

Abstract
Chronic myelomonocytic leukemia is a clonal stem cell disorder that is characterized mainly by absolute peripheral monocytosis. This disease can present myeloproliferative and myelodysplastic characteristics. According to the classification established by the World Health Organization, chronic myelomonocytic leukemia is inserted in a group of myeloproliferative/myelodysplastic disorders; its diagnosis requires the presence of persistent monocytosis and dysplasia involving one or more myeloid cell lineages. Furthermore, there should be an absence of the Philadelphia chromosome and the BCR/ABL fusion gene and less than 20% blasts in the blood or bone marrow. Phenotypically, the cells in chronic myelomonocytic leukemia can present myelomonocytic antigens, such as CD33 and CD13, overexpressions of CD56 and CD2 and variable expressions of HLA-DR, CD36, CD14, CD15, CD68 and CD64. The increase in the CD34 expression may be associated with a transformation into acute leukemia. Cytogenetic alterations are frequent in chronic myelomonocytic leukemia, and molecular mutations such as NRAS have been identified. The present article reports on a case of chronic myelomonocytic leukemia, diagnosed by morphologic and phenotypical findings that, despite having been suggestive of acute monocytic leukemia, were differentiated through a detailed analysis of cell morphology. Furthermore, typical cells of chronic lymphocytic leukemia were found, making this a rare finding.
Keywords: Leukemia, myelomonocytic, chronic; Leukemia, myelomonocytic, acute; Leukemia, lymphocytic, chronic, B-Cell; Case reports
Introduction
In the 80s, chronic myelomonocytic leukemia (CMML) was classified by the FAB (French-American-British) system as a subcategory of myelodysplastic syndromes. However, the presence of characteristics that were common to myeloproliferative diseases was noticed(1), and in 2001, the World Health Organization (WHO), together with Hematopathology Society and the European Hematology Association published the Classification of Tumors of Hematopoietic and Lymphoid Tissues which included CMML in the new group of Myeloproliferative/Myelodysplastic Diseases(2).
CMML is a clonal hematopoietic stem cell disease characterized by absolute monocytosis in peripheral blood, in conjunction with signs of dysplasia and cell proliferation. The diagnostic criteria include: persistent monocytosis (peripheral = 1 x 109/L) in peripheral blood, absence of the Philadelphia chromosome and the BCR/ABL fusion gene, < 20% blasts (including myeloblasts, monoblasts and promonocytes) in peripheral blood or bone marrow, dysplasia in one or more cell lineages, and the absence of the reordering of PDGFRA or PDGFRB (should be excluded in cases that present eosinophilia)(3,4). In the majority of cases, the monocyte count corresponds to more than 10% of the total number of leucocytes, possibly reaching 80 x 109/L(3).
This disease can be classified according to the number of blasts and promonocytes present in the peripheral blood and bone marrow in the following way: CMML-1 presents < 5% blasts in peripheral blood and < 10% in bone marrow; CMML-2 presents between 5 and 19% blasts in peripheral blood or between 10 and 19% in the bone marrow, or the presence of Auer Rods and < 20% blasts in peripheral blood and bone marrow; and CMML-1 or CMML-2 with eosinophilia presents an eosinophil count in the peripheral blood higher than 1.5 x 109/L(2). The bone marrow may present monocytic or granulocytic hyperplasia. When granulocytic hyperplasia is prominent, it becomes difficult to distinguish between the population of normal monocytes and myelocytes, as these cells appear as hypo or agranular. In this case, cytochemical stains can help to identify monocytes(4). The presence of mild anemia and giant platelets, as well as dysplastic alterations can also occur in these cells(3).
Regarding immunophenotyping, some markers are used to assist in the diagnosis of CMML such as the expression of myelomonocytic antigens including CD33 and CD13, variable expressions of CD68 and CD64 and decreased expression of CD14. The morphologic differentiation between CMML and acute monocytic leukemia can be difficult(3). Phenotypically, CMML presents an atypical expression of CD56 in cells of the monocytic lineage as in acute monocytic leukemia; however, this atypical expression combined with the diminished expression of a myeloid marker (HLA-DR, CD13, CD15, CD36 or CD64) is specific to CMML, just as the atypical expression of CD2 is. Apart from this marker, the presence of higher proportions of granulocytic cell lineages and smaller proportions of monocytic lineages are detected in CMML when compared to acute monocytic leukemia(5).
Non-specific cytogenetic clonal alterations, such as trisomy of chromosome 8 and monosomy 7/deletion7q, occur in 20 to 40% of patients. Furthermore, they are frequently related to genetic alterations with point mutations in the RAS genes(2,4). For a differential diagnosis of this neoplasia, all diseases that cause monocytosis should be considered, such as chronic and acute infections, chronic inflammatory processes and some neoplasias such as Hodgkin's lymphoma and chronic myeloid leukemia(3).
The average age at diagnosis of this disease is 70 years and its etiology is unknown, but it is possibly related to carcinogenic and ionizing radiation factors. CMML involves the peripheral blood and bone marrow as well as other organs such as the spleen, liver, skin and lymph nodes. It presents a survival rate of only one year; however, factors such as low hemoglobin doses, low platelet count, leukocytosis, monocytosis, lymphocytosis, presence of immature myeloid cells in peripheral blood, high percentage of blasts and decreasing erythroid series in the bone marrow, cytogenetic alterations and high levels of lactate dehydrogenase (LDH) and ß2-microglobuline are responsible for further drops in survival rates. Furthermore, approximately one third of patients evolve to acute leukemia, which means a worse prognosis(6-8). This paper presents a case study about the laboratory diagnosis of CMML and progression to acute leukemia that demonstrated the simultaneous occurrence of cells typical of chronic lymphocytic leukemia (CLL).
Case report
A specimen of a 72-year-old patient arrived in the laboratory with a clinical indication of acute leukosis and fever. The biochemical exams showed LDH 1713 U/L, PCR 12.94 mg/L, creatinine 1.60 mg/dL and uric acid 7.5 mg/dL. The peripheral blood analysis presented a total leukocyte count of 193,270 x 109/L, hemoglobin 6.1 g/dL, 9 erythroblasts and a platelet count of 79 x 109/L with 48% of blasts presenting a high nuclear:cytoplasm ratio, reticular chromatin and visible nucleoli, presence of 38% of granulocytes that had delayed maturation and some dysplastic characteristics such as hypogranular granulocytes, hypersegmented nucleus, vacuolated monocytes with irregular nuclei, and giant and hypogranular platelets (Figure 1).
Figure 1.
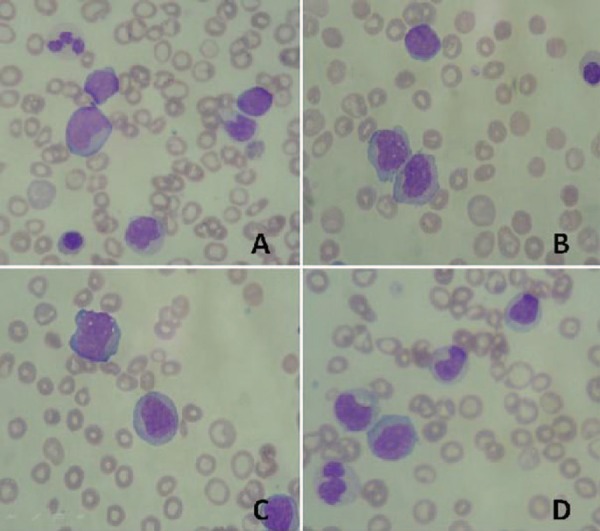
A - Presence of blastic cells in peripheral blood; B and C - Vacuolated monocytes with lobular or nuclear chromatin alterations and the presence of young cells of monocytic lineage; D - Presence of dysplasia in the granulocytic series
The myelogram appeared to be hypercellular, showing hypocellular erythroid series with maintained maturation, hypercellular granulocytic series (51% granulocytes) with delayed maturation and dysgranulopoiesis (hypogranular and hypersegmented segmentations), 53% blasts, hypocellular lymphoplasmacytic series with prevalence of mature lymphocytes and hypocellular megakaryocytic series with maintained maturation.
A cytogenetic study of the bone marrow was performed by 24/48 hours of culturing with ethidium bromide staining, which did not present metaphase, making it impossible to define a chromosomal diagnosis.
Immunophenotyping demonstrated the presence of 50% of myeloid blasts in the bone marrow, which were divided into two populations. Population 1 was formed by cells that were less mature and of smaller size; population 2, by cells that were larger, and more mature. Both presented phenotypical characteristics and a maturation pattern of the monocytic lineage (Figure 2). Furthermore, 20% of the granulocytic series presented dysplastic alterations such as hypogranularity with loss of CD10 and CD16 expressions, abnormal expressions of CD11b and CD13, and a weak expression of MPO in the mature cells. Weak expressions of CD36 and CD64 were also present, demonstrating that these cells lean towards the monocytic/dendritic lineage.
Figure 2.

A - The myeloid blasts are divided into two CD34++ populations. B - The red population expresses CD117+ and HLA-DR, showing that these cells may be producing a granulocytic lineage. C - Yet, the green cell population is CD117- and HLA-DR++. D and E - Markers of the second blastic population demonstrating CD36+ and CD64-
The presence of 15% of monocytes with mature cell phenotypes (CD45+++) and a phenotype alteration with a weak expression of HLA-DR, 40% of which were CD14-, were also found.
Furthermore, 1.8% pathologic B-lymphocytes (CD19+) with restriction of kappa light chains and suggestive antigenic expression of CLL (CD23 and CD5+, weak IgM+, weak CD79b+, FMC7-) were also found.
Hence, through morphologic and phenotypical findings, the diagnosis of acute monocytic leukemia was considered, but different to CMML, this acute leukemia is more commonly found in young individuals(2). Therefore, the dysplastic background and monocytosis suggest the myeloproliferative/myelodysplastic disorder CMML and progression to acute leukemia in parallel with the presence of 1.8% of clonal lymphocytes which are frequently found in CLL.
Discussion
In CMML, the increase in the CD34 expression may be associated with a transformation to acute leukemia(4). In the flow cytometry analysis, the presence of two blast populations with expressions of CD34 was demonstrated, one of them (CD117, HLA-DR++, CD36+, CD64-/+) being derived from the monocytic/dendritic lineage. Acute leukemias of monocytic lineage are characterized by having more than 80% of cells from the monocytic lineage, including monoblasts, promonocytes and monocytes, and less than 20% from the granulocytic chain(9). However, in this case, the occurrence of monocytes with phenotypical alterations in the bone marrow, dysgranulopoiesis and dysplasia in the monocytic series of the peripheral blood point to a diagnosis of CMML, which presented this aberrant phenotype associated with a blastic population (50% myeloid blasts), is associated with a transformation into an acute form(3,7).
This case study is important to demonstrate the importance of a detailed analysis of cellular morphology, because the description of dysplastic alterations in the peripheral blood is fundamental for the diagnosis of myelodysplastic/myeloproliferative syndromes, the category of this type of leukemia. In CMML, circulating monocytes are usually mature, but some may present granular alterations in lobular or nuclear chromatin. Dysplasia may be apparent in neutrophils, causing alterations in nuclear lobulation and cytoplasmic granulation. As previously demonstrated, the patient presented granulocytes with dysplastic characteristics, such as hypogranular neutrophils and hypersegmented nuclei; vacuolated monocytes with irregular nuclei and giant and hypogranular platelets. Also, in myeloproliferative/myeloblastic diseases, the bone marrow is hypercellular and shows some signs of dysplasia, accompanied by organ involvement, leukocytosis, and elevated LDH(4). In the current case, the patient showed an elevation in LDH, hypercellular activity with dysplasia of the granulocytic series of the bone marrow and leukocytosis in the peripheral blood.
Additionally, by flow cytometry, the presence of pathologic cells which are characteristic of chronic lymphocytic leukemia was proven; these cells express superficial immunoglobulins IgM/IgG of weak intensity, and CD20+, CD22+, CD5+, CD19+ CD79a+, CD23+, CD43+ and CD11c+ (weak expression). The CD10 is negative and the FMC7 and CD79b are usually negative or weakly expressed in typical CLL(10). In this case, immunophenotyping was fundamental to demonstrate the presence of cell clones of pathologic B-Cells.
In conclusion, this is a rare case as it demonstrated the presence of two hematologic neoplasms that do not have a clinical relationship.
Footnotes
Conflict-of-interest disclosure: The authors declare no competing financial interest
References
- 1. Tefferi A.Is chronic myelomonocytic leukemia more akin to myelodysplastic or myeloproliferative neoplasms and does it matter? Leuk Lymphoma. 2008; 49(7): 1225-7 Comment in: Leuk Lymphoma. 2008;49(7):1228-9. Comment on: Leuk Lymphoma. 2008;49(7):1292-6. [DOI] [PubMed] [Google Scholar]
- 2. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009; 114(5): 937-51 Comment in: Blood. 2010;115(3):748-9; author reply 749-50 [DOI] [PubMed] [Google Scholar]
- 3. Orazi A, Greming U.The myelodysplastic/myeloproliferative neoplasms: myeloproliferative diseases with dysplastic features. Leukemia. 2008; 22(7): 1308-19 [DOI] [PubMed] [Google Scholar]
- 4. Bacher U, Haferlach T, Schnittger S, Kreipe H, Krogen N.Recent advances in diagnosis, molecular pathology and therapy of chronic myelomonocytic leukaemia. Br J Haematol. 2011; 153(2): 149-67 [DOI] [PubMed] [Google Scholar]
- 5. Beran M, Wen S, Shen S, Onida F, Jelinek J, Cortes J, et al. Prognostic factors and risk assessment in chronic myelomonocytic leukemia: Validation study of the M.D. Anderson Prognostic Scoring System. Leuk Lymphoma. 2007; 48(6): 1150-60 [DOI] [PubMed] [Google Scholar]
- 6. Tkachuk D, Hirschmann JV.Wintrobe Atlas Colorido de Hematologia. Rio de Janeiro: Revinter; 2010. p. 135-8 [Google Scholar]
- 7. Onida F, Kantarjian HM, Smith TL, Ball G, Keating MJ, Estey EH, et al. Prognostic factors and scoring systems in chronic myelomonocytic leukemia: a retrospective analysis of 213 patients. Blood. 2002; 99(3): 840-9 [DOI] [PubMed] [Google Scholar]
- 8. Mori Y, Yoshimoto G, Kumano T, Miyamoto T, Iino T, Takenaka K, et al. Distinctive expression of myelomonocytic markers and down-regulation of CD34 in acute myelogenous leukaemia with FLT3 tandem duplication and nucleophosmin mutation. Eur J Haematol. 2007; 79(1): 17-24 [DOI] [PubMed] [Google Scholar]
- 9. Brunning RD, Matutes E, Flandrin G, Vardiman J, Bennett J, Head D.Acute myeloid leukaemias not otherwise categorised In: Jaffe ES, Harris NL, Stein H, Vardiman JW.The WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon: IARC Press; 2001. p. 94-6 [Google Scholar]
- 10. Muller Hermelink HK, Catovsky D, Montserrat E, Harris NL.Chronic lymphocytic leukaemia/Small lymphocytic lymphoma In: Jaffe ES, Harris NL, Stein H, Vardiman JW.The WHO Classification of tumours of haematopoietic and lymphoid tissues. Lyon: IARC Press; 2001. p. 127-30 [Google Scholar]