). The impact of metapopulation processes on genetic diversity has been discussed recently in an excellent review by Pannell and Charlesworth (54).
A comparison of the amount of genetic differentiation between viruses from different white pulps of the spleen with the total amount of viral genetic variation clearly demonstrated spatial structure, although the extent of population structuring varied between individuals. Although the genetic diversity, both within subpopulations and as a whole, is likely to be lower than their equilibrium values [because of the relatively genetically homogeneous viral population at the initial bottleneck of infection (6, 55–58)], the ratio of the two, as measured by FST, reaches its equilibrium value relatively rapidly, and hence estimates of FST, and our conclusions based on them are not expected to be greatly affected by the bottleneck of infection.
The use of Tajima's D statistic as a summary statistic describing the shape of the viral phylogeny within pulps showed that low genetic diversity was associated with low values of D, which we concluded were because of founder effects. Although selection can also result in deviations of D from 0, selection within pulps is likely to be weak because of the very small within-pulp population size, and hence any significant departures are likely to reflect founder effects. Interestingly, when Tajima's D was calculated for the pooled sequences from each individual, low levels of genetic differentiation were associated with significantly negative values of D, especially when rate heterogeneity in mutation rate along the sequence was taken into account (Table 2). There are several plausible explanations. First, the age structure of subpopulations, which arises as a consequence of founding and turnover, has been hypothesized to result in negative values of D when there is sufficient within-population diversity (59), although the size of this effect requires knowledge of the distribution of coalescence times under a metapopulation model. This is currently an open problem, although progress is being made in this area.‖ Second, the viruses in different pulps in the spleen may themselves have gone through a bottleneck. In virus isolated from peripheral blood, it has been shown that a bottleneck effect is present early in infection that wanes as infection progresses (60), and this may reflect a similar process in solid tissue. Third, negative values of D can arise when neutral variation is linked to selectively advantageous mutations that sweep through the population (61). Fourth, negative values of D may arise because of purifying selection on deleterious mutations (26). Although selection on a single deleterious mutation has little or no effect on phylogenetic tree shape (62), this effect may be greater if selection acts on multiple mutations.
Table 2.
Estimates of average pairwise diversity, number of mutations, and Tajima's D (no rate heterogeneity) and Misawa and Tajima's D (corrected for rate heterogeneity) for the pooled sequences
| Patient | No. of sequences (length in base pairs) | Average no. of differences (%) | Number of mutations | Tajima's D | Misawa and Tajima's D (α = 0.3) |
|---|---|---|---|---|---|
| B | 55 (257 bp) | 10.63 (4.14) | 48 | 0.045 (P = 0.53) | 0.154 (P = 0.57) |
| L | 81 (217 bp) | 8.68 (4.00) | 75 | −1.414 (P = 0.072) | −1.678 (P = 0.036) |
| M | 38 (268 bp) | 11.98 (4.47) | 79 | −1.328 (P = 0.091) | −1.547 (P = 0.054) |
| N | 30 (265 bp) | 3.524 (1.33) | 33 | −2.099 (P = 0.007) | −2.237 (P = 0.003) |
| P | 90 (285 bp) | 11.076 (3.89) | 110 | −1.633 (P = 0.04) | −1.932 (P = 0.015) |
| S | 77 (229 bp) | 6.734 (2.94) | 82 | −2.001 (P = 0.011) | −2.289 (P = 0.003) |
Metapopulation dynamics are likely to affect the impact of recombination on viral genetic variation through its effects on subpopulation diversity. We demonstrated a positive correlation across pulps between pairwise diversity and the estimated minimum number of recombination events based on the pattern of “four-types,” pairs of sites where all four combinations of two mutants are present. We interpreted this correlation as showing that low diversity arising because of founder effects restricts the impact of recombination, which occurs on a local rather than a global scale. Although “four-types” can also be generated by convergent evolution, their clustered spatial pattern along the sequence suggests that they were more likely to be generated by a few recombination events rather than by a large number of multiple substitutions. A more rigorous model-based test of whether these patterns arise because of recombination rather than multiple substitutions requires knowledge of the distribution of coalescent times under a metapopulation model. Although a positive correlation between genetic diversity and the number of recombination events could be generated if recombinants formed part of a founding viral population rather than being produced locally, this would also require the presence of the parental strains in the founding population. This seems biologically unlikely, and the number of four-types is likely to reflect recombination events, which occur after founding of the subpopulation.
Although spatial models have been applied to the question of HIV population structure, these models have been of the “island” type, which does not allow for extinction of subpopulations, and where dissemination of virus is assumed to occur only between extant subpopulations, such as between different parts of the brain (63) and between the plasma and cerebrospinal fluid.** Although this may be a reasonable assumption when looking at large-scale population structure (as large subpopulations are unlikely to go extinct), or when looking at infection of glial cells in the brain (which undergo slow turnover), turnover of productively infected T cells in solid lymphoid tissue is high because of local exhaustion of target cells and/or to the infiltration of HIV-specific cytotoxic T cells (37, 38) and with low levels of mixing will lead to extinction of foci of infection. Under the island model, the large differences in FST seen between individuals would have to be generated by large differences in the migration rate, rather than subtle differences in the number of founders, as in the metapopulation model. The low level of diversity and the presence of founder effects and local recombinant viruses in subpopulations of infected cells suggest they have been recently generated, rather than being set up early in infection, with continuous migration between subpopulations.
Small-scale spatial variation of HIV has important implications for within-host HIV evolution. The effective population size, Ne, under a metapopulation model is lower than the actual number of productively infected cells, NT, whereas Ne is greater than NT under the island model (64). For the metapopulation model, the ratio of effective population size to total population size is given by
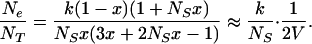 |
2 |
The approximation is for moderate amounts of turnover, x ≫ 1/NS, and V is the variance in fitness between subpopulations, which for our simple metapopulation model is given by V = x/(1 − x). Eq. 2 clearly illustrates that the more dramatic the founder effect and the higher the variance in fitness between subpopulations, the lower the effective population size is relative to the census size. Thus, if k = 2, x = 0.1, and NS = 100, Ne is approximately an order of magnitude lower than the actual population size and would be even lower if there were differences in productivity between subpopulations, which would increase V further.
Metapopulation structure could play a central role in the evolution of HIV populations within infected patients via its effect on Ne. Population subdivision can lead to a dramatic reduction in Ne when subpopulations turn over, because of the disproportionately high fitness of founders (64). Hence, local extinction of subpopulations could contribute to the highly variable way in which drug resistance evolves in different individuals (25, 26), even though the absolute number of infected cells is high (107–108) (4). Rouzine and Coffin (28) estimated the effective population size of HIV at 104–105 based on the frequency of all four combinations of mutations at variable sites in virus isolated from peripheral blood mononuclear cells, on the assumption that these combinations are produced by independent substitutions rather than recombination. We have demonstrated recombination at the local level, and although it is not known how recombination at a subpopulation level translates to the effective recombination rate in the body as a whole, estimates of Ne based on the level of linkage disequilibrium are likely to be overestimates. Nevertheless, metapopulation structure is unlikely to be the sole cause of differences between the actual and effective population sizes as estimates of Ne based on the level of diversity (24, 36) and the rate of divergence of the C2-V3 region of the env gene (27) are of the order 103–104, which is lower than could realistically be produced under our simple metapopulation model. Other factors, such as the process of “genetic draft” (61), where a reduction in effective population size arises because of selection on linked viral mutations, may play an important role.
The consequences of a metapopulation of infected cells extend beyond the impact on effective population size. Although they did not use the term “metapopulation,” Grossman et al. (65, 66) suggested that local short-lived bursts of replication could aid persistence of the virus during highly active antiretroviral therapy (HAART), and that changes in the dynamics of these bursts during therapy could account for the “biphasic” decay of virus in the blood observed during HAART. Metapopulation structure is imposed by the spatial structure of lymphoid tissues, which has been hypothesized to play a key role in determining how the immune system “chooses” an appropriate response against parasites (67), and the compartmentalization of specific immune cells in HIV infection (37) is also likely to have important implications for the way in which viral immune escape mutants evolve. Like viral infection of target cells, many other processes that play a role in viral replication, such as the effects of cytokines and bystander effects, (68, 69) are also local in nature, and an understanding of their role in viral dynamics may benefit from consideration of virally infected cells as a “population of populations.”
Acknowledgments
Thanks go to John Wakeley and two anonymous referees for comments on the manuscript. This work was funded in part by a Medical Research Council Fellowship (681/298) to S.D.W.F. and by an unrestricted gift from Roche Molecular Systems, Alameda, CA.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Wakeley, J., 8th Annual Discussion Meeting on HIV Dynamics and Evolution, Paris, April 2001.
Wang, Y., Marra, C. M., Learn, G. H., Rodrigo, A. G., Collier, A. C., Coombs, R. W., He, X., Zhao, H., & Mullins, J. I., Abstract no. 307, 7th Conference of Retroviruses and Opportunistic Infections, San Francisco, January 2000.
References
- 1.Mansky L M, Temin H M. J Virol. 1995;69:5087–5094. doi: 10.1128/jvi.69.8.5087-5094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ho D D, Neumann A U, Perelson A S, Chen W, Leonard J M, Markowitz M. Nature (London) 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 3.Wei X, Ghosh S K, Taylor M E, Johnson V A, Emini E A, Deutsch P, Lifson J D, Bonhoeffer S, Nowak M A, Hahn B H, et al. Nature (London) 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 4.Chun T W, Carruth L, Finzi D, Shen X, DiGiuseppe J A, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn T C, et al. Nature (London) 1997;387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 5.Simmonds P, Zhang L Q, McOmish F, Balfe P, Ludlam C A, Leigh Brown A J. J Virol. 1991;65:6266–6276. doi: 10.1128/jvi.65.11.6266-6276.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolfs T F W, Zwart G, Bakker M, Goudsmit J. Virology. 1992;189:103–110. doi: 10.1016/0042-6822(92)90685-i. [DOI] [PubMed] [Google Scholar]
- 7.Cichutek K, Merget H, Norley S, Linde R, Kreuz W, Gahr M, Kurth R. Proc Natl Acad Sci USA. 1992;89:7365–7369. doi: 10.1073/pnas.89.16.7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonhoeffer S, Holmes E C, Nowak M A. Nature (London) 1995;376:125. doi: 10.1038/376125a0. [DOI] [PubMed] [Google Scholar]
- 9.Leigh Brown A J, Cleland A. AIDS. 1996;10:1067–1073. [PubMed] [Google Scholar]
- 10.Liu S L, Schacker T, Musey L, Shriner D, McElrath M J, Corey L, Mullins J I. J Virol. 1997;71:4284–4295. doi: 10.1128/jvi.71.6.4284-4295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDonald R A, Mayers D L, Chung R C Y, Wagner K F, RattoKim S, Birx D L, Michael N L. J Virol. 1997;71:1871–1879. doi: 10.1128/jvi.71.3.1871-1879.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamaguchi Y, Gojobori T. Proc Natl Acad Sci USA. 1997;94:1264–1269. doi: 10.1073/pnas.94.4.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nielsen R, Yang Z. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Z H, Nielsen R, Goldman N, Pedersen A M K. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamaguchi-Kabata Y, Gojobori T. J Virol. 2000;74:4335–4350. doi: 10.1128/jvi.74.9.4335-4350.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frost, S. D. W., Günthard, H. F., Wong, J. K., Havlir, D., Richman, D. D. & Leigh Brown, A. J. (2001) Virology, in press. [DOI] [PubMed]
- 17.Boucher C A B, O'Sullivan E, Mulder J W, Ramautarsing C, Kellam P, Darby G, Lange J M A, Goudsmit J, Larder B A. J Infect Dis. 1992;165:105–110. doi: 10.1093/infdis/165.1.105. [DOI] [PubMed] [Google Scholar]
- 18.Condra J H, Holder D J, Schleif W A, Blahy O M, Danovich R M, Gabryelski L J, Graham D J, Laird D, Quintero J C, Rhodes A, et al. J Virol. 1996;70:8270–8276. doi: 10.1128/jvi.70.12.8270-8276.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Molla A, Korneyeva M, Gao Q, Vasavanonda S, Schipper P J, Mo H M, Markowitz M, Chernyavskiy T, Niu P, Lyons N, et al. Nat Med. 1996;2:760–766. doi: 10.1038/nm0796-760. [DOI] [PubMed] [Google Scholar]
- 20.Crandall K A, Kelsey C R, Imamichi H, Lane H C, Slazman N P. Mol Biol Evol. 1999;16:372–382. doi: 10.1093/oxfordjournals.molbev.a026118. [DOI] [PubMed] [Google Scholar]
- 21.Havlir D V, Eastman S, Gamst A, Richman D D. J Virol. 1996;70:7894–7899. doi: 10.1128/jvi.70.11.7894-7899.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eastman P S, Mittler J, Kelso R, Gee C, Boyer E, Kolberg J, Urdea M, Leonard J M, Norbeck D W, Mo H, Markowitz M. J Virol. 1998;72:5154–5164. doi: 10.1128/jvi.72.6.5154-5164.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frost S D W, Nijhuis M, Schuurman R, Boucher C A B, Leigh Brown A J. J Virol. 2000;74:6262–6268. doi: 10.1128/jvi.74.14.6262-6268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nijhuis M, Boucher C A B, Schipper P, Leitner T, Schuurman R, Albert J. Proc Natl Acad Sci USA. 1998;95:14441–14446. doi: 10.1073/pnas.95.24.14441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leigh Brown A J, Richman D D. Nat Med. 1997;3:268–271. doi: 10.1038/nm0397-268. [DOI] [PubMed] [Google Scholar]
- 26.Leigh Brown A J. Proc Natl Acad Sci USA. 1997;94:1862–1865. doi: 10.1073/pnas.94.5.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodrigo A G, Shpaer E G, Delwart E L, Iversen A K N, Gallo M V, Brojatsch J, Hirsch M S, Walker B D, Mullins J I. Proc Natl Acad Sci USA. 1999;96:2187–2191. doi: 10.1073/pnas.96.5.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rouzine I M, Coffin J M. Proc Natl Acad Sci USA. 1999;96:10758–10763. doi: 10.1073/pnas.96.19.10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ribeiro R M, Bonhoeffer S. Proc Natl Acad Sci USA. 2000;97:7681–7686. doi: 10.1073/pnas.97.14.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coffin J M. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- 31.Epstein L G, Kuiken C, Blumberg B M, Hartman S, Sharer L R, Clement M, Goudsmit J. Virology. 1991;180:583–590. doi: 10.1016/0042-6822(91)90072-j. [DOI] [PubMed] [Google Scholar]
- 32.Delassus S, Meyerhans A, Cheynier R, Wain-Hobson S. Virology. 1992;188:811–818. doi: 10.1016/0042-6822(92)90536-x. [DOI] [PubMed] [Google Scholar]
- 33.Hughes E S, Bell J E, Simmonds P. J Gen Virol. 1997;78:2871–2882. doi: 10.1099/0022-1317-78-11-2871. [DOI] [PubMed] [Google Scholar]
- 34.Wong J K, Ignacio C C, Torriani F, Havlir D, Fitch N J S, Richman D D. J Virol. 1997;71:2059–2071. doi: 10.1128/jvi.71.3.2059-2071.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schrager L K, D'Souza M P. J Am Med Assoc. 1998;280:67–71. doi: 10.1001/jama.280.1.67. [DOI] [PubMed] [Google Scholar]
- 36.Kashuba A D M, Dyer J R, Kramer L M, Raasch R H, Eron J J, Cohen M S. Antimicrob Agents Chemother. 1999;43:1817–1826. doi: 10.1128/aac.43.8.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheynier R, Henrichwark S, Hadida F, Pelletier E, Oksenhendler E, Autran B, Wain Hobson S. Cell. 1994;78:373–387. doi: 10.1016/0092-8674(94)90417-0. [DOI] [PubMed] [Google Scholar]
- 38.Gratton S, Cheynier R, Dumaurier M-J, Oksenhendler E, Wain- Hobson S. Proc Natl Acad Sci USA. 2000;97:14566–14571. doi: 10.1073/pnas.97.26.14566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Levins R. Bull Ent Soc Am. 1969;5:237–240. [Google Scholar]
- 40.Thomson J D, Higgins D G, Gibson T J. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wright S. Ann Eugen. 1951;15:323–354. doi: 10.1111/j.1469-1809.1949.tb02451.x. [DOI] [PubMed] [Google Scholar]
- 42.Schneider S, Roessli D, Excoffier L. arlequin: A Software for Population Genetics Analysis. Department of Anthropology, University of Geneva, Geneva: Genetics and Biometry Laboratory; 2000. [Google Scholar]
- 43.Jukes T H, Cantor C R. In: Mammalian Protein Metabolism. Munro H N, editor. New York: Academic; 1969. pp. 21–132. [Google Scholar]
- 44.Rozas J, Rozas R. Bioinformatics. 1999;15:174–175. doi: 10.1093/bioinformatics/15.2.174. [DOI] [PubMed] [Google Scholar]
- 45.Tajima F. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simonsen K L, Churchill G A, Aquadro C F. Genetics. 1995;141:413–429. doi: 10.1093/genetics/141.1.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Misawa K, Tajima F. Genetics. 1997;147:1959–1964. doi: 10.1093/genetics/147.4.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leitner T, Kumar S, Albert J. J Virol. 1997;71:4761–4770. doi: 10.1128/jvi.71.6.4761-4770.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sawyer S A. geneconv: A Computer Package for the Statistical Detection of Gene Conversion. Washington University, St. Louis, MO: Department of Mathematics; 1999. [Google Scholar]
- 50.Hudson R R, Kaplan N L. Genetics. 1985;111:147–164. doi: 10.1093/genetics/111.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Slatkin M. Theor Popul Biol. 1977;12:253–262. doi: 10.1016/0040-5809(77)90045-4. [DOI] [PubMed] [Google Scholar]
- 52.Pannell J R, Charlesworth B. Evolution (Lawrence, KS) 1999;53:664–676. doi: 10.1111/j.1558-5646.1999.tb05362.x. [DOI] [PubMed] [Google Scholar]
- 53.Hanski I, Gilpin M E, editors. Metapopulation Biology: Ecology, Genetics, and Evolution. San Diego: Academic; 1997. [Google Scholar]
- 54.Pannell J R, Charlesworth B. Philos Trans R Soc London B. 2000;355:1851–1864. doi: 10.1098/rstb.2000.0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McNearney T, Hornickova Z, Markham R, Birdwell A, Arens M, Saah A, Ratner L. Proc Natl Acad Sci USA. 1992;89:10247–10251. doi: 10.1073/pnas.89.21.10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu T F, Hunag Y X, Wang N, Connor R, Cao Y Z, Ho D D. J Cell Biochem. 1993;S17E:25. [Google Scholar]
- 57.Zhang L Q, Mackenzie P, Cleland A, Holmes E C, Leigh Brown A J, Simmonds P. J Virol. 1993;67:3345–3356. doi: 10.1128/jvi.67.6.3345-3356.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Long E M, Martin H L, Kreiss J K, Rainwater S M J, Lavreys L, Jackson D J, Rakwar J, Mandaliya K, Overbaugh J. Nat Med. 2000;6:71–75. doi: 10.1038/71563. [DOI] [PubMed] [Google Scholar]
- 59.Charlesworth B, Morgan M T, Charlesworth D. Genetics. 1993;134:1289–1303. doi: 10.1093/genetics/134.4.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crandall K A, Vasco D A, Posada D, Imamichi H. AIDS. 1999;13:S39–S47. [PubMed] [Google Scholar]
- 61.Gillespie J H. Genetics. 2000;155:909–919. doi: 10.1093/genetics/155.2.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Golding G B. In: Progress in Population Genetics and Human Evolution. Donnelly P, Tavaré S, editors. New York: Springer; 1997. pp. 271–285. [Google Scholar]
- 63.Shapshak P, Segal D M, Crandall K A, Fujimura R K, Zhang B T, Xin K Q, Okuda K, Petito C K, Eisdorfer C, Goodkin K. AIDS Res Hum Retr. 1999;15:811–820. doi: 10.1089/088922299310719. [DOI] [PubMed] [Google Scholar]
- 64.Whitlock M C, Barton N H. Genetics. 1997;146:427–441. doi: 10.1093/genetics/146.1.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grossman Z, Feinberg M B, Paul W E. Proc Natl Acad Sci USA. 1998;95:6314–6319. doi: 10.1073/pnas.95.11.6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grossman Z, Polis M, Feinberg M B, Grossman Z, Levi I, Jankelevich S, Yarchoan R, Boon J, de Wolf F, Lange J M, et al. Nat Med. 1999;5:1099–1104. doi: 10.1038/13410. [DOI] [PubMed] [Google Scholar]
- 67.Segel L A, Bar-Or R L. J Immunol. 1999;163:1342–1349. [PubMed] [Google Scholar]
- 68.Frost S D W, Michie C A. Trends Microbiol. 1996;4:77–82. doi: 10.1016/0966-842X(96)81516-2. [DOI] [PubMed] [Google Scholar]
- 69.Krakauer D C, Nowak M A. Proc R Soc London Ser B. 1999;266:1069–1075. doi: 10.1098/rspb.1999.0745. [DOI] [PMC free article] [PubMed] [Google Scholar]