

Abstract
We used complementation analysis as a probe for the detection of genetic heterogeneity within a single locus affected in a human disease, argininosuccinate lyase (L-argininosuccinate arginine-lyase, EC 4.3.2.1) deficiency. Fibroblasts cultured from 28 unrelated patients were fused in all possible pairwise combinations, and the argininosuccinate lyase activity in heterokaryons was assayed by measuring the incorporation of 14C from L-[ureido-14C]citrulline into acid-precipitable material. Partial complementation was observed in fusions involving 20 of the 28 strains, with the lyase activity increasing from 2- to 10-fold. Thirteen of the mutants were identified by the complementation analysis as being phenotypically unique. Of the 20 complementing strains, 3 were remarkable because they participated in all but 2 of the 32 positive complementation tests; 2 others constituted a unique subgroup that produced the highest increases in argininosuccinate lyase activity of all fusions. The 8 strains that did not complement any others consisted of two types: 3 mutants with the highest residual argininosuccinate lyase activity of all strains and 5 mutants with low residual activity. All of the mutants mapped to a single major complementation group. The data could be summarized as a circular complementation map with an attached linear tail, the mutants being distributed among 12 subgroups in a complex pattern. We conclude that all of these mutants are affected at a single locus, that extensive genetic heterogeneity is present in the mutant population, and that the affected locus in argininosuccinate lyase deficiency is likely to be the structural gene coding for that enzyme.
Full text
PDF



Images in this article
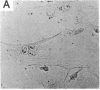
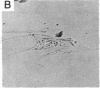

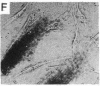
Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- COSTELLO W. P., BEVAN E. A. COMPLEMENTATION BETWEEN AD-5/7 ALLELES IN YEAST. Genetics. 1964 Dec;50:1219–1230. doi: 10.1093/genetics/50.6.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathelineau L., Pham Dinh D., Briand P., Kamoun P. Studies on complementation in argininosuccinate synthetase and argininosuccinate lyase deficiencies in human fibroblasts. Hum Genet. 1981;57(3):282–284. doi: 10.1007/BF00278945. [DOI] [PubMed] [Google Scholar]
- DORFMAN B. ALLELIC COMPLEMENTATION AT THE AD-5/7 LOCUS IN YEAST. Genetics. 1964 Dec;50:1231–1243. doi: 10.1093/genetics/50.6.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FINCHAM J. R., STADLER D. R. COMPLEMENTATION RELATIONSHIP OF NEUROSPORA AM MUTANTS IN RELATION TO THEIR FORMATION OF ABNORMAL VARIETIES OF GLUTAMATE DEHYDROGENASE. Genet Res. 1965 Feb;6:121–129. doi: 10.1017/s0016672300003980. [DOI] [PubMed] [Google Scholar]
- Galjaard H., Hoogeveen A., de Wit-Verbeek H. A., Reuser A. J., Keijzer W., Westerveld A., Bootsma D. Tay-Sachs and Sandhoff's disease: intergenic complementation after somatic cell hybridization. Exp Cell Res. 1974 Aug;87(2):444–448. doi: 10.1016/0014-4827(74)90515-1. [DOI] [PubMed] [Google Scholar]
- Glick N. R., Snodgrass P. J., Schafer I. A. Neonatal argininosuccinic aciduria with normal brain and kidney but absent liver argininosuccinate lyase activity. Am J Hum Genet. 1976 Jan;28(1):22–30. [PMC free article] [PubMed] [Google Scholar]
- Gravel R. A., Lam K. F., Scully K. J., Hsia Y. Genetic complementation of propionyl-CoA carboxylase deficiency in cultured human fibroblasts. Am J Hum Genet. 1977 Jul;29(4):378–388. [PMC free article] [PubMed] [Google Scholar]
- Gravel R. A., Mahoney M. J., Ruddle F. H., Rosenberg L. E. Genetic complementation in heterokaryons of human fibroblasts defective in cobalamin metabolism. Proc Natl Acad Sci U S A. 1975 Aug;72(8):3181–3185. doi: 10.1073/pnas.72.8.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill H. Z., Goodman S. I. Detection of inborn errors of metabolism. III. Defects in urea cycle metabolism. Clin Genet. 1974;6(2):79–81. [PubMed] [Google Scholar]
- Jacoby L. B., Littlefield J. W., Milunsky A., Shih V. E., Wilroy R. S., Jr A microassay for argininosuccinase in cultured cells. Am J Hum Genet. 1972 May;24(3):321–324. [PMC free article] [PubMed] [Google Scholar]
- Kraemer K. H., De Weerd-Kastelein E. A., Robbins J. H., Keijzer W., Barrett S. F., Petinga R. A., Bootsma D. Five complementation groups in xeroderma pigmentosum. Mutat Res. 1975 Dec;33(2-3):327–340. doi: 10.1016/0027-5107(75)90208-0. [DOI] [PubMed] [Google Scholar]
- Nadler H. L., Chacko C. M., Rachmeler M. Interallelic complementation in hybrid cells derived from human diploid strains deficient in galactose-1-phosphate uridyl transferase activity. Proc Natl Acad Sci U S A. 1970 Oct;67(2):976–982. doi: 10.1073/pnas.67.2.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien W. E., Barr R. H. Argininosuccinate lyase: purification and characterization from human liver. Biochemistry. 1981 Mar 31;20(7):2056–2060. doi: 10.1021/bi00510a049. [DOI] [PubMed] [Google Scholar]
- Palekar A. G., Mantagos S. Human liver arginiosuccinase purification and partial characterization. J Biol Chem. 1981 Sep 10;256(17):9192–9194. [PubMed] [Google Scholar]
- Perry T. L., Wirtz M. L., Kennaway N. G., Hsia Y. E., Atienza F. C., Uemura H. S. Amino acid and enzyme studies of brain and other tissues in an infant with argininosuccinic aciduria. Clin Chim Acta. 1980 Aug 4;105(2):257–267. doi: 10.1016/0009-8981(80)90468-4. [DOI] [PubMed] [Google Scholar]
- Rattazzi M. C., Brown J. A., Davidson R. G., Shows T. B. Studies on complementation of beta hexosaminidase deficiency in human GM2 gangliosidosis. Am J Hum Genet. 1976 Mar;28(2):143–154. [PMC free article] [PubMed] [Google Scholar]
- Robertson A., Hill W. G. Identity of different mutations for deleterious genes. Nature. 1983 Jan 13;301(5896):176–177. doi: 10.1038/301176a0. [DOI] [PubMed] [Google Scholar]
- Stanners C. P., Eliceiri G. L., Green H. Two types of ribosome in mouse-hamster hybrid cells. Nat New Biol. 1971 Mar 10;230(10):52–54. doi: 10.1038/newbio230052a0. [DOI] [PubMed] [Google Scholar]
- Welply J. K., Fowler A. V., Zabin I. beta-Galactosidase alpha-complementation. Overlapping sequences. J Biol Chem. 1981 Jul 10;256(13):6804–6810. [PubMed] [Google Scholar]
- Willard H. F., Mellman I. S., Rosenberg L. E. Genetic complementation among inherited deficiencies of methylmalonyl-CoA mutase activity: evidence for a new class of human cobalamin mutant. Am J Hum Genet. 1978 Jan;30(1):1–13. [PMC free article] [PubMed] [Google Scholar]
- Zabin I., Villarejo M. R. Protein complementation. Annu Rev Biochem. 1975;44:295–313. doi: 10.1146/annurev.bi.44.070175.001455. [DOI] [PubMed] [Google Scholar]