

Abstract
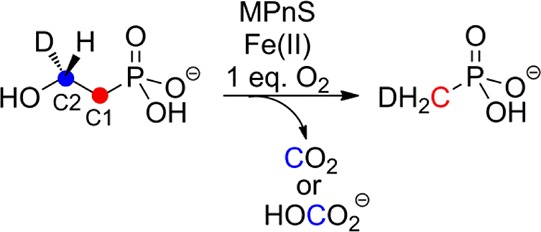
Methylphosphonate synthase is a non-heme iron-dependent oxygenase that converts 2-hydroxyethylphosphonate (2-HEP) to methylphosphonate. On the basis of experiments with two enantiomers of a substrate analog, 2-hydroxypropylphosphonate, catalysis is proposed to commence with stereospecific abstraction of the pro-S hydrogen on C2 of the substrate. Experiments with isotopologues of 2-HEP indicate stereospecific hydrogen transfer of the pro-R hydrogen at C2 of the substrate to the methyl group of methylphosphonate. Kinetic studies with these substrate isotopologues reveal that neither hydrogen transfer is rate limiting under saturating substrate conditions. A mechanism is proposed that is consistent with the available data.
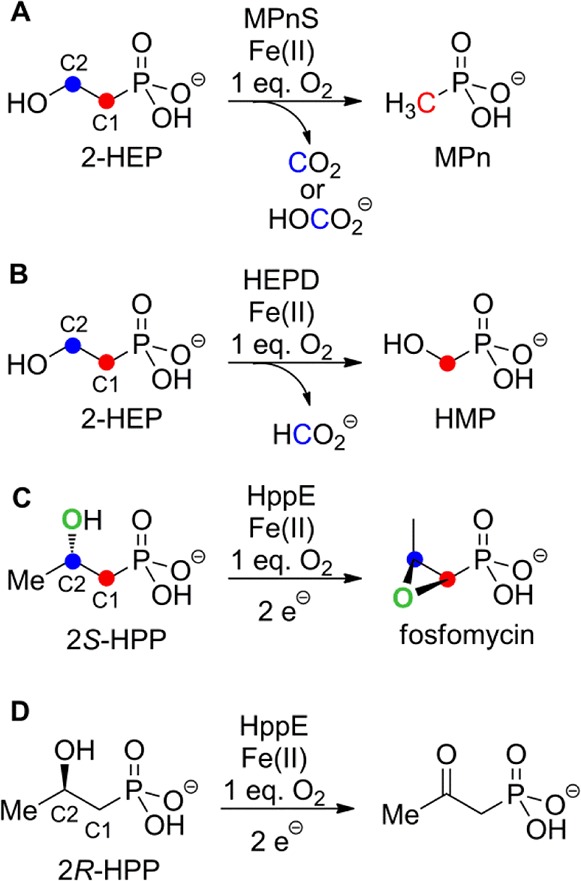
Phosphonate natural products are characterized by a stable carbon–phosphorus bond and are produced by a variety of organisms.1 The first example of phosphonate biosynthesis by an archaeal species was recently reported for Nitrosopumilus maritimus, a highly abundant marine organism that has been linked to the carbon and phosphorus cycles in the ocean.2N. maritimus was shown to produce methylphosphonate (MPn) from PEP in a four-step enzymatic process. The last reaction in this sequence is the conversion of 2-hydroxyethylphosphonate (2-HEP) to MPn catalyzed by methylphosphonate synthase (MPnS, Scheme 1A).2 MPnS has sequence homology with 2-hydroxyethylphosphonate dioxygenase (HEPD), an enzyme that converts 2-HEP to hydroxymethylphosphonate (HMP) during the biosynthesis of the commercial herbicide phosphinothricin (Scheme 1B).3−7 The two enzymes share low sequence identity (21%), but key common residues include two His that are part of the canonical 2-His-1-carboxylate facial triad8 in HEPD (Figure S1). The Asp/Glu of this triad in HEPD is not conserved in MPnS and the identity of its third metal ligand, if any, has yet to be identified. Neither enzyme requires external electrons for catalysis, but whereas HEPD transfers the oxygen atoms from O2 to C1 and C2 of 2-HEP,4 MPnS oxidizes only the C2 carbon of its substrate. This unprecedented reaction requires the removal of both hydrogens at C2. In addition to the unusual nature of the chemistry catalyzed by MPnS, the enzyme may also represent a key step in methane production in the oceans.2 A third enzyme that is structurally related to HEPD is 2-hydroxypropylphosphonate epoxidase (HppE), which removes the pro-R hydrogen from C1 of 2S-hydroxypropylphosphonate to achieve the unusual dehydrogenative epoxidation reaction shown in Scheme 1C during the biosynthesis of fosfomycin.9−11
Scheme 1. Reactions Catalyzed by MPnS, HEPD, and HppE.
To understand the mechanism of MPnS, first the ratio of O2 consumed to MPn produced was determined. An assay mixture containing 1 μM MPnS, which was anaerobically reconstituted with 1 equiv of Fe(II), was allowed to equilibrate in the chamber of a Clark-type oxygen electrode. Catalysis was initiated by the addition of 100 μM 2-HEP, and the consumption of oxygen was monitored. An aliquot of the assay mixture was removed once about 25 μM O2 had been consumed. The sample was quenched with sulfuric acid and the mixture was analyzed by LC-MS to determine the amount of MPn produced using a standard curve (Figure S2). The ratio of MPn produced to O2 consumed was 1.05 ± 0.07 based on three independent experiments.
With the stoichiometry of O2 consumption established, kinetic parameters were determined. The dependence of the rate of O2 consumption on the concentration of O2 was determined by a continuous assay using the oxygen electrode, establishing a Km,oxygen of 11 ± 3 μM and a kcat of 0.25 ± 0.03 s–1 at a concentration of 500 μM 2-HEP (Figure S3). The dependence of O2 consumption on the concentration of 2-HEP determined by initial rate measurements also exhibited Michaelis–Menten kinetics with a Km of 4.5 μM for 2-HEP and a kcat of 0.18 ± 0.01 s–1 at an oxygen concentration of 260 μM (Table 1, Figure S4). To provide information on the timing of the removal of the two hydrogens at C2 and to investigate whether either hydrogen transfer occurred in a (partially) rate-limiting step, the kinetics of three previously synthesized isotopologues,4,7 (R)- and (S)-[2-2H1]-HEP, and 2-[2-2H2]-HEP were determined (Figure S3). The isotopologues displayed rates at saturating 2-HEP concentrations that were not significantly different from that of unlabeled 2-HEP at close to identical O2 concentrations (250 μM), indicating that hydrogen transfers do not contribute substantially to the overall rate equation for kcat under these conditions. The errors on Km are relatively large,12 making it difficult to draw strong conclusions, but the values of kcat/Km suggest that there may be a small kinetic isotope effect (KIE) on the bimolecular rate constant for both enantiomers of 2-[2H]-2-HEP. One possible explanation for these observations is that the rate of kcat may be limited by product release, whereas the steps involved in removal of each of the hydrogen atoms at C2 contribute to kcat/Km.
Table 1. Kinetic Constants of MPnS with 2-HEP and Its Isotopologues.
| substrate | Km,HEP (μM) | kcat (s–1) | kcat/Km,HEP(M–1 s–1) |
|---|---|---|---|
| 2-HEP | 4.5 ± 1.1 | 0.18 ± 0.01 | (4.0 ± 1.0) × 104 |
| (R)-[2-2H1]-HEP | 7.1 ± 0.8 | 0.17 ± 0.01 | (2.4 ± 0.3) × 104 |
| (S)-[2-2H1]-HEP | 6.5 ± 1.5 | 0.17 ± 0.01 | (2.6 ± 0.6) × 104 |
| 2-[2-2H2]-HEP | 7.2 ± 1.4 | 0.17 ± 0.01 | (2.4 ± 0.5) × 104 |
To obtain additional information regarding the fate of the two hydrogen atoms at C2 of 2-HEP, MPnS was incubated with each enantiomer of 2-hydroxypropylphosphonate (HPP)13 under identical conditions (50 μM enzyme/1.5 mM (2R)- or (2S)-HPP) and oxygen consumption was monitored. Negligible oxygen consumption was observed with (2S)-HPP, suggesting that this enantiomer is not a substrate for MPnS. In contrast, when (2R)-HPP was presented as a substrate, oxygen consumption was observed but the activity ceased well before complete consumption of the substrate. Furthermore, the recovered enzyme was inactive toward 2-HEP. These findings suggest a time-dependent inactivation of MPnS by (2R)-HPP. Next, an assay containing 1.5 mM (2R)-HPP and 0.5 mM MPnS was analyzed by 31P NMR spectroscopy, revealing three new resonances (Figure 1). Nearly all of the substrate was converted to 2-oxopropylphosphonate (OPP; 10.8 ppm), with small amounts of MPn (23.1 ppm) and inorganic phosphate (Pi, 3 ppm) also formed. Very little substrate remained (20.3 ppm). The identity of all products was confirmed by spiking with authentic synthetic materials.4,5 Oxidation of an alcohol group in a substrate analog to a ketone is also observed with HppE (Scheme 1D)11,14 and HEPD.5 The fact that only the R enantiomer of 2-HPP is a substrate for MPnS suggests that the pro-S hydrogen on C2 of 2-HEP is removed initially, consistent with a similar first step of HEPD. The stereochemical removal of the pro-S hydrogen of 2-HEP is also congruent with the X-ray structure of HEPD4 and the sequence homology of HEPD with MPnS (Figure S1). (2S)-HPP is likely not a substrate because the hydrogen is not positioned correctly in the active site for abstraction.
Figure 1.

31P NMR spectrum of the products from incubation of (2R)-HPP with MPnS.
Next, the fate of the pro-R hydrogen on C2 of 2-HEP was determined. The conversion of 2-HEP to MPn requires the acquirement of a proton or hydrogen atom at C1 during or following C–C bond cleavage. This hydrogen can come either from bulk solvent (or enzyme) or from the substrate itself. Both possibilities were examined by assaying MPnS in 99.9% D2O with 2-HEP, and in H2O with (S)-[2-2H1]-HEP or (R)-[2-2H1]-HEP. The resulting MPn products were analyzed by FT-MS to determine whether 2H1-MPn (m/zcalc = 95.99663) or unlabeled MPn (m/zcalc = 94.99036) was produced (Figure 2). The assays containing D2O and (S)-[2-2H1]-HEP produced unlabeled MPn, signifying that the new hydrogen on the carbon of MPn does not originate from solvent (or solvent exchangeable residues on the enzyme) or from the pro-S hydrogen on C2 of 2-HEP. In the assay containing (R)-[2-2H1]-HEP, however, only deuterium-labeled MPn was observed by FT-MS (Figure 2B), indicating that the pro-R hydrogen of 2-HEP is transferred to MPn during catalysis.1531P NMR analysis of the MPn formed confirmed transfer of the pro-R hydrogen on C2 of 2-HEP to the methyl group of the MPn product (Figure S5).
Figure 2.

FT-MS analysis (negative mode) after incubation of MPnS with (A) (S)-[2-2H1]-HEP in H2O, (B) 2-HEP in D2O, and (C) (R)-[2-2H1]-HEP in H2O, to determine the origin of the third hydrogen on methylphosphonate (MPn). The ion at m/z 95.02508 in panel C is not unlabeled MPn but an unknown impurity.15 These MS data are corroborated by 31P NMR analyses of the products (Figure S5).
The experiments described here allow a proposal for the mechanism of MPnS (Scheme 2). Following substrate binding, O2 activation results in the formation of a ferric-superoxo species (I), which is poised to stereospecifically abstract the pro-S hydrogen on C2 of 2-HEP.16,17 The radical anion (II) then transfers an electron to the Fe(III), generating phosphonoacetaldehyde ligated to the iron;18 these two steps could also occur in a concerted process with electron transfer commencing during hydrogen atom abstraction. Next, nucleophilic attack by the hydroperoxide onto the carbonyl would result in a bridged alkylperoxo intermediate (III). Subsequent homolytic O–O bond cleavage generates a gem-diol radical,18 which results in C–C bond cleavage, forming a MPn radical and formate bound to the ferric-iron center (IV). The formation of this radical species has also been proposed for HEPD,18 and is supported by recent studies showing racemization at the C1 carbon during conversion to HMP.7 In HEPD, the MPn radical is believed to combine with the ferric hydroxide in a step reminiscent of the rebound step during metal-catalyzed hydroxylations. In MPnS, however, the MPn radical is proposed to abstract the hydrogen atom on formate, resulting in MPn and a CO2 radical anion. The latter is a very strong reductant19 that can reduce the metal center back to its ferrous state, preparing the enzyme for another round of catalysis following release of CO2 and MPn. Alternatively, hydrogen atom abstraction from formate and electron transfer to the Fe(III) could occur simultaneously. Understanding the determinants that govern the different fates of the MPn radical for each enzyme will require structural information for MPnS.
Scheme 2. Proposed Mechanism for MPnS-Catalyzed Conversion of 2-HEP to MPn.

In summary, this study provides the first mechanistic insights into catalysis by methylphosphonate synthase. Our data suggest that the pro-S hydrogen is removed first and show that the pro-R hydrogen is transferred from C2 of 2-HEP to the methyl group of MPn. Neither step is rate limiting under saturating substrate concentrations. Further elucidation of the details of the early steps requires either spectroscopic characterization of the intermediates16 or possibly a combination of 2H and 18O-kinetic isotope effects.20,21 MPnS is another addition to the impressive array of transformations catalyzed by non-heme iron-dependent enzymes.8,22
Acknowledgments
This work was supported by the National Institutes of Health (PO1 GM077596 to W.A.v.d.D.) and the Robert C. and Carolyn J. Springborn Endowment (predoctoral fellowship to S.C.P.). H.A.C was supported by an NRSA postdoctoral fellowship (F32 GM 95024-01).
Supporting Information Available
Experimental procedures, Supporting Figures, and kinetic data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Metcalf W. W.; van der Donk W. A. Annu. Rev. Biochem. 2009, 78, 65–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf W. W.; Griffin B. M.; Cicchillo R. M.; Gao J.; Janga S.; Cooke H. A.; Circello B. T.; Evans B. S.; Martens-Habbena W.; Stahl D. A.; van der Donk W. A. Science 2012, 337, 1104–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z.; Blodgett J. A.; Circello B. T.; Eliot A. C.; Woodyer R.; Li G.; van der Donk W. A.; Metcalf W. W.; Zhao H. J. Biol. Chem. 2008, 283, 23161–23168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchillo R. M.; Zhang H.; Blodgett J. A. V.; Whitteck J. T.; Li G.; Nair S. K.; van der Donk W. A.; Metcalf W. W. Nature 2009, 459, 871–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitteck J. T.; Cicchillo R. M.; van der Donk W. A. J. Am. Chem. Soc. 2009, 131, 16225–16232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blodgett J. A.; Thomas P. M.; Li G.; Velasquez J. E.; van der Donk W. A.; Kelleher N. L.; Metcalf W. W. Nat. Chem. Biol. 2007, 3, 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitteck J. T.; Malova P.; Peck S. C.; Cicchillo R. M.; Hammerschmidt F.; van der Donk W. A. J. Am. Chem. Soc. 2011, 133, 4236–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costas M.; Mehn M. P.; Jensen M. P.; Que L. Jr. Chem. Rev. 2004, 104, 939–986. [DOI] [PubMed] [Google Scholar]
- Liu P.; Murakami K.; Seki T.; He X.; Yeung S. M.; Kuzuyama T.; Seto H.; Liu H. W. J. Am. Chem. Soc. 2001, 123, 4619–4620. [DOI] [PubMed] [Google Scholar]
- Higgins L. J.; Yan F.; Liu P.; Liu H. W.; Drennan C. L. Nature 2005, 437, 838–844. [DOI] [PubMed] [Google Scholar]
- Yun D.; Dey M.; Higgins L. J.; Yan F.; Liu H. W.; Drennan C. L. J. Am. Chem. Soc. 2011, 133, 11262–11269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Km values for 2-HEP and its isotopologues are small and difficult to determine by initial rate measurements using the oxygen electrode because of the inherently small readings and limits in the detection sensitivity. The Km values were consistently larger for the labeled compounds in independent experiments, but conclusions based on the apparent KIEs on kcat/Km must be considered tentative.
- Hammerschmidt F. Monatsh. Chem. 1991, 122, 389–398. [Google Scholar]
- Zhao Z. B.; Liu P. H.; Murakami K.; Kuzuyama T.; Seto H.; Liu H. W. Angew. Chem., Int. Ed. 2002, 41, 4529–4532. [DOI] [PubMed] [Google Scholar]
- Observed mass of 95.02508 is outside of the range of error (10 ppm) for the calculated mass of MPn.
- van der Donk W. A.; Krebs C.; Bollinger J. M. Jr. Curr. Opin. Struct. Biol. 2010, 20, 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollinger J. M. Jr.; Diao Y.; Matthews M. L.; Xing G.; Krebs C. Dalton Trans. 2009, 905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao H.; Morokuma K. J. Am. Chem. Soc. 2010, 132, 17901–17909. [DOI] [PubMed] [Google Scholar]
- Stubbe J.; van der Donk W. A. Chem. Rev. 1998, 98, 705–762. [DOI] [PubMed] [Google Scholar]
- Roth J. P. Curr. Opin. Chem. Biol. 2007, 11, 142–150. [DOI] [PubMed] [Google Scholar]
- Mirica L. M.; McCusker K. P.; Munos J. W.; Liu H. W.; Klinman J. P. J. Am. Chem. Soc. 2008, 130, 8122–8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovaleva E. G.; Lipscomb J. D. Nat. Chem. Biol. 2008, 4, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.