

Abstract
Objective
To identify disease-causing mutations in two consanguineous Pakistani families with fundus albipunctatus.
Methods
Affected individuals in both families underwent a thorough clinical examination including funduscopy and electroretinography. Blood samples were collected from all participating members and genomic DNA was extracted. Exclusion analysis was completed with microsatellite short tandem repeat markers that span all reported loci for fundus albipunctatus. Two-point logarithm of odds (LOD) scores were calculated, and coding exons and exon–intron boundaries of RLBP1 were sequenced bi-directionally.
Results
The ophthalmic examination of affected patients in both families was consistent with fundus albipunctatus. The alleles of markers on chromosome 15q flanking RLBP1 segregated with the disease phenotype in both families and linkage was further confirmed by two-point LOD scores. Bi-directional sequencing of RLBP1 identified a nonsense mutation (R156X) and a missense mutation (G116R) that segregated with the disease phenotype in their respective families.
Conclusions
These results strongly suggest that mutations in RLBP1 are responsible for fundus albipunctatus in the affected individuals of these consanguineous Pakistani families.
INTRODUCTION
Fundus albipunctatus (FA) is a rare form of apparently stationary night blindness that is also characterised by the presence of symmetrical round white dots in the fundus with a greater concentration in the midperiphery but with normal retinal arterioles, disc and visual fields.1 In FA patients, electroretinogram (ERG) recordings yield poor responses after retinal bleaching and profoundly delayed dark adaptation improves upon prolonged exposure to darkness.1 A clinically distinct disorder, retinitis punctata albescens (RPA) is characterised by nyctalopia, reduced visual acuity, multiple round white deposits in the retina, progressive attenuation of retinal arterioles, and non-detectable or severely reduced ERG recordings.2
RPA and FA are genetically heterogeneous disorders: mutations in RLBP1 and occasionally in RHO, RDS and RDH5 have been causally associated with RPA, whereas mutations in RLBP1 and RDH5 have been identified with patients diagnosed with FA.3–10 Previously, Katsanis and colleagues reported a missense mutation in RLBP1 (R150Q) that segregated with the disease phenotype in a Saudi family with both RPA and FA.10
RLBP1 encodes a 317 amino acid cytosolic cellular retinaldehyde binding protein (CRALBP) that has been reported in the developing ciliary body and iris.11,12 CRALBP has been localised in adult species to the retinal pigment epithelium, Müller cells of neural retina and ocular ciliary epithelium.13–15 It can select 11-cis-retinaldehyde from a mixture of retinoids and protect it from photoisomerisation, suggesting a possible role in the generation of 11-cis-retinoids in the visual cycle.16
Here we report on two consanguineous families from the Punjab province of Pakistan that included multiple affected individuals diagnosed with FA. Exclusion analyses with closely spaced short tandem repeat (STR) markers localised the critical interval to a region on chromosome 15q that harbours RLBP1. Sequencing identified novel mutations that segregated with the disease phenotype in their respective families and were not present in ethnically matched controls.
METHODS
Clinical assessment
In total, 125 consanguineous Pakistani families with inherited retinal dystrophies were recruited to participate in a collaborative study between the National Centre of Excellence in Molecular Biology (NCEMB), Lahore, Pakistan, and the National Eye Institute, Bethesda, Maryland, USA. A detailed medical history was obtained by interviewing family members. Fundus photographs were obtained at the Layton Rahmatulla Benevolent Trust Hospital, Lahore. ERG measurements were recorded with equipment manufactured by LKC (Gaithersburg, Maryland, USA). Rod responses were determined through an incident flash attenuated by −25 dB and the rod–cone response was measured at 0 dB. Isolated cone responses were recorded at 0 dB with a 30 Hz flicker over a background illumination of 17–34 cd/m2. Blood samples were collected from all participating members and genomic DNA was extracted by the non-organic method described previously.17,18
Genotype analysis
Exclusion analyses were performed with highly polymorphic fluorescent STR markers spanning all reported loci for FA. A multiplex PCR was completed in a GeneAmp PCR System 9700 thermocycler (Applied Biosystems, Foster city, CA, USA) as described previously.19,20 PCR products from each DNA sample were pooled and mixed with a loading cocktail containing HD-400 size standards (Applied Biosystems). The resulting PCR products were separated in an ABI 3100 DNA Analyzer (Applied Biosystems) and genotypes were assigned with GeneMapper software (Applied Biosystems).
Linkage analysis
Two-point linkage analyses were performed using the FASTLINK version of MLINK from the LINKAGE Program Package (provided in the public domain by the Human Genome Mapping Project Resources Centre, Cambridge, UK).21,22 Maximum two-point LOD scores were calculated using ILINK. Autosomal recessive FA was analysed as a fully penetrant trait with an affected allele frequency of 0.001. The marker order and distances between the markers were obtained from the Marshfield database (http://research.marshfieldclinic.org/genetics/) and the National Center for Biotechnology Information chromosome 15 sequence maps.
Mutation screening
Primer pairs for individual exons were designed using the Primer3 program (http://primer3.sourceforge.net/). The sequences and annealing temperatures are available upon request. Amplifications were performed in 25 μl reactions as described previously.19,20 PCR products were separated on a 2% agarose gel and purified by ethanol precipitation. The PCR primers for each exon were used for bidirectional sequencing using BigDye Terminator Ready reaction mix (Applied Biosystems), according to the manufacturer's instructions. Sequencing products were resuspended in 10 μl of formamide (Applied Biosystems) and denatured at 95°C for 5 min. The sequencing products were separated in an ABI PRISM 3100 DNA Analyser (Applied Biosystems). The results were assembled with ABI PRISM sequencing analysis software version 3.7 and analysed with SeqScape software (Applied Biosystems).
RESULTS
Family PKRP064 recruited from the Punjab province of Pakistan included six affected individuals in three consanguineous loops (figure 1). This family resides in remote villages of the Punjab province of Pakistan, and so the exact age of disease onset is not known. However, the medical records available to us along with communications with the family elders were suggestive of an early, most probably a congenital onset. The fundus photographs of affected individuals illustrate multiple round white deposits in the mid and peripheral regions of the retina (figure 2). The scotopic ERG responses were non-detectable, whereas the photopic response was severely reduced in affected individuals (figure 3). Taken together, the ophthalmological examination in PKRP064 identified typical features of FA with an early onset form of night blindness (table 1).
Figure 1.

Pedigree drawing of family PKRP064 with haplotypes of six adjacent 15q markers and segregation of c.466C/T variation with the disease phenotype. Squares are males, circles are females; filled symbols are affected individuals; a double line between individuals indicates consanguinity and a diagonal line through a symbol is deceased family member. The alleles forming the risk haplotype are shaded black, alleles co-segregating with disease haplotype but not showing homozygosity are shaded grey and alleles not co-segregating with fundus albipunctatus are shown in white.
Figure 2.

Fundus photographs of individuals of family PKRP064. (A) Individual 11: OD and OS; (B) unaffected individual 14: OD and OS. The fundus photographs show multiple round white deposits with relative absence in the central macula. OD, oculus dexter (right eye); OS, oculus sinister (left eye).
Figure 3.

Electroretinographic (ECG) responses of family PKRP064. Individual 11: (A) OD combined rod and cone response, and cone flicker response; (B) OS combined rod and cone response, and cone flicker response. Unaffected individual 14: (C) OD combined rod and cone response, and cone flicker response; (D) OS combined rod and cone response, and cone flicker response. ERG recordings illustrate absence of the rod and cone response in affected individuals whereas the photopic flicker responses are severely reduced in amplitude. OD, oculus dexter (right eye (R)); OS, oculus sinister (left eye (L)).
Table 1.
Clinical characteristics of affected individuals of family PKRP064
| Visual acuity |
|||||
|---|---|---|---|---|---|
| Individual ID | Age at first examination (years) | OD | OS | Fundus findings | Electroretinography recording |
| 7 | 15 | 6/18 | 6/12 | Multiple round white deposits | NA |
| 11 | 18 | 6/16 | 6/18 | Multiple round white deposits | a- and b-waves are absent under scotopic condition, whereas the cone responses are reduced under photopic condition |
| 12 | 20 | 6/36 | 6/45 | Multiple round white deposits | a- and b-waves are absent under scotopic condition, whereas the cone responses are reduced under photopic condition |
| 13 | 22 | 6/60 | 6/40 | Multiple round white deposits | NA |
| 19 | 14 | 6/18 | 6/12 | Multiple round white deposits | NA |
| 22 | 16 | 6/36 | 6/60 | Multiple round white deposits | NA |
NA, results not available; OD, oculus dexter (right eye); OS, oculus sinister (left eye).
Genomic DNA of affected individuals was investigated with closely spaced STR markers that span all reported loci associated with FA (data not shown). Two-point linkage analysis with alleles of chromosome 15q STR markers was suggestive of linkage with significant LOD scores. Maximum two-point LOD scores of 4.77 and 3.19 were obtained with markers D15S1045, and D15S202, at θ=0, respectively (table 2).
Table 2.
Two point limit of detection (LOD) scores of chromosome 15q markers of families PKRP064 and PKRP107
| Marker | cM | Mb | 0.00 | 0.01 | 0.05 | 0.09 | 0.10 | 0.20 | 0.30 | Zmax | θ max |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PKRP064 | |||||||||||
| D15S152 | 80.04 | 85.88 | − ∞ | −1.67 | −0.19 | 0.24 | 0.30 | 0.50 | 0.32 | 0.50 | 0.20 |
| D15S979 | 83.40 | 88.83 | − ∞ | −0.07 | 0.46 | 0.53 | 0.53 | 0.34 | 0.10 | 0.53 | 0.10 |
| D15S1045 | 85.64 | 89.59 | 4.77 | 4.66 | 4.19 | 3.72 | 3.60 | 2.42 | 1.29 | 4.77 | 0.00 |
| D15S202 | 85.64 | 90.00 | 3.19 | 3.08 | 2.65 | 2.23 | 2.13 | 1.18 | 0.39 | 3.19 | 0.00 |
| D15S116 | 85.64 | 90.01 | −1.33 | 1.05 | 1.57 | 1.60 | 1.59 | 1.25 | 0.76 | 1.61 | 0.07 |
| D15S207 | 102.21 | 96.21 | − ∞ | −0.55 | 0.12 | 0.30 | 0.32 | 0.34 | 0.20 | 0.34 | 0.20 |
| PKRP107 | |||||||||||
| D15S152 | 80.04 | 85.88 | 3.04 | 2.95 | 2.67 | 2.43 | 2.36 | 1.76 | 1.16 | 3.04 | 0.00 |
| D15S979 | 83.40 | 88.83 | 3.09 | 2.98 | 2.71 | 2.46 | 2.39 | 1.79 | 1.17 | 3.09 | 0.00 |
| D15S1045 | 85.64 | 89.59 | 3.01 | 2.91 | 2.64 | 2.39 | 2.32 | 1.72 | 1.10 | 3.01 | 0.00 |
| D15S202 | 85.64 | 90.00 | 1.47 | 1.45 | 1.35 | 1.24 | 1.21 | 0.93 | 0.64 | 1.47 | 0.00 |
| D15S116 | 85.64 | 90.01 | 1.62 | 1.59 | 1.47 | 1.24 | 1.31 | 0.98 | 0.64 | 1.62 | 0.00 |
| D15S207 | 102.21 | 96.21 | − ∞ | −2.28 | −0.89 | −0.44 | −0.37 | −0.01 | 0.05 | 0.05 | 0.30 |
LOD scores were calculated with the FASTLINK version of MLINK from the LINKAGE program package. Maximum LOD scores for each marker were calculated using ILINK.
Haplotype analysis supported linkage to the chromosome 15q. There are recombination events in individuals 12 and 22 at D15S152 and D15S979, respectively, which defines the proximal boundary (figure 1). The distal boundary is defined by recombination in individual 19 at marker D15S207 (figure 1). In addition, lack of homozygosity at D15S116 in individual 22 suggests that the pathogenic mutation resides proximal to marker D15S116 in a 2.24 cM (1.18 Mb) region flanked by D15S979 proximally and D15S116 distally.
The critical interval harbours RLBP1, a gene previously associated with RPA and FA, and sequencing of the coding exons of RLPB1 identified a C to T transition c.466C→T (figure 4A), which leads to premature termination of the protein R156X. This variation segregated with the disease phenotype in family PKRP064: all affected individuals were homozygous for the mutation, whereas unaffected individuals were either heterozygous carriers or were homozygous for the wild type allele (figure 1). The mutation was not present in 192 ethnically matched control chromosomes.
Figure 4.

Forward and reverse sequence chromatograms of family PKRP064. (A) Individual 15 harbouring the wild type allele; (B) individual 14 heterozygous and (C) affected individual 12 homozygous for C to T transition c.466 C→T, resulting in a p.R156X. All affected members of family PKRP064 are homozygous for the mutation, whereas the unaffected individuals are either heterozygous carriers or homozygous for the wild type allele.
Subsequently, we identified a second family with typical clinical characteristics of FA (figure 5). Exclusion analyses localised the disease phenotype to chromosome 15q with suggestive LOD scores. Two-point scores of 3.04, 3.09, and 3.01 at θ=0 were obtained with markers D15S152, D15S979 and D15S1045, respectively (table 2). Bi-directional sequencing identified a missense mutation c.346G/C, resulting in glycine to arginine substitution: p.G116R (figure 5). The mutation segregated with the disease phenotype in the family (figure 6) and was not present in 192 ethnically matched control chromosomes. The mutation segregated with the disease phenotype in the family and was not present in 192 ethnically matched control chromosomes. We investigated the evolutionary conservation of glycine 116 in other RLBP1 orthologues using the UCSC genome browser (http://genome.ucsc.edu/cgi-bin/ hgGateway) and as shown in figure 6, the amino acid glycine at position 116 is conserved in other RLBP1 orthologues.
Figure 5.

Sequence chromatograms of family PKRP107 and alignment of G116 in RLBP1 orthologues. (A) Individual 15 harbouring the wild type allele; (B) individual 7 heterozygous and (C) affected individual 8 homozygous for G to C transition c.346G→C, resulting in p. G116R. (D) Conservation of G116 in other RLBP1 orthologues is illustrated with primates coloured green; placental mammals blue; vertebrates are purple. The arrow points to the amino acid glycine 116 (G116).
Figure 6.
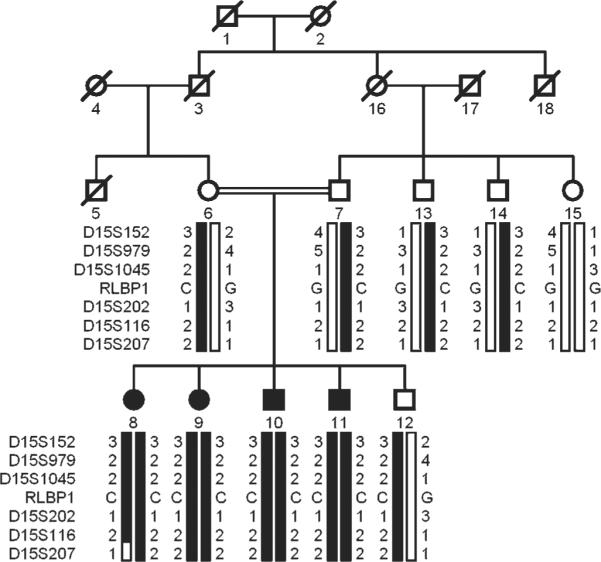
Pedigree drawing of family PKRP107 with haplotypes of six adjacent 15q markers and segregation of c.346G→C variation with the disease phenotype. Symbols are as described in figure 1. The alleles forming the risk haplotype are shaded black, and alleles not co-segregating with fundus albipunctatus (FA) are shown in white.
DISCUSSION
Here, we report pathogenic mutations in RLBP1 associated with FA in two consanguineous Pakistani families. Exclusion analysis localised the critical interval to chromosome 15q harbouring RLBP1 and subsequent sequencing identified causal mutations in RLBP1. These mutations segregated with the disease phenotype in their respective families and were not present in ethnically matched controls. These results strongly suggest that pathogenic mutations in RLBP1 are responsible for FA in both Pakistani families. Identification of novel pathogenic mutations reaffirms the diverse allelic heterogeneity of RLBP1.
The p.R156X mutation is expected to produce an unstable transcript that will be degraded by nonsense-mediated decay.9 If somehow the mutant mRNA escapes nonsense mediated decay, the protein thus produced will lack the 161 amino acids of the C-terminal domain. On the other hand, the evolutionary conservation of amino acid glycine 116 in other RLBP1 orthologues suggests that p.G116R will have a deleterious effect on RLBP1 structure; however, the molecular mechanism of causality still remains unknown. Additional functional analysis will help us better understand how these mutations affect the ability of RLBP1 protein and lead to the disease phenotype.
Acknowledgements
The authors are grateful to all family members for their participation in this study.
Funding This work was supported in part by Higher Education Commission, Islamabad, Pakistan and Ministry of Science and Technology, Islamabad, Pakistan.
Footnotes
Competing interests None to declare.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the Institutional Review Board of National Centre of Excellence in Molecular Biology and the National Eye Institute.
Provenance and peer review Not commissioned; externally peer reviewed.
REFERENCES
- 1.Hajali M, Fishman GA, Dryja TP, et al. Diagnosis in a patient with fundus albipunctatus and atypical fundus changes. Doc Ophthalmol. 2009;118:233–8. doi: 10.1007/s10633-008-9151-8. [DOI] [PubMed] [Google Scholar]
- 2.Fishman GA, Roberts MF, Derlacki DJ, et al. Novel mutations in the cellular retinaldehyde-binding protein gene (RLBP1) associated with retinitis punctata albescens: evidence of interfamilial genetic heterogeneity and fundus changes in heterozygotes. Arch Ophthalmol. 2004;122:70–5. doi: 10.1001/archopht.122.1.70. [DOI] [PubMed] [Google Scholar]
- 3.Maw MA, Kennedy B, Knight A, et al. Mutation of the gene encoding cellular retinaldehyde-binding protein in autosomal recessive retinitis pigmentosa. Nat Genet. 1997;17:198–200. doi: 10.1038/ng1097-198. [DOI] [PubMed] [Google Scholar]
- 4.Morimura H, Berson EL, Dryja TP. Recessive mutations in the RLBP1 gene encoding cellular retinaldehyde-binding protein in a form of retinitis punctata albescens. Invest Ophthalmol Vis Sci. 1999;40:1000–4. [PubMed] [Google Scholar]
- 5.Nakamura M, Lin J, Ito Y, et al. Novel mutation in RLBP1 gene in a Japanese patient with retinitis punctata albescens. Am J Ophthalmol. 2005;139:1133–5. doi: 10.1016/j.ajo.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 6.Humbert G, Delettre C, Senechal A, et al. Homozygous deletion related to Alu repeats in RLBP1 causes retinitis punctata albescens. Invest Ophthalmol Vis Sci. 2006;47:4719–24. doi: 10.1167/iovs.05-1488. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto H, Simon A, Eriksson U, et al. Mutations in the gene encoding 11-cis retinol dehydrogenase cause delayed dark adaptation and fundus albipunctatus. Nat Genet. 1999;22:188–91. doi: 10.1038/9707. [DOI] [PubMed] [Google Scholar]
- 8.Wada Y, Abe T, Fuse N, et al. A frequent 1085delC/insGAAG mutation in the RDH5 gene in Japanese patients with fundus albipunctatus. Invest Ophthalmol Vis Sci. 2000;41:1894–7. [PubMed] [Google Scholar]
- 9.Wada Y, Abe T, Sato H, et al. A novel Gly35Ser mutation in the RDH5 gene in a Japanese family with fundus albipunctatus associated with cone dystrophy. Arch Ophthalmol. 2001;119:1059–63. doi: 10.1001/archopht.119.7.1059. [DOI] [PubMed] [Google Scholar]
- 10.Katsanis N, Shroyer NF, Lewis RA, et al. Fundus albipunctatus and retinitis punctata albescens in a pedigree with an R150Q mutation in RLBP1. Clin Genet. 2001;59:424–9. doi: 10.1034/j.1399-0004.2001.590607.x. [DOI] [PubMed] [Google Scholar]
- 11.Eisenfeld AJ, Bunt-Milam AH, Saari JC. Immunocytochemical localization of retinoid-binding proteins in developing normal and RCS rats. Prog Clin Biol Res. 1985;190:231–40. [PubMed] [Google Scholar]
- 12.De Leeuw AM, Gaur VP, Saari JC, et al. Immunolocalization of cellular retinol-, retinaldehyde- and retinoic acid-binding proteins in rat retina during pre- and postnatal development. J Neurocytol. 1990;19:253–64. doi: 10.1007/BF01217303. [DOI] [PubMed] [Google Scholar]
- 13.Bunt-Milam AH, Saari JC. Immunocytochemical localization of two retinoid-binding proteins in vertebrate retina. J Cell Biol. 1983;97:703–12. doi: 10.1083/jcb.97.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bridges CD, Foster RG, Landers RA, et al. Interstitial retinol-binding protein and cellular retinal-binding protein in the mammalian pineal. Vision Res. 1987;27:2049–60. doi: 10.1016/0042-6989(87)90119-2. [DOI] [PubMed] [Google Scholar]
- 15.Martin-Alonso JM, Ghosh S, Hernando N, et al. Differential expression of the cellular retinaldehyde-binding protein in bovine ciliary epithelium. Exp Eye Res. 1993;56:659–9. doi: 10.1006/exer.1993.1083. [DOI] [PubMed] [Google Scholar]
- 16.Intres R, Goldflam S, Cook JR, et al. Molecular cloning and structural analysis of the human gene encoding cellular retinaldehyde-binding protein. J Biol Chem. 1994;269:25411–18. [PubMed] [Google Scholar]
- 17.Kaul H, Riazuddin SA, Yasmeen A, et al. A new locus for autosomal recessive congenital cataract identified in a Pakistani family. Mol Vis. 2010;16:240–5. [PMC free article] [PubMed] [Google Scholar]
- 18.Kaul H, Riazuddin SA, Shahid M, et al. Autosomal recessive congenital cataract linked to EPHA2 in a consanguineous Pakistani family. Mol Vis. 2010;16:511–17. [PMC free article] [PubMed] [Google Scholar]
- 19.Yasmeen A, Riazuddin SA, Kaul H, et al. Autosomal recessive congenital cataract in consanguineous Pakistani families is associated with mutations in GALK1. Mol Vis. 2010;16:682–8. [PMC free article] [PubMed] [Google Scholar]
- 20.Shahzadi A, Riazuddin SA, Ali S, et al. Nonsense mutation in MERTK causes autosomal recessive retinitis pigmentosa in a consanguineous Pakistani family. Br J Ophthalmol. 2010;94:1094–9. doi: 10.1136/bjo.2009.171892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lathrop GM, Lalouel JM. Easy calculations of LOD scores and genetic risks on small computers. Am J Hum Genet. 1984;36:460–5. [PMC free article] [PubMed] [Google Scholar]
- 22.Schaffer AA, Gupta SK, Shriram K, et al. Avoiding recomputation in linkage analysis. Hum Hered. 1994;44:225–37. doi: 10.1159/000154222. [DOI] [PubMed] [Google Scholar]