

Abstract
Epithelial ovarian cancers (EOCs) often exhibit morphologic features of embryonic Müllerian duct–derived tissue lineages and colonize peritoneal surfaces that overlie connective and adipose tissues. However, the mechanisms that enable EOC cells to readily adapt to the peritoneal environment are poorly understood. In this study, we show that expression of HOXA9, a Müllerian-patterning gene, is strongly associated with poor outcomes in patients with EOC and in mouse xenograft models of EOC. Whereas HOXA9 promoted EOC growth in vivo, HOXA9 did not stimulate autonomous tumor cell growth in vitro. On the other hand, expression of HOXA9 in EOC cells induced normal peritoneal fibroblasts to express markers of cancer-associated fibroblasts (CAFs) and to stimulate growth of EOC and endothelial cells. Similarly, expression of HOXA9 in EOC cells induced normal adipose- and bone marrow–derived mesenchymal stem cells (MSCs) to acquire features of CAFs. These effects of HOXA9 were due in substantial part to its transcriptional activation of the gene encoding TGF-β2 that acted in a paracrine manner on peritoneal fibroblasts and MSCs to induce CXCL12, IL-6, and VEGF-A expression. These results indicate that HOXA9 expression in EOC cells promotes a microenvironment that is permissive for tumor growth.
Introduction
Approximately 70% of patients with epithelial ovarian cancer (EOC) present with disease that involves the peritoneal cavity (1). Unlike many other types of cancers, EOC rarely spreads via hematogenous routes. EOC cells typically shed into the peritoneal fluid and implant on the mesothelial linings of the omentum and other peritoneal surfaces that overlie connective and white adipose tissues (1, 2). Because of their propensity for peritoneal involvement, EOC cells are likely to be programmed to dynamically adapt to the peritoneal environment, but the underlying mechanisms are unknown.
A striking feature of EOCs is their morphologic resemblance to tissue lineages that derive from the Müllerian ducts. Serous EOC, the most common EOC subtype, histologically resembles fallopian tube cancers. Endometrioid EOCs often exhibit endometrial-like glandular structures, whereas mucinous EOCs are composed of endocervical-like or intestinal-like cells (3). Homeobox genes constitute a gene superfamily that controls cell differentiation and specification of the body plan during embryonic development (4–7). Homeobox genes encode transcription factors, but only few bona fide transcriptional targets have been identified (4–7). Of the homeobox gene families, the mammalian HOX family is the largest and comprises 39 genes that are organized in 4 clusters and are aligned in 13 paralogous groups (4). Patterning of the Müllerian system is coordinated by members of the HOXA gene cluster (8–10). We previously identified a broad trend in expression of this Müllerian HOX gene program in the major subtypes of EOC, according to the patterns of Müllerian-like differentiation of these tumors. Whereas HOXA11 was preferentially expressed in mucinous EOCs, HOXA10 was expressed in endometrioid and mucinous EOCs and HOXA9 was expressed in both serous and nonserous EOCs (11). Furthermore, we demonstrated that this HOX gene program was responsible for the Müllerian-like differentiation patterns of the EOC subtypes (11). However, the functional significance of Müllerian HOX genes to the clinical behavior of EOC is not known.
The expansion and functional features of epithelial and stromal cell populations are dynamically regulated by cross-talk between these cells during normal tissue morphogenesis. Tumor growth is increasingly recognized to be similarly orchestrated by interplay between tumor cells and the stroma (12, 13). Cancer-associated fibroblasts (CAFs) are a predominant component of the tumor stroma and have a profoundly negative impact on clinical outcomes (12, 13). Whereas the ability of CAFs to stimulate growth of tumor cells has been extensively studied (14–18), it is not clear whether CAFs are reciprocally controlled by developmental programs that are activated in tumor cells. We speculated that the dominance of Müllerian phenotypes in EOCs reflects an evolutionary strategy by these cancers to adapt to their microenvironment and that developmental pathways that are activated in EOC cells modulate interactions of EOC cells with the stroma. In this study, we investigated the significance of Müllerian HOX genes to EOC growth, the tumor stroma, and clinical outcomes. We found that expression of HOXA9, but not HOXA10 or HOXA11, was associated with poor survival in patients with EOC and in mouse xenograft models of EOC. HOXA9 promoted EOC growth by inducing normal peritoneal fibroblasts and adipose- and bone marrow–derived mesenchymal stem cells (MSCs) to acquire molecular and functional features of CAFs in substantial part via its induction of tumor-derived TGF-β2. Our findings support a model in which HOXA9 expression in EOC cells “educates” the stroma to become permissive for tumor growth and demonstrate for the first time to our knowledge the significance of a Müllerian-patterning gene in the aggressive behavior of EOC.
Results
HOXA9 is associated with poor survival of patients with EOC.
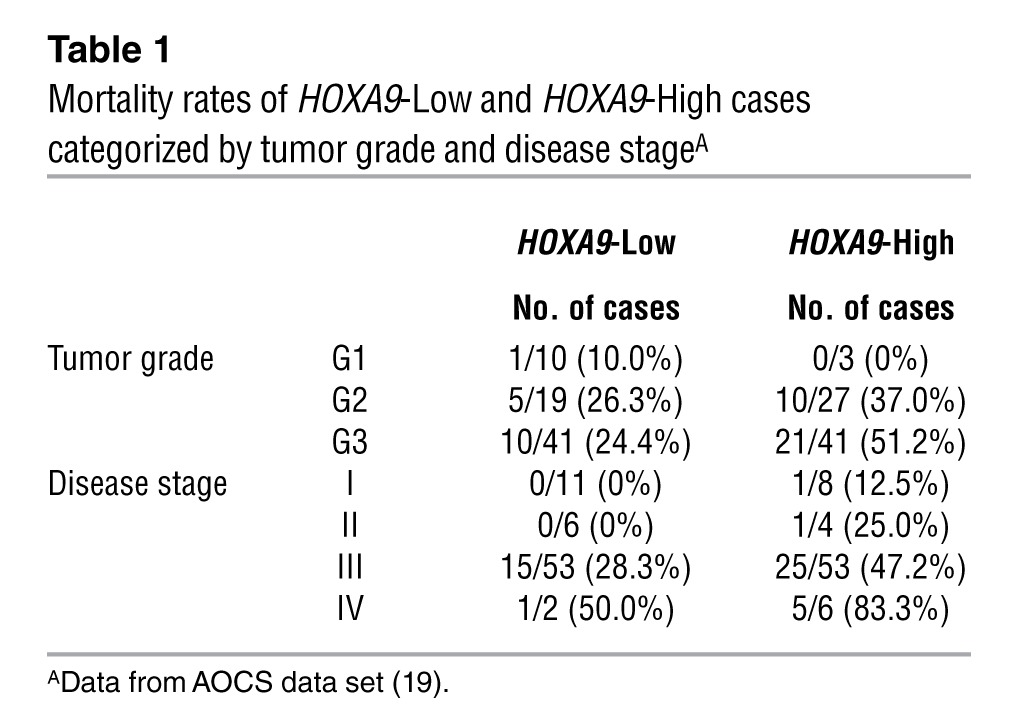
We initially evaluated the clinical significance of Müllerian HOX genes in the Australian Ovarian Cancer Study (AOCS) gene expression data set (19). HOXA9 transcript levels did not significantly differ among serous carcinomas of ovarian, tubal, and peritoneal origin or between serous and nonserous EOCs (Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI62229DS1). These observations were consistent with our findings of HOXA9 protein levels in an independent cohort (11). High HOXA9 expression in tumors was significantly associated with poor overall survival of patients in the AOCS cohort (P = 0.0004 by log-rank test, hazard ratio (HR) of death = 2.90, 95% confidence interval (CI) = 1.61 to 5.19; Figure 1). Mortality rates of cases with high HOXA9 expression (referred to herein as HOXA9-High cases) were higher than those of cases with low HOXA9 expression (referred to herein as HOXA9-Low cases), irrespective of tumor grade and disease stage (Table 1). High HOXA9 expression was also significantly associated with poor survival in the Cancer Genome Atlas (TCGA) EOC data set (P = 0.01, HR of death = 1.56, 95% CI = 1.11 to 2.18; Supplemental Figure 1). No association with survival was found for HOXA10, HOXA11, other neighboring HOXA genes, or paralogs of HOXA9 (Figure 1).
Figure 1. High HOXA9 expression is associated with reduced survival of patients with EOC.

Kaplan-Meier plot analysis of overall survival times of patients in the AOCS data set stratified by transcript levels of each indicated HOX gene in tumors. For each HOX gene, transcript levels were defined as High (≥ upper quartile) and Low (≤ lower quartile) (n = 72 cases per group). Significance values were determined by log-rank test.
Table 1.
Mortality rates of HOXA9-Low and HOXA9-High cases categorized by tumor grade and disease stageA
HOXA9 is associated with poor survival in EOC xenograft models.
We previously generated mouse EOC (MOSEC) lines that stably express Hoxa9, Hoxa10, and Hoxa11 (11). Female nude mice that were inoculated i.p. with MOSEC cells that overexpressed Hoxa9 (referred to herein as +HOXA9 MOSEC cells) developed larger implants (Supplemental Figure 2, A and B) and had significantly shorter survival times than mice inoculated with vector-control MOSEC cells (P = 0.003, Figure 2A). In contrast, tumor burden and survival rates of mice that were inoculated i.p. with +HOXA10 or +HOXA11 MOSEC cells were not significantly different from those of the vector-control group (Supplemental Figure 2, A and B, and Figure 2, B and C). These observations were consistent with the association of HOXA9, but not HOXA10 or HOXA11, with poor survival of patients with EOC (Figure 1). The increased growth of +HOXA9 tumors, as compared with that of +HOXA10 and +HOXA11 tumors, was not due to differences in ectopic HOX levels between the MOSEC lines (Supplemental Figure 3A) or to nonphysiological levels, as the HOXA9 level in +HOXA9 MOSEC cells was within the range of HOXA9 levels detected in human EOC clinical specimens (Supplemental Figure 3, B–D).
Figure 2. HOXA9 promotes tumor growth in mouse xenograft models of EOC.

(A–C) Comparison of survival rates of female nude mice inoculated i.p. with vector-control and with (A) +HOXA9, (B) +HOXA10, and (C) +HOXA11 MOSEC lines (n = 10 per group). (D) Western blot of HOXA9 in SKOV3ip and ES-2 lines stably expressing empty pGFP-V-RS vector, nontargeting shRNA, and shRNAs targeting different sites of HOXA9 (shA9-A, shA9-B). Full uncut gels are shown in the Supplemental Material. (E and F) Growth rates of s.c. tumors derived from +HOXA9 control (Empty vector, Nontargeting) and HOXA9-knockdown (shA9-A, shA9-B) (E) SKOV3ip and (F) ES-2 lines (n = 5 per group). *P < 0.0005. (G and H) Mice were inoculated i.p. with GFP-expressing (G) SKOV3ip and (H) ES-2 lines and sacrificed at 4 weeks and 20 days, respectively. Implants were viewed under a fluorescence stereoscope. Omental implants in +HOXA9 control groups are indicated by arrows. Original magnification, ×0.8.
HOXA9 promotes EOC growth in vivo but not in vitro.
To confirm that HOXA9 promotes EOC growth, we evaluated the effect of inhibiting HOXA9 expression in human EOC lines. Endogenous HOXA9 levels in the SKOV3ip (serous EOC) and ES-2 (nonserous EOC) cell lines were similar to HOXA9 levels in several EOC clinical specimens (Supplemental Figure 3, B–D). Two shRNAs that targeted different sites of HOXA9 (shA9-A, shA9-B) were equally effective in knocking down HOXA9 in SKOV3ip and ES-2 cells (Figure 2D). Knockdown of HOXA9 markedly inhibited the volumes of s.c. and i.p. tumors derived from SKOV3ip and ES-2 lines (Figure 2, E–H) and significantly reduced mitotic activity in tumor xenografts (P < 0.001, Supplemental Figure 4A). However, in vitro growth rates of HOXA9-knockdown and control EOC lines were identical, irrespective of whether these cells were cultured on plastic, in 3-dimensional Matrigel cultures, or under serum-deprived or anchorage-independent conditions (Supplemental Figure 4, B–E). Similarly, enforced expression of Hoxa9 increased proliferative activity of MOSEC cells in vivo but not in vitro (Supplemental Figure 5, A and B), indicating that the ability of HOXA9 to promote tumor growth might depend on interactions with host cells.
HOXA9 is associated with increased abundance of CAFs.
Tothill et al. classified tumors in the AOCS cohort into molecular subtypes by their gene expression signatures (19). The C1 subtype was associated with the poorest outcomes and was characterized by a desmoplastic or “reactive” stromal gene signature (19). Tumors that were classified as being of the C1 subtype constituted 39% of cases in the HOXA9-High group, but only 4% of cases in the HOXA9-Low group in the AOCS cohort (Table 2). Conversely, the frequency of the C4 subtype (characterized by a low stromal response signature) was higher in the HOXA9-Low group than in the HOXA9-High group (Table 2). CAFs are often characterized by their expression of α–smooth muscle actin (αSMA) and fibroblast activation protein (FAP) (13). Expression of ACTA2 (the gene encoding αSMA) and FAP was significantly higher in HOXA9-High tumors than in HOXA9-Low tumors in the AOCS cohort (ACTA2, P = 0.026; FAP, P = 10–7, Figure 3A). αSMA+ cells were abundant in tumors derived from +HOXA9 control SKOV3ip and ES-2 lines but were sparse in respective HOXA9-knockdown tumors (P < 0.0001, Figure 3, B and C). As compared with those in vector-control MOSEC tumors, αSMA+ cells were more abundant in +HOXA9 but not in +HOXA10 or +HOXA11 MOSEC tumors (Supplemental Figure 5C). These results suggest that the association between HOXA9 and poor survival could be linked to the ability of HOXA9 to promote a CAF-rich microenvironment.
Table 2.
Frequency of molecular subtypes classified by Tothill et al. among HOXA9-Low and HOXA9-High casesA
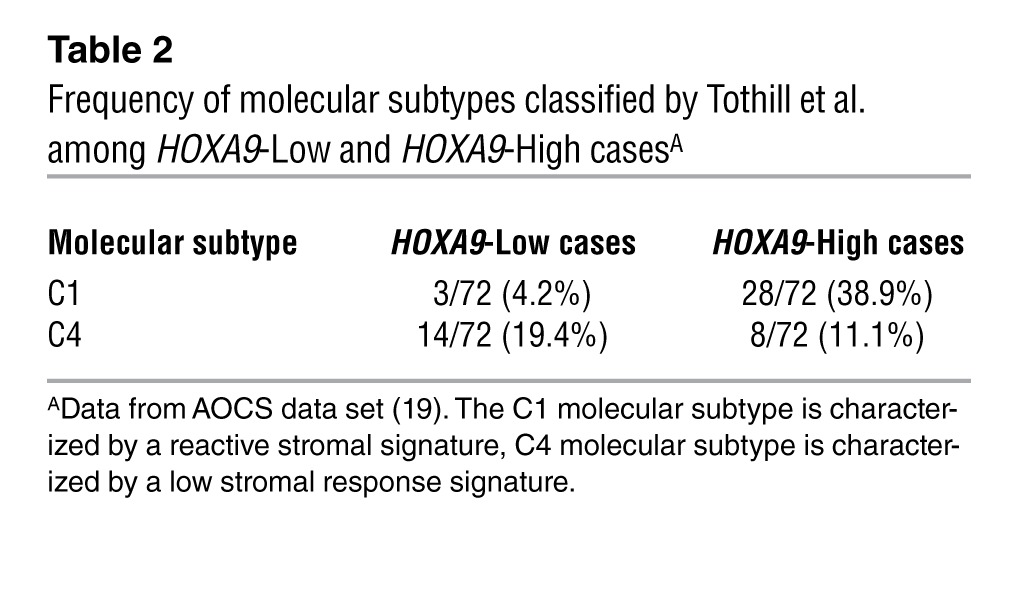
Figure 3. HOXA9 expression in EOC is associated with increased CAF abundance.

(A) Differences in transcript levels of ACTA2 (encoding αSMA) and FAP, between HOXA9-Low and HOXA9-High tumors in the AOCS data set, as estimated by Mann Whitney U test. In box-and-whisker plots, horizontal bars indicate the medians, boxes indicate 25th to 75th percentiles, and whiskers indicate minimum and maximum values. (B–D) αSMA expression was evaluated in tumors of mice sacrificed at 20 days after inoculation with SKOV3ip and ES-2 lines. (B) The average number of αSMA+ cells per 1,000 tumor cells was calculated by scoring 5 random fields of stained tissue sections of each mouse (n = 5 mice per group). *P < 0.0001. Immunofluorescence staining of GFP-expressing tumor cells (green) and αSMA (red) in tumors of mice inoculated with (C) +HOXA9 control and HOXA9-knockdown SKOV3ip lines (scale bar: 100 μm) and (D) +HOXA9 control (nontargeting) SKOV3ip and ES-2 lines (scale bar: 50 μm). Nuclei were visualized by staining with DAPI (blue).
HOXA9 does not induce CAF-like features in EOC cells.
Some CAFs can derive from tumor cells that have undergone epithelial-to-mesenchymal transition (EMT) (20). However, expression of genes encoding transcription factors that orchestrate EMT, such as SNAI1 and SNAI2, was not altered by HOXA9 in EOC cells in vitro or in vivo (Supplemental Figure 6, A and B). Furthermore, immunofluorescence staining of xenografts demonstrated no overlap between +HOXA9 EOC cells and cells that expressed αSMA (Figure 3D), indicating that αSMA+ cells do not derive from tumor cells. These findings indicate that increased abundance of CAFs in +HOXA9 tumors is unlikely to be due to transdifferentiation of tumor cells into CAF-like cells.
HOXA9 expression in EOC cells induces omental fibroblasts to acquire CAF features.
Increasing evidence indicates that CAFs derive either from MSCs or tissue-resident fibroblasts (14–18). Because EOC frequently involves the omentum, we investigated whether HOXA9 expression in EOC cells induces normal omental fibroblasts to acquire features of CAFs. HOXA9 expression in EOC cells did not affect proliferation of omental fibroblasts (Supplemental Figure 7A). ACTA2 mRNA and αSMA protein levels were strongly induced in fibroblasts following incubation (i.e., “priming”) in medium conditioned by +HOXA9 control SKOV3ip cells but not following incubation in medium conditioned by HOXA9-knockdown SKOV3ip cells (Figure 4, A and B, and Supplemental Figure 7B). A similar difference in FAP induction was also observed (Supplemental Figure 7B). CAFs are a source of various mitogenic factors for tumor cells (15, 17). Expression of CXCL12 and IL-6 was strongly induced in omental fibroblasts following priming in medium conditioned by control SKOV3ip cells but not following priming in medium conditioned by HOXA9-knockdown SKOV3ip cells (Figure 4C and Supplemental Figure 7B). Tumor cell proliferation was more highly stimulated by fibroblast-derived factors where fibroblasts had been primed in medium conditioned by control than by HOXA9-knockdown SKOV3ip cells (P < 0.001, Figure 4D). The ability of medium conditioned by +HOXA9 tumor-primed fibroblasts to stimulate tumor cell proliferation was inhibited when fibroblast-conditioned medium was depleted of CXCL12 and IL-6 (Figure 4E). These findings indicate that HOXA9 induces tumor-derived factors that stimulate normal fibroblasts to acquire CAF features and to express CXCL12 and IL-6 that act in a paracrine manner to promote tumor cell proliferation.
Figure 4. HOXA9 expression in EOC cells induces CAF features in normal omental fibroblasts.

(A) Diagram of fibroblast-priming assays. +HOXA9 control and HOXA9-knockdown SKOV3ip cells were cultured for 2 days to generate tumor-conditioned media (shown in pink). Normal omental fibroblasts were incubated for 5 days in SKOV3ip-conditioned medium (i.e., primed) or nonconditioned medium (i.e., unprimed) and then analyzed by Western blot and qRT-PCR. Fresh nonconditioned medium was added to washed fibroblasts. At 2 days thereafter, medium conditioned by fibroblasts (shown in light blue) was analyzed by ELISA and used for incubating control (nontargeting) SKOV3ip cells. (B) Western blot of αSMA levels in unprimed and primed fibroblasts. Full uncut gels are shown in the Supplemental Material. (C) Levels of growth factors in media conditioned by unprimed and primed fibroblasts. (D) Growth rates of control SKOV3ip cells incubated in fibroblast-conditioned medium. Average results of assays using 3 independent sets of each type of fibroblast-conditioned medium are shown in C and D. (E) Relative growth of control SKOV3ip cells at 6 days after incubation in medium conditioned by fibroblasts that were initially primed in +HOXA9 control SKOV3ip-conditioned medium, where fibroblast-conditioned medium was left untreated, treated with IgG, or depleted by IP with Abs to CXCL12 and IL-6. *P < 0.001; #P < 0.005.
To confirm these findings, we evaluated omental fibroblasts that were primed in medium conditioned by MOSEC cells. αSMA, FAP, CXCL12, and IL-6 expression was more strongly induced in fibroblasts following priming in medium conditioned by +HOXA9 MOSEC cells than that following priming in medium conditioned by parental or vector-control MOSEC cells. However, increased induction was not observed when fibroblasts were primed in media conditioned by +HOXA10 or +HOXA11 MOSEC cells (Supplemental Figures 7, C–E). Tumor cell proliferation was more strongly stimulated by fibroblast-derived factors where fibroblasts had been primed in medium conditioned by +HOXA9 than by control MOSEC cells (P < 0.001), whereas priming of fibroblasts in media conditioned by +HOXA10 or +HOXA11 MOSEC cells had little stimulatory effect (Supplemental Figure 7F). These findings indicate that HOXA9 expression in EOC cells specifically induces normal fibroblasts to acquire CAF features and that this capability is not shared by HOXA10 or HOXA11.
HOXA9 expression in EOC cells promotes the ability of fibroblasts to stimulate endothelial cell growth.
Because CAF abundance in EOC correlates with increased microvessel density (21), we investigated the effect of HOXA9 expression in EOC cells on endothelial cell growth. HOXA9-knockdown SKOV3ip tumors had lower microvessel density than +HOXA9 control SKOV3ip tumors (P < 0.005, Figure 5A). However, expression of genes encoding the angiogenic factors IL-6 and VEGF-A in SKOV3ip cells was not altered by HOXA9 in vitro or in vivo (Figure 5B). Endothelial cells grew at identical rates when cultured in medium conditioned by HOXA9-knockdown or by control SKOV3ip cells (Figure 5C). On the other hand, endothelial cell growth was more strongly stimulated by omental fibroblast-derived factors where fibroblasts were primed in medium conditioned by control SKOV3ip cells than by HOXA9-knockdown SKOV3ip cells (P < 0.005, Figure 5D). As observed for IL-6, VEGF-A expression was strongly induced in fibroblasts following priming in medium conditioned by control but not by HOXA9-knockdown SKOV3ip cells (Figure 4C and Supplemental Figure 7B). The ability of medium conditioned by fibroblasts that were primed by HOXA9-knockdown SKOV3ip cells to stimulate endothelial cell growth was restored when this conditioned medium was reconstituted with IL-6 and VEGF-A at concentrations released by fibroblasts that were primed by +HOXA9 control SKOV3ip cells (Supplemental Figure 8A). Expression of Il6 and Vegfa in mouse host cells was significantly higher in omental tumors of control SKOV3ip models than in tumors of HOXA9-knockdown models (Il6, P = 0.03; Vegfa, P = 0.007; Figure 5B). We confirmed our findings in studies using MOSEC lines. As compared with that in vector-control tumors, microvessel density was significantly higher in +HOXA9 tumors (P < 0.005) but not in +HOXA10 or +HOXA11 tumors (Supplemental Figure 8B). Expression of HOXA9, but not HOXA10 or HOXA11, in MOSEC cells induced omental fibroblasts to express IL-6 and VEGF-A and to stimulate endothelial cell growth (Supplemental Figure 7, C and E, and Supplemental Figure 8C). These findings indicate that HOXA9 expression in EOC cells promotes tumor microvessel density by inducing normal omental cells to express IL-6 and VEGF-A and that this capability is not shared by other Müllerian HOX genes.
Figure 5. HOXA9 expression in EOC cells promotes the ability of fibroblasts to stimulate endothelial cell growth.

(A) The average number of microvessels per 104 tumor cells was calculated in tumors derived from +HOXA9 control (nontargeting) and HOXA9-knockdown (shA9-B) SKOV3ip lines by scoring 5 random fields of CD34-stained tissue sections of each mouse (n = 5 mice per group). *P < 0.005. (B) Relative mRNA levels of IL6 and VEGFA in cultured SKOV3ip cells and of IL6 and VEGFA (in human EOC cells) and Il6 and Vegfa (in mouse host cells) in omental tumors of mice that were inoculated with SKOV3ip lines (n = 5 mice per group). *P = 0.03; †P = 0.007. P values > 0.05 were considered not significant. Evaluation of specificity of human- and mouse- specific qRT-PCR primers is shown in Supplemental Figure 6B. (C) Growth rates of mouse endothelial cells incubated in nonconditioned medium and in SKOV3ip-conditioned media. **P < 0.005. (D) Normal omental fibroblasts were left unprimed or primed with SKOV3ip-conditioned media (shown in pink) for 5 days. Fresh nonconditioned medium was added to washed fibroblasts. Two days thereafter, medium conditioned by fibroblasts (shown in light blue) was collected. Growth rates of endothelial cells incubated in fibroblast-conditioned medium were measured. *P < 0.005. Average results of assays using 3 independent sets of each type of conditioned medium are shown in C and D.
HOXA9 expression in EOC cells also induces CAF features in MSCs.
MSCs are another potential source of CAFs in many types of tumors, including EOC (14, 16, 17, 22). Bone marrow is the most well-characterized source of MSCs, but MSCs reside in most tissues and are abundant in white adipose tissues (23, 24). As was observed in omental fibroblasts, ACTA2, FAP, IL6, CXCL12 and VEGFA mRNA levels were strongly induced in normal bone marrow– and adipose-derived MSCs following incubation in medium conditioned by +HOXA9 control SKOV3ip cells but not in medium conditioned by HOXA9-knockdown SKOV3ip cells (Figure 6). HOXA9 expression in EOC cells did not affect MSC proliferation (data not shown). These results indicate that HOXA9 induces tumor-derived factors that not only stimulate omental fibroblasts but also MSCs from various sources to acquire CAF features.
Figure 6. HOXA9 expression in EOC cells induces CAF features in bone marrow– and adipose-derived MSCs.

Levels of ACTA2, FAP, IL6, CXCL12, and VEGFA transcripts were assayed in bone marrow MSCs and adipose MSCs at 5 days after incubation in media conditioned by +HOXA9 control (Nontargeting) and HOXA9-knockdown (shA9-B) SKOV3ip cells. The mRNA level of each gene, assayed by qRT-PCR, is expressed relative to its level in MSCs incubated in nonconditioned medium.
HOXA9 correlates with TGF-β2 expression.
To investigate the mechanism of HOXA9, we initially evaluated the effect of HOXA9 on expression of growth factors in SKOV3ip cells. HOXA9 did not alter expression of genes encoding several fibroblast-stimulatory factors, such as TGF-β1, which is known to induce transition of normal fibroblasts and MSCs into CAFs (refs. 17, 18, Figure 7, A and B, Supplemental Figure 9, A and B). However, knockdown of HOXA9 significantly downregulated levels of TGFB2 RNA and secreted, activated TGF-β2 protein (P < 0.001, Figure 7, A and B). Conversely, enforced expression of HOXA9, but not HOXA10 or HOXA11, induced TGF-β2 expression in MOSEC cells (Supplemental Figure 9, B and C). HOXA9, HOXA10, and HOXA11 are normally coexpressed in the posterior uterine segment (10, 11). To additionally evaluate whether TGF-β2 expression is selectively controlled by HOXA9, we analyzed TGF-β2 expression in the posterior uterine segment of Hox knockout mice. As compared with that of wild-type littermates, glandular and stromal staining of TGF-β2 was markedly weaker in uterine tissues of Hoxa9–/– mice but not of Hoxa10–/– or Hoxa11–/– mice (Supplemental Figure 9D). Levels of HOXA9 transcripts significantly correlated with TGFB2 transcript levels in EOC tissue specimens (r = 0.65, P = 0.00004), but no significant correlation was observed for TGFB1 (r = 0.11, P = 0.54) (Figure 7C). High HOXA9 protein levels were also associated with high TGF-β2 protein levels in clinical specimens of omental implants (Supplemental Figure 9, E and F). These findings raise the possibility that HOXA9 controls TGFB2 expression at the transcriptional level.
Figure 7. HOXA9 induces TGF-β2 expression in EOC cells.

(A) Relative TGFB1 and TGFB2 mRNA levels in SKOV3ip lines. *P < 0.001; **P < 0.005. (B) TGF-β1 and TGF-β2 levels in media conditioned by SKOV3ip lines. *P < 0.001. (C) Relative HOXA9, TGFB1, and TGFB2 mRNA levels in primary ovarian tumors of 33 cases. Correlations were determined by Spearman test. (D) Representation of the mouse Tgfb2 promoter. Locations of 5 putative HOXA9-binding sites evaluated by chromatin IP (S1 to S5) and regions evaluated in luciferase reporter assays (pA, pB, pC, pC-mtS4) relative to the transcription start site (TSS) are indicated. Wild-type sequences within the S4 site (in pA and pC) and mutant sequences (in pC-mtS4) are shown. (E) Chromatin IP analysis of interactions of FLAG-tagged HOXA9 in MOSEC cells with sites S1 to S5. Immunoprecipitated DNA was assayed by qPCR and is expressed as a percentage of total chromatin input. (F) Chromatin IP analysis of interactions of endogenous HOXA9 in SKOV3ip cells with conserved sites S4 and S5 in the human TGFB2 promoter. The input corresponds to 1% of chromatin solution before IP. IP using cells expressing FLAG-tag alone or cells expressing HOXA9 shRNA (shA9-B) as well as IP with IgG and amplification of Gapdh and GAPDH as irrelevant genes are included as negative controls in E and F. Full uncut gels are shown in the Supplemental Material. (G) qPCR analysis of immunoprecipitated DNA from assays in F, expressed as a percentage of total chromatin input. (H) Activity of Tgfb2 promoter regions shown in D was assayed in +HOXA9 control (black bar) and HOXA9-knockdown (white bar) SKOV3ip cells. Average relative luciferase activities of 3 independent assays are shown.
TGFB2 is a direct transcriptional target of HOXA9.
Five putative HOXA9-binding sites were identified in the mouse Tgfb2 promoter (Figure 7D). Binding of ectopic HOXA9 in MOSEC cells was detected to 2 of these sites (S4 and S5) in chromatin IP assays (Figure 7E). Binding of endogenous HOXA9 to the conserved S4 and S5 sites in the human TGFB2 promoter was detected in +HOXA9 control SKOV3ip cells, but no binding was detected in HOXA9-knockdown SKOV3ip cells (Figure 7, F and G). Luciferase reporter assays using deletion constructs of the Tgfb2 promoter indicated that the S4 site, but not the S5 site, was essential for HOXA9-induced promoter activity (Figure 7H). Activation by HOXA9 through the S4 binding site was confirmed in reporter assays by using a Tgfb2 promoter construct in which the S4 site was mutated (Figure 7H). These results indicate that TGFB2 is a direct transcriptional target of HOXA9.
HOXA9 expression in EOC cells also increases stromal TGF-β expression.
TGF-β1 not only induces transition of normal fibroblasts and MSCs into CAFs but is also highly expressed by CAFs (18). We investigated whether HOXA9 could stimulate stromal TGF-β expression through its induction of tumor-derived TGF-β2. Omental fibroblasts that were primed in medium conditioned by +HOXA9 control but not by HOXA9-knockdown SKOV3ip cells had significantly elevated expression of TGF-β1 and TGF-β2 (P < 0.005, Figure 8A and Supplemental Figure 10A). Similarly, TGF-β1 and TGF-β2 were more strongly induced in fibroblasts when primed in medium conditioned by +HOXA9 MOSEC cells than when primed in medium conditioned by vector-control, +HOXA10, or +HOXA11 MOSEC cells (Supplemental Figure 10, B and C). TGFB1 and TGFB2 mRNA levels were also induced in adipose MSCs by +HOXA9 tumor–conditioned medium, whereas TGFB1 but not TGFB2 mRNA was induced in bone marrow MSCs (Supplemental Figure 10A). Tgfb1 and Tgfb2 mRNA levels in mouse host cells were lower in omental tumors of HOXA9-knockdown models than in those of +HOXA9 control models (Figure 8B). TGFB1 and TGFB2 expression was induced in human omental fibroblasts following stimulation with recombinant TGF-β1 and TGF-β2 at concentrations released by +HOXA9 tumor cells and by fibroblasts primed by +HOXA9 tumor cells (Figure 8C). TGFB1 and TGFB2 expression in fibroblasts was inhibited when +HOXA9 tumor–conditioned medium was depleted of TGF-β2 (Figure 8D). These findings suggest that HOXA9 expression in EOC cells can induce autostimulatory production of TGF-β ligands in the stroma.
Figure 8. TGF-β2 induces expression of TGF-β ligands and CAF markers in omental fibroblasts.

(A) Levels of TGF-β1 and TGF-β2 released by omental fibroblasts that were primed in SKOV3ip-conditioned medium or left unprimed, as in Figure 4C. Average results of ELISAs of 3 independent sets of each type of fibroblast-conditioned medium are shown. *P < 0.005; #P < 0.001. (B) Relative levels of TGFB1 and TGFB2 mRNAs (in human EOC cells) and Tgfb1 and Tgfb2 mRNAs (in mouse host cells) in omental tumors of mice that were inoculated with +HOXA9 control (Nontargeting) and HOXA9-knockdown (shA9-B) SKOV3ip lines (n = 5 mice per group). Specificity of human- and mouse-specific qRT-PCR primers is shown in Supplemental Figure 6B. *P = 0.025; †P = 0.018. (C and D) Relative TGFB1, TGFB2, ACTA2, and FAP mRNA levels in omental fibroblasts at 5 days after incubation with (C) recombinant TGF-β1 and TGF-β2 at the indicated concentrations and (D) media conditioned by +HOXA9 control SKOV3ip cells, where SKOV3ip-conditioned medium was left untreated, treated with IgG, or depleted by IP with Ab to TGF-β2.
Effects of HOXA9 on fibroblasts are mediated by its induction of TGF-β2 expression in EOC cells.
We investigated whether TGF-β2 recapitulates the stimulatory effects of HOXA9 on fibroblasts and whether the effects of HOXA9 could be reversed by inhibiting tumor-derived TGF-β2. Expression of ACTA2, FAP, IL6, CXCL12, and VEGFA was induced in omental fibroblasts following stimulation with recombinant TGF-β2 at concentrations released by +HOXA9 tumor cells (Figure 8C and Supplemental Figure 11A). Conversely, depletion of TGF-β2 from +HOXA9 tumor–conditioned medium inhibited expression of CAF markers and growth factors in fibroblasts (Figure 8D and Supplemental Figure 11B). Identical results were obtained in MSCs (data not shown). TGF-β2 did not alter proliferation of fibroblasts and MSCs (data not shown).
To determine whether the ability of HOXA9 to activate fibroblasts and thereby promote proliferation of tumor and endothelial cells is mediated by its induction of tumor-derived TGF-β2, we evaluated the effects of specifically inhibiting TGF-β2 in +HOXA9 tumor cells. Stable expression of TGFB2 shRNA in SKOV3ip cells decreased the TGF-β2 level to the same low level seen in HOXA9-knockdown SKOV3ip cells (Figure 9A). In addition, TGFB2 cDNA was stably expressed in HOXA9-knockdown SKOV3ip cells to test whether reconstituting TGF-β2 in tumor cells restores the effects of HOXA9 (Figure 9A). Neither knockdown nor overexpression of TGF-β2 affected growth rates of SKOV3ip cells cultured in nonconditioned medium (Supplemental Figure 11C). However, fibroblasts that were primed by TGF-β2–knockdown SKOV3ip cells had a weaker stimulatory effect on proliferation of tumor cells and of endothelial cells than fibroblasts primed by control SKOV3ip cells (Supplemental Figure 11, D and E). This weaker stimulatory effect was identical to that of fibroblasts primed by HOXA9-knockdown SKOV3ip cells (Supplemental Figure 11, D and E). Conversely, reconstitution of TGF-β2 in HOXA9-knockdown SKOV3ip cells increased the ability of fibroblasts to stimulate growth of tumor and endothelial cells (Supplemental Figure 11, D and E). These findings indicate that HOXA9 expression in EOC cells promotes the ability of fibroblasts to stimulate growth of tumor and endothelial cells through its induction of tumor-derived TGF-β2.
Figure 9. Effects of HOXA9 on growth of EOC xenografts are mediated via its induction of tumor-derived TGF-β2.

(A) TGF-β2 levels released by control (Nontargeting), TGF-β2–knockdown (shTGF-β2), and HOXA9-knockdown (shA9-B) SKOV3ip lines and a HOXA9-knockdown line stably expressing TGF-β2 (shA9-B + TGF-β2). *P < 0.001. (B and C) Female nude mice were inoculated i.p. with SKOV3ip lines and sacrificed at 4 weeks thereafter. (B) Implants viewed under a fluorescence stereoscope. The arrow indicates the omental implant in the control group. Original magnification, ×0.7. (C) The average number of Ki-67+ tumor cells, αSMA+ cells, and microvessels in tumors was calculated by scoring 5 random fields of stained tumor tissue sections of each mouse (n = 5 mice per group). *P < 0.005; †P < 0.01. (D) Survival rates of mice inoculated i.p. with SKOV3ip lines (n = 10 per group). Significance values for each group as compared with the Nontargeting control group are indicated. (E) Survival rates of patients in the AOCS data set stratified by TGFB2 expression in tumors, where TGFB2 transcript levels were defined as High (≥ upper quartile) and Low (≤ lower quartile) (n = 72 cases per group).
Stimulation by HOXA9 of EOC growth in vivo is mediated by its induction of tumor-derived TGF-β2.
To validate our in vitro findings, we first evaluated whether reconstituting TGF-β2 in HOXA9-knockdown EOC cells restores the tumor growth–promoting phenotype of HOXA9 in i.p. xenograft models. As compared with that in mice inoculated with HOXA9-knockdown SKOV3ip cells, mice that were inoculated with HOXA9-knockdown SKOV3ip cells stably expressing TGF-β2 developed substantially larger tumors (Figure 9B). Reconstitution of TGF-β2 in HOXA9-knockdown tumor cells markedly increased mitotic activity, abundance of αSMA+ cells, and microvessel density in tumors to the levels seen in +HOXA9 control tumors (Figure 9C). As compared with that of mice with HOXA9-knockdown tumors, mice with HOXA9-knockdown tumors stably expressing TGF-β2 had significantly shorter survival times (P = 0.003, Figure 9D). The poor survival rate of these mice was almost identical to that of the +HOXA9 control group (Figure 9D). Conversely, tumor growth was reduced in mice that were inoculated with TGF-β2–knockdown SKOV3ip cells as compared with that in mice with control tumors (Figure 9B). Mitotic activity, αSMA+ cells, and microvessel density were markedly reduced in TGF-β2–knockdown tumors, similar to that observed in HOXA9-knockdown tumors (Figure 9C). As compared with that of mice with control tumors, mice with TGF-β2–knockdown tumors had significantly longer survival times (P = 0.003, Figure 9D). The higher survival rate of mice with TGF-β2–knockdown tumors and the reduced size of omental tumors in these mice were very similar to observations in the HOXA9-knockdown group (Figure 9, B and D). When patients with EOC were stratified according to expression of TGFB2 in tumors, high TGFB2 expression was significantly associated with poor overall survival (P = 0.005, HR of death = 2.03, 95% CI = 1.24 to 3.34; Figure 9E). This association of high TGFB2 expression with poor survival in the AOCS data set was consistent with the association of high HOXA9 expression with poor survival in the same patient cohort (Figure 1) and with observations in SKOV3ip xenograft models.
To confirm our findings, we evaluated the effect of inhibiting TGF-β2 in +HOXA9 MOSEC cells in vivo (Supplemental Figure 12A). As compared with those of mice inoculated with +HOXA9 MOSEC cells, mice that were inoculated with +HOXA9 MOSEC cells in which TGF-β2 was knocked down developed smaller i.p. tumors (Supplemental Figures 12, B and C) and had significantly longer survival times (P = 0.0002, Supplemental Figure 12D). Knockdown of TGF-β2 in +HOXA9 MOSEC cells substantially reduced mitotic activity, abundance of αSMA+ cells, and microvessel density in tumors (Supplemental Figure 12E). Together, our findings support a model in which HOXA9 promotes tumor growth by inducing tumor-derived TGF-β2 that acts in a paracrine manner to stimulate the ability of fibroblasts to support tumor growth (Figure 10).
Figure 10. Bidirectional signaling between EOC cells and CAFs.

Control of growth factor expression and interactions among EOC cells, CAFs, and endothelial cells by HOXA9-induced, tumor-derived TGF-β2.
Discussion
As is the case in normal organogenesis, it is increasingly recognized that tumor growth is orchestrated by dynamic interactions between epithelial and stromal cells (12, 13). Whereas the tumor growth–promoting ability of CAFs has been extensively studied, it is not clear whether CAFs are reciprocally controlled by developmental pathways that are activated in tumor cells. EOC is an ideal disease site to investigate this question, because its histogenesis remarkably recapitulates patterning of the Müllerian system. Here, we show that expression of HOXA9, a Müllerian-patterning gene, in EOC cells is strongly associated with poor outcomes in patients with EOC and in xenograft models and induces normal resident fibroblasts and MSCs to acquire molecular and functional features of CAFs that promote EOC growth. Our finding that HOXA9 promotes CAFs by inducing expression of TGF-β2 in EOC cells supports evidence that tumor-derived factors play important roles in governing the composition of the tumor stroma (13). Our studies demonstrated that inhibiting TGF-β2 in EOC cells that express HOXA9 substantially blocks the stimulatory effects of HOXA9 on CAFs and tumor growth. Conversely, reconstituting TGF-β2 in EOC cells in which HOXA9 is inhibited restored the CAF-activating, tumor growth–promoting phenotype of HOXA9. These findings indicate that the tumor growth–promoting effect of HOXA9 is mediated, in substantial part, by stimulating CAFs via its induction of tumor-derived TGF-β2 (Figure 10). However, since progression of EOC is driven by multiple autocrine and paracrine mechanisms (1, 2), we cannot exclude the possibility that HOXA9 also promotes progression of this disease by stimulating other signaling pathways in tumor or host cells.
A significant consequence of the induction by HOXA9 of tumor-derived TGF-β2 is the reinforcement of tumor-stroma cross-talk via increased stromal production of CXCL12, IL-6, and VEGF-A that act in a paracrine manner to stimulate proliferation of tumor and endothelial cells (Figure 10). Elevated stromal CXCL12 levels might also increase angiogenesis and CAF abundance in +HOXA9 tumors, as CXCL12 stimulates recruitment of endothelial progenitor cells and MSCs to tumors (15, 17). We have found that TGF-β2 also stimulates chemotaxis of MSCs toward tumor cells (S.Y. Ko and H. Naora, unpublished observations). Increased tumor-derived TGF-β2 levels could additionally sustain CAFs by stimulating autoinduction of TGF-β ligands in the stroma. The significance of TGF-β autoinduction in wound healing has long been recognized (25). Acquisition of a TGF-β autocrine signaling loop in resident mammary fibroblasts has been reported to drive transition of these cells into CAFs (18), but a mechanism for initiation of this loop was not identified. Our findings support the existence of a self-sustaining TGF-β autoregulatory loop in CAFs and indicate that this loop is triggered in EOC, at least in part, by tumor-derived TGF-β2. TGF-β1, TGF-β2, and TGF-β3 bind the TGF-β type II receptor (TβRII), but these ligands have many non-compensatory functions (26). To our knowledge, our study is the first to demonstrate the significance of TGF-β2 in promoting CAFs. Our finding that TGFB2 but not TGFB1 is a HOXA9 target gene supports evidence that these ligands are differentially regulated by developmental cues (27). Although TGFB3 transcripts have been detected in EOCs, secreted TGF-β3 protein has not been detected (28, 29). We did not detect secreted TGF-β3 protein in our models or in 17 other EOC lines that we tested. Two independent studies have found higher incidence of TGF-β2 than TGF-β1 expression in EOC (28, 29). Anti–TGF-β2 therapies have been developed to circumvent immunosuppression in patients with cancer (30). Our findings raise the intriguing possibility that anti–TGF-β2 therapies or small molecule TGF-β receptor inhibitors could be used to inhibit CAFs in EOC.
Whereas recent evidence indicates that CAFs derive from normal tissue-resident fibroblasts and MSCs (14–18), tumor cells that have undergone EMT are thought to account for only a small fraction of CAFs (13). Because TGF-β signaling induces EMT in many cell types (31), CAFs in +HOXA9 tumors might also derive from tumor cells. This possibility cannot be completely eliminated but is negligible, as we found virtually no overlap between +HOXA9 tumor cells and cells that expressed αSMA in situ. Furthermore, HOXA9 did not alter expression of genes encoding transcription factors that are induced by TGF-β and orchestrate EMT. Whereas TGF-β1 and TGF-β3 bind to TβRII with high affinity, TGF-β2 requires the accessory molecule betaglycan to facilitate receptor binding (32). Loss of betaglycan expression has been reported in 73% of high-grade EOCs (33). It is thus unlikely that CAFs in +HOXA9 tumors derive substantially from tumor cells that undergo EMT in response to induction of TGF-β2 by HOXA9.
It is thought that CAFs derive from bone marrow MSCs at advanced stages of disease, whereas tissue-resident MSCs (and fibroblasts) are the predominant source of CAFs at early stages of tumor development (34). Whereas the number of bone marrow MSCs declines with age (35), visceral white adipose tissue increases with age. Because EOC occurs more frequently in postmenopausal women, adipose MSCs might significantly contribute to the EOC stroma. Adipose MSCs display multipotency comparable to bone marrow MSCs (24) and have been reported to stimulate growth of tumor cells (22). Our finding that adipose MSCs can be more effectively educated by +HOXA9 cells than –HOXA9 EOC cells to acquire CAF features could explain the greater propensity of +HOXA9 tumors to develop large implants on the omentum that is a major repository of visceral white adipose tissue. Normal cells expressing CAF markers have been detected in omental tissues of patients with EOC without overt omental metastasis (21). Because TGF-β2 is a soluble factor, the propensity of +HOXA9 tumors for omental involvement might stem from fertilization of the omental “soil” by tumor-derived TGF-β2.
Although many homeobox genes are aberrantly expressed in tumors, their mechanisms in tumorigenesis are poorly understood and only a few bona fide transcriptional target genes have been identified (5–7). Some homeobox genes promote angiogenesis by inducing expression of angiogenic factors or chemotactic factors for endothelial progenitor cells (36, 37). However, many of the homeobox genes studied to date alter tumor cell proliferation or survival in vitro (38–40), implying that these genes modulate tumor growth via tumor cell-autonomous mechanisms. In contrast, our study demonstrates that the ability of HOXA9 to promote EOC growth is mediated by its transcriptional activation of TGFB2 in tumor cells and the paracrine effects of tumor-derived TGF-β2 on CAFs. However, CAFs might also be activated by other mechanisms, since not all EOCs express HOXA9. HOXA9 expression has been detected in EOCs at a range of levels in 2 independent studies (11, 41) and also widely varies among patients with EOC in the AOCS and TCGA cohorts. HOXA9 promoter methylation has been reported in EOCs (42, 43) but has been detected at significantly lower frequency in late-stage than in early-stage EOC (43). This is consistent with our findings that HOXA9 promotes progression of EOC. Although some HOX genes have overlapping functions (4), our study demonstrates that the ability of HOXA9 expression in EOC cells to induce normal fibroblasts and MSCs to acquire CAF features is not shared by HOXA10 or HOXA11. Interestingly, HOXA10 has both tumor-promoting and tumor-suppressive properties. HOXA10 induces expression of β3-integrin, which increases ovarian epithelial cell proliferation by promoting direct contact with fibroblasts (44, 45), but HOXA10 and β3-integrin also inhibit invasiveness (46, 47). Consistent with these prior reports, we observed a modest increase in mitotic activity in +HOXA10 MOSEC xenografts, as compared with that in vector-controls, but found no significant association of HOXA10 with poor survival in xenograft models or in patients with EOC.
The precise mechanisms of HOXA9 in controlling Müllerian patterning are unknown. Whereas HOXA10 and HOXA11 are upregulated in the endometrium during luteal phase, HOXA9 is downregulated (48–50), implicating a function of HOXA9 in proliferative control in the Müllerian system. There is evidence that TGF-β2 is required for development of the female reproductive tract (26), but it is not known whether HOXA9 controls Müllerian patterning by regulating TGF-β2. Our analysis of Hox knockout mice suggest that TGF-β2 is selectively induced by HOXA9, but not by HOXA10 or HOXA11, in normal Müllerian tissue as well as in EOC cells. HOXA9 has been identified to be the single most highly correlated gene for poor prognosis in acute myeloid leukemia (AML) (51). Overexpression of Hoxa9 in primary bone marrow cells enhances hematopoietic stem cell regeneration, expands the myeloid progenitor pool, and collaborates with Meis1a to induce AML in mice (52, 53). Very few target genes have been identified that explain the leukemogenic potential of HOXA9. Interestingly, TGF-β2, like HOXA9, is preferentially expressed in primitive hematopoietic cells and stimulates the repopulating capacity of hematopoietic stem cells (54). It is possible that HOXA9, via its induction of TGF-β2, might similarly control expansion of progenitor cells in the Müllerian system. However, HOX genes function in a context-dependent manner (4), and the function of a HOX gene cannot be directly inferred from studies in different cell types. Indeed, HOXA9 has been reported to be downregulated in breast and lung cancer cells and to have tumor-suppressive properties in these tumor cell types (55, 56).
In summary, our findings support a model in which HOXA9 expression in EOC cells educates the stroma to become permissive for tumor growth. Whereas targeting HOXA9 is therapeutically challenging, inhibiting its effector TGF-β2 could be a promising therapeutic strategy. This is significant for advanced-stage EOC, which is rarely cured by conventional chemotherapies. Moreover, because distinct sets of homeobox genes control expansion and specification of different cell lineages, further studies of these patterning regulators could provide new insights into the control of tumor-stroma interactions in other disease sites.
Methods
Reagents.
Abs, growth factors, and plasmids are described in Supplemental Methods.
Human tissue specimens and cells.
Archived human EOC tissue specimens, that were not necessary for diagnosis and were delinked from patient identifiers, were obtained from gynecologic tumor banks at The University of Texas MD Anderson Cancer Center and University of Chicago. All cases were stage III/IV serous EOC. Fibroblasts were isolated from normal omental tissues of women undergoing surgery for benign conditions as previously described (57), with informed patient consent. Normal human bone marrow MSCs were provided through the Tulane Center for Gene Therapy, MSC cell distribution center (Darwin Prockop, Tulane University, New Orleans, Louisiana, USA). Normal human adipose MSCs isolated from lipoaspirate were purchased from ATCC. Immortalized, nontumorigenic human ovarian surface epithelial cells (TNOE072) were provided by Jinsong Liu (The University of Texas MD Anderson Cancer Center). Cell culture media are described in Supplemental Methods.
Mouse cell cultures, EOC cell lines, and transfection.
Immortalized mouse endothelial cells (58) were provided by Robert Langley (The University of Texas MD Anderson Cancer Center). L929 mouse skin fibroblasts were purchased from ATCC. Stable MOSEC lines are described in previous studies (11). The serous EOC line SKOV3ip and clear-cell EOC line ES-2 were provided by Gordon Mills (The University of Texas MD Anderson Cancer Center) and Patrice Morin (National Institute on Aging, Baltimore, Maryland, USA), respectively. Cell culture media are described in Supplemental Methods. SKOV3ip, ES-2, and MOSEC cells were transfected with plasmids by using FuGENE6 reagent (Roche) and selected with puromycin (0.5 μg/ml).
Conditioned media and in vitro cell growth assays.
Generation of conditioned media and in vitro growth assays are described in Supplemental Methods.
Animal studies.
Four-week-old female nude mice were purchased from National Cancer Institute. Mice were inoculated i.p. with cells of MOSEC (1.5 × 106), SKOV3ip (2 × 106), and ES-2 (1 × 106) lines or s.c. with cells of SKOV3ip (1 × 106) and ES-2 (6 × 105) lines. For survival studies, mice were euthanized by CO2 asphyxiation when morbid ascites had developed. Volumes of s.c. tumors were calculated from tumor diameters measured daily using calipers. GFP-expressing i.p. xenografts were visualized in euthanized mice under a Leica MZML III fluorescence stereomicroscope. Tissues from Hox knockout mice were provided by Steven Potter (Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio, USA) and Deneen Wellik (University of Michigan, Ann Arbor, Michigan, USA).
Immunohistochemistry.
Formalin-fixed, paraffin-embedded tissue sections were used for staining with H&E and with Abs. Staining was detected by streptavidin-biotin-peroxidase and 3, 3′-diaminobenzidine (Dako). Staining using Alexa Fluor–conjugated Abs was performed on frozen tissue sections. Evaluation of staining with each Ab is described in the figure legends.
Immunoblotting and ELISA.
Whole cell extracts were separated by SDS-PAGE and transferred on to PVDF membrane (GE Healthcare). ELISAs kits were purchased from R&D Systems (for mouse TGF-β1 and human IL-6, CXCL12, VEGF-A, TGF-β1, TGF-β2) and LifeSciences Advanced Technologies (for mouse TGF-β2). Levels of each growth factor were assayed in 3 independent sets of each type of conditioned medium and normalized to total cellular protein content.
qRT-PCR, chromatin IP, and luciferase reporter assays.
These assays are described in Supplemental Methods. Primers are listed in Supplemental Table 2.
Bioinformatic analysis.
Analysis of gene expression data from the AOCS and TCGA studies is described in Supplemental Methods.
Statistics.
Statistical analysis was performed using STATISTICA6 software (StatSoft Inc.). Statistically significant data in in vitro and in vivo assays were assessed by unpaired 2-tailed Student’s t test unless noted otherwise. Data represent mean ± SD. P values of less than 0.05 were considered significant. Significance of differences in gene expression among groups of patients were assessed by Mann-Whitney U test. Survival times of groups were compared by Kaplan-Meier plot analysis, and intergroup differences were assessed by log-rank test. Cox’s F test was used in mouse survival studies. Correlation coefficients were determined by Spearman test.
Study approval.
Studies using human tissues and cells were reviewed and approved by The University of Texas MD Anderson Cancer Center Institutional Research Board and University of Chicago Institutional Research Board, and informed patient consent was obtained. Animal studies were reviewed and approved by The University of Texas MD Anderson Cancer Center Institutional Committee on Use and Care of Animals.
Supplementary Material
Acknowledgments
This work was supported by grants from the Cancer and Prevention Research Institute of Texas (RP120390) (to H. Naora) and US NIH (CA101826, CA141078) (to H. Naora) and an MD Anderson Cancer Center Institutional Research Grant (to H. Naora).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(10):3603–3617. doi:10.1172/JCI62229.
References
- 1.Lengyel E. Ovarian cancer development and metastasis. Am J Pathol. 2010;177(3):1053–1064. doi: 10.2353/ajpath.2010.100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naora H, Montell DJ. Ovarian cancer metastasis: Integrating insights from disparate model organisms. Nat Rev Cancer. 2005;5(5):355–366. doi: 10.1038/nrc1611. [DOI] [PubMed] [Google Scholar]
- 3.Clements PB, Young RH. Atlas of Gynecologic Surgical Pathology. Philadelphia, Pennsylvania, USA: WB Saunders; 2000. [Google Scholar]
- 4.Pearson JC, Lemons D, McGinnis W. Modulating HOX gene functions during animal body patterning. . Nat Rev Genet. 2005;6(12):893–904. doi: 10.1038/nrg1726. [DOI] [PubMed] [Google Scholar]
- 5.Abate-Shen C. Deregulated homeobox gene expression in cancer: cause or consequence? Nat Rev Cancer. 2002;2(10):777–785. doi: 10.1038/nrc907. [DOI] [PubMed] [Google Scholar]
- 6.Samuel S, Naora H. Homeobox gene expression in cancer: Insights from developmental regulation and deregulation. Eur J Cancer. 2005;41(16):2428–2437. doi: 10.1016/j.ejca.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 7.Robson EJ, He SJ, Eccles MR. A panorama of PAX genes in cancer and development. . Nat Rev Cancer. 2006;6(1):52–62. doi: 10.1038/nrc1778. [DOI] [PubMed] [Google Scholar]
- 8.Hsieh-Li HM, et al. Hoxa 11 structure, extensive antisense transcription, and function in male and female fertility. . Development. 1995;121(5):1373–1385. doi: 10.1242/dev.121.5.1373. [DOI] [PubMed] [Google Scholar]
- 9.Benson GV, et al. Mechanisms of reduced fertility in Hoxa-10 mutant mice: uterine homeosis and loss of maternal Hoxa-10 expression. . Development. 1996;122(9):2687–2696. doi: 10.1242/dev.122.9.2687. [DOI] [PubMed] [Google Scholar]
- 10.Taylor HS, Vanden Heuvel GB, Igarashi P. A conserved Hox axis in the mouse and human female reproductive system: late establishment and persistent adult expression of the Hoxa cluster genes. . Biol Reprod. 1997;57(6):1338–1345. doi: 10.1095/biolreprod57.6.1338. [DOI] [PubMed] [Google Scholar]
- 11.Cheng W, Liu J, Yoshida H, Rosen D, Naora H. Lineage infidelity of epithelial ovarian cancers is controlled by HOX genes that specify regional identity in the reproductive tract. . Nat Med. 2005;11(5):531–537. doi: 10.1016/0002-9343(51)90210-0. [DOI] [PubMed] [Google Scholar]
- 12.Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol. 2006;1:119–150. doi: 10.1146/annurev.pathol.1.110304.100224. [DOI] [PubMed] [Google Scholar]
- 13.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 14.Direkze NC, et al. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004;64(23):8492–8495. doi: 10.1158/0008-5472.CAN-04-1708. [DOI] [PubMed] [Google Scholar]
- 15.Orimo A, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 16.Spaeth EL, et al. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One. 2009;4(4):e4992. doi: 10.1371/journal.pone.0004992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quante M, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19(2):257–272. doi: 10.1016/j.ccr.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kojima Y, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;107(46):20009–20014. doi: 10.1073/pnas.1013805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tothill RW, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res. 2008;14(16):5198–5208. doi: 10.1158/1078-0432.CCR-08-0196. [DOI] [PubMed] [Google Scholar]
- 20.Petersen OW, et al. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am J Pathol. 2003;162(2):391–402. doi: 10.1016/S0002-9440(10)63834-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, et al. Ovarian cancer-associated fibroblasts contribute to epithelial ovarian carcinoma metastasis by promoting angiogenesis, lymphangiogenesis and tumor cell invasion. Cancer Lett. 2011;303(1):47–55. doi: 10.1016/j.canlet.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 22.McLean K, et al. Human ovarian carcinoma-associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J Clin Invest. 2011;121(8):3206–3219. doi: 10.1172/JCI45273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.da Silva Meirelles L, Chagastelles PC, Nardi NB. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J Cell Sci. 2006;119(11):2204–2213. doi: 10.1242/jcs.02932. [DOI] [PubMed] [Google Scholar]
- 24.Zuk PA, et al. Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell. 2002;13(12):4279–4295. doi: 10.1091/mbc.E02-02-0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bascom CC, et al. Complex regulation of transforming growth factor β1, β2 and β3 mRNA expression in mouse fibroblasts and keratinocytes by transforming growth factors β1 and β2. Mol Cell Biol. 1989;9(12):5508–5515. doi: 10.1128/mcb.9.12.5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanford LP, et al. TGFβ2 knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development. 1997;124(13):2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pelton RW, Saxena B, Jones M, Moses HL, Gold LI. Immunohistochemical localization of TGFβ1, TGFβ2 and TGFβ3 in the mouse embryo: Expression patterns suggest multiple roles during embryonic development. J Cell Biol. 1991;115(4):1091–1105. doi: 10.1083/jcb.115.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartlett JM, et al. Transforming growth factor-β isoform expression in human ovarian tumors. Eur J Cancer. 1997;33(14):2397–2403. doi: 10.1016/S0959-8049(97)00304-3. [DOI] [PubMed] [Google Scholar]
- 29.Abendstein B, et al. Regulation of transforming growth factor-β secretion by human peritoneal mesothelial and ovarian carcinoma cells. Cytokine. 2000;12(7):1115–1119. doi: 10.1006/cyto.1999.0632. [DOI] [PubMed] [Google Scholar]
- 30.Nemunaitis J, et al. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. J Clin Oncol. 2006;24(29):4721–4730. doi: 10.1200/JCO.2005.05.5335. [DOI] [PubMed] [Google Scholar]
- 31.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 32.Lopez-Casillas F, Wrana JL, Massague J. Betaglycan presents ligand to the TGFβ signaling receptor. Cell. 1993;73(7):1435–1444. doi: 10.1016/0092-8674(93)90368-Z. [DOI] [PubMed] [Google Scholar]
- 33.Hempel N, et al. Loss of betaglycan expression in ovarian cancer: role in motility and invasion. Cancer Res. 2007;67(11):5231–5238. doi: 10.1158/0008-5472.CAN-07-0035. [DOI] [PubMed] [Google Scholar]
- 34.Zhao M, Dumur CI, Holt SE, Beckman MJ, Elmore LW. Multipotent adipose stromal cells and breast cancer development: think globally, act locally. Mol Carcinog. 2010;49(11):923–927. doi: 10.1002/mc.20675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stenderup K, Justesen J, Clausen C, Kassem M. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone. 2003;33(6):919–926. doi: 10.1016/j.bone.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 36.Hara F, et al. A homeobox gene related to Drosophila Distal-less promotes ovarian tumorigenicity by inducing expression of vascular endothelial growth factor and fibroblast growth factor-2. . Am J Pathol. 2007;170(5):1594–1606. doi: 10.2353/ajpath.2007.061025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mace KA, et al. HOXA3 modulates injury-induced mobilization and recruitment of bone marrow-derived cells. Stem Cells. 2009;27(7):1654–1665. doi: 10.1002/stem.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muratovska A, Zhou C, He S, Goodyer P, Eccles MR. Paired-Box genes are frequently expressed in cancer and often required for cancer cell survival. . Oncogene. 2003;22(39):7989–7997. doi: 10.1038/sj.onc.1206766. [DOI] [PubMed] [Google Scholar]
- 39.Coletta RD, et al. The Six1 homeoprotein stimulates tumorigenesis by reactivation of cyclin A1. Proc Natl Acad Sci U S A. 2004;101(17):6478–6483. doi: 10.1073/pnas.0401139101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trinh BQ, Barengo N, Naora H. Homeodomain protein DLX4 counteracts key transcriptional control mechanisms of the TGF–β_ cytostatic program and blocks the antiproliferative effect of TGF-β. Oncogene. 2011;30(24):2718–2729. doi: 10.1038/onc.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bahrani-Mostafavi Z, et al. Correlation analysis of HOX, ErbB and IGFBP family gene expression in ovarian cancer. Cancer Invest. 2008;26(10):990–998. doi: 10.1080/07357900802074349. [DOI] [PubMed] [Google Scholar]
- 42.Montavon C, et al. Prognostic and diagnostic significance of DNA methylation patterns in high-grade serous ovarian cancer. Gynecol Oncol. 2012;124(3):582–588. doi: 10.1016/j.ygyno.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 43.Wu Q, et al. DNA methylation profiling of ovarian carcinomas and their in vitro models identifies HOXA9, HOXB5, SCGB3A1 and CRABP1 as novel targets. . Mol Cancer. 2007;6:45. doi: 10.1186/1476-4598-6-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daftary GS, Troy PJ, Bagot CN, Young SL, Taylor HS. Direct regulation of β3-integrin subunit gene expression by HOXA10 in endometrial cells. Mol Endocrinol. 2002;16(3):571–579. doi: 10.1210/me.16.3.571. [DOI] [PubMed] [Google Scholar]
- 45.Ko SY, Lengyel E, Naora H. The Müllerian HOXA10 gene promotes growth of ovarian surface epithelial cells by stimulating epithelial–stromal interactions. . Mol Cell Endocrinol. 2010;317(1–2):112–119. doi: 10.1016/j.mce.2009.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoshida H, Broaddus R, Cheng W, Xie S, Naora H. Deregulation of the HOXA10 homeobox gene in endometrial carcinoma: Role in epithelial-mesenchymal transition. . Cancer Res. 2006;66(2):889–897. doi: 10.1158/0008-5472.CAN-05-2828. [DOI] [PubMed] [Google Scholar]
- 47.Kaur S, et al. Beta3-integrin expression on tumor cells inhibits tumor progression, reduces metastasis, and is associated with a favorable prognosis in patients with ovarian cancer. Am J Pathol. 2009;175(5):2184–2196. doi: 10.2353/ajpath.2009.090028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taylor HS, Arici A, Olive D, Igarashi P. HOXA10 is expressed in response to sex steroids at the time of implantation in the human endometrium. . J Clin Invest. 1998;101(7):1379–1384. doi: 10.1172/JCI1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taylor HS, Igarashi P, Olive DL, Arici A. Sex steroids mediate HOXA11 expression in the human peri-implantation endometrium. . J Clin Endocrinol Metab. 1999;84(3):1129–1135. doi: 10.1210/jc.84.3.1129. [DOI] [PubMed] [Google Scholar]
- 50.Carson DD, et al. Changes in gene expression during the early to mid-luteal (receptive phase) transition in human endometrium detected by high-density microarray screening. Mol Hum Reprod. 2002;8(9):871–879. doi: 10.1093/molehr/8.9.871. [DOI] [PubMed] [Google Scholar]
- 51.Golub TR, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286(5439):531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 52.Kroon E, et al. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J. 1998;17(13):3714–3725. doi: 10.1093/emboj/17.13.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thorsteinsdottir U, et al. Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. . Blood. 2002;99(1):121–129. doi: 10.1182/blood.V99.1.121. [DOI] [PubMed] [Google Scholar]
- 54.Langer JC, Henckaerts E, Orenstein J, Snoeck HW. Quantitative trait analysis reveals transforming growth factor-β2 as a positive regulator of early hematopoietic progenitor and stem cell function. J Exp Med. 2004;199(1):5–14. doi: 10.1084/jem.20030980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gilbert PM, et al. HOXA9 regulates BRCA1 expression to modulate human breast tumor phenotype. J Clin Invest. 2010;120(5):1535–1550. doi: 10.1172/JCI39534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Son JW, et al. Genome-wide combination profiling of DNA copy number and methylation for deciphering biomarkers in non-small cell lung cancer patients. Cancer Lett. 2011;311(1):29–37. doi: 10.1016/j.canlet.2011.06.021. [DOI] [PubMed] [Google Scholar]
- 57.Kenny HA, Krausz T, Yamada SD, Lengyel E. Use of a novel 3D culture model to elucidate the role of mesothelial cells, fibroblasts and extracellular matrices on adhesion and invasion of ovarian cancer cells to the omentum. Int J Cancer. 2007;121(7):1463–1472. doi: 10.1002/ijc.22874. [DOI] [PubMed] [Google Scholar]
- 58.Langley RR, et al. Tissue-specific microvascular endothelial cell lines from H-2Kb-tsA58 mice for studies of angiogenesis and metastasis. . Cancer Res. 2003;63(11):2971–2976. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.