

Abstract
During myocardial ischemia/reperfusion, lipid peroxidation leads to the formation of toxic aldehydes that contribute to ischemic dysfunction. Mitochondrial aldehyde dehydrogenase type 2 (ALDH2) alleviates ischemic heart damage and reperfusion arrhythmias via aldehyde detoxification. Because excessive norepinephrine release in the heart is a pivotal arrhythmogenic mechanism, we hypothesized that neuronal ALDH2 activation might diminish ischemic norepinephrine release. Incubation of cardiac sympathetic nerve endings with acetaldehyde, at concentrations achieved in myocardial ischemia, caused a concentration-dependent increase in norepinephrine release. A major increase in norepinephrine release also occurred when sympathetic nerve endings were incubated in hypoxic conditions. ALDH2 activation substantially reduced acetaldehyde- and hypoxia-induced norepinephrine release, an action prevented by inhibition of ALDH2 or protein kinase Cε (PKCε). Selective activation of Gi/o-coupled adenosine A1, A3, or histamine H3 receptors markedly inhibited both acetaldehyde- and hypoxia-induced norepinephrine release. These effects were also abolished by PKCε and/or ALDH2 inhibition. Moreover, A1-, A3-, or H3-receptor activation increased ALDH2 activity in a sympathetic neuron model (differentiated PC12 cells stably transfected with H3 receptors). This action was prevented by the inhibition of PKCε and ALDH2. Our findings suggest the existence in sympathetic neurons of a protective pathway initiated by A1-, A3-, and H3-receptor activation by adenosine and histamine released in close proximity of these terminals. This pathway comprises the sequential activation of PKCε and ALDH2, culminating in aldehyde detoxification and inhibition of hypoxic norepinephrine release. Thus, pharmacological activation of PKCε and ALDH2 in cardiac sympathetic nerves may have significant protective effects by alleviating norepinephrine-induced life-threatening arrhythmias that characterize myocardial ischemia/reperfusion.
Introduction
Acetaldehyde, the product of ethanol oxidation by alcohol dehydrogenase, has been known for quite some time to display sympathomimetic effects (Eade, 1959). Direct perfusion of the canine sinus node with acetaldehyde has been reported to elicit tachycardia that was inhibited by β-adrenergic blockade (James and Bear, 1967). Likewise, intravenous infusion of acetaldehyde in the rat caused tachycardia whose time course mimicked that of its blood level (Hellström and Tottmar, 1982). These effects were attributed to catecholamine release, because acetaldehyde seemed to enhance catecholamine secretion from bovine adrenal medulla (Schneider, 1971) and promote norepinephrine (NE) release from rat brain (Thadani and Truitt, 1977).
Of the toxic aldehydes known to be formed by lipid peroxidation during ischemia/reperfusion (Esterbauer et al., 1991; Cordis et al., 1993; Eaton et al., 1999), acetaldehyde and 4-hydroxynonenal have been shown to degranulate mast cells (Koda et al., 2010) and release NE from cardiac sympathetic nerve endings (Morrey et al., 2010). Hence, we asked whether toxic aldehydes might be causally involved in the release of NE, which characterizes ischemia/reperfusion (Imamura et al., 1994, 1996) and whether selective activation of neuronal mitochondrial aldehyde dehydrogenase type 2 (ALDH2) (Chen et al., 2008) might bring about a beneficial decrease in NE release.
Our findings suggest that, in ischemic conditions, adenosine and histamine formed in close proximity of cardiac sympathetic nerve endings activate Gi/o-coupled receptors, such as A1, A3, and H3. This initiates a signaling sequence involving activation/translocation of PKCε and phosphorylation/activation of mitochondrial ALDH2, which disposes of toxic aldehydes and their NE-releasing effects.
Materials and Methods
NE Release from Cardiac Synaptosomes.
Male Hartley guinea pigs weighing 300 to 350 g (Charles River Laboratories, Kingston, NY) were killed by cervical dislocation under light anesthesia with CO2 vapor in accordance with institutional guidelines. The ribcage was dissected away, and the heart was rapidly excised, freed from fat and connective tissue, and transferred to a Langendorff apparatus. Spontaneously beating hearts were perfused through the aorta for 15 min at constant pressure (40 cm of H2O) with Ringer's solution at 37°C saturated with 5% CO2 and 95% O2. Ringer's solution composition was 154 mM NaCl, 5.6 mM KCl, 2.2 mM CaCl2, 6.0 mM NaHCO3, and 5.6 mM dextrose. This procedure ensured that no blood traces remained in the coronary circulation. At the end of the perfusion, the hearts were minced in ice-cold HEPES-buffered saline solution (HBS), which contained 50 mM HEPES, pH 7.4, 144 mM NaCl, 5 mM KCl, 1.2 mM CaCl2, 1.2 mM MgCl2, and 10 mM glucose. Synaptosomes were isolated as described previously (Seyedi et al., 1997). Minced tissue was digested with 40 mg of collagenase (Type II; Worthington Biochemicals, Freehold, NJ) per 10 ml of HBS per gram of wet heart weight for 1 h at 37°C. HBS contained 1 mM pargyline to prevent enzymatic destruction of NE. After low-speed centrifugation (10 min at 120g and 4°C), the resulting pellet was suspended in 10 volumes of 0.32 M sucrose and homogenized with a Teflon/glass homogenizer. The homogenate was spun at 650g for 10 min at 4°C, and the pellet was then rehomogenized and respun. The pellet containing cellular debris was discarded, and the supernatants from the last two spins were combined and equally subdivided into tubes. Each tube was centrifuged for 20 min at 20,000g at 4°C. This pellet, which contained cardiac synaptosomes, was resuspended in HBS to a final volume of 1 ml in the presence or absence of acetaldehyde for a total of 10 min in a water bath at 37°C. Each suspension functioned as an independent sample and was used only once. In every experiment, one sample was untreated (control, basal NE release), and others were incubated with drugs for 10 to 30 min. When antagonists were used, samples were incubated with the antagonists before incubation with the agonist. Controls were incubated for an equivalent length of time without drugs. At the end of the incubation period, each sample was centrifuged (20 min; 20,000g; 4°C). The supernatant was assayed for NE content by high-pressure liquid chromatography with electrochemical detection (Seyedi et al., 1997). The pellet was assayed for protein content by a modified Lowry procedure (Seyedi et al., 1997).
Induction of Ischemia-Like Conditions in Cardiac Synaptosomes.
Ischemia-like conditions (i.e., hypoxia) were generated by incubating synaptosomes for 30 min in glucose-free HBS bubbled with 95% N2 and 5% CO2, containing sodium dithionite (3 mM; PO2 ≈0 mm Hg; pH 7.4; ischemic-like release) (Sesti et al., 2003). Matched synaptosomes were incubated for an equivalent period with oxygenated (95% O2 and 5% CO2) HBS (normoxic basal release).
After incubation, each sample was centrifuged (20 min; 20,000g; 4°C), and the supernatant was assayed for NE content by high-pressure liquid chromatography. The pellet was assayed for protein content by a modified Lowry assay using a Bio-Rad DC Protein Assay kit (Bio-Rad Laboratories, Hercules, CA).
Cell Culture.
Rat pheochromocytoma PC12 cells were transfected with the human histamine H3 receptor (donated by Dr. T. W. Lovenberg, Johnson and Johnson Pharmaceutical R&D, LLC, San Diego, CA) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) following the manufacturer's protocol. PC12-H3 cell lines were selected and maintained in selection media containing 500 μg/ml G-418 sulfate (Mediatech, Herndon, VA). PC12-H3 cells were maintained in Dulbecco's modified Eagle's medium plus 10% fetal bovine serum, 5% donor horse serum, 1% l-glutamine, and antibiotics at 37°C in 5% CO2. The differentiating protocol involved plating PC12-H3 cells on tissue culture plates coated with collagen (rat tail type VII; Sigma-Aldrich, St. Louis, MO) combined with exposure to low serum medium containing 1% fetal bovine serum, 0.5% donor horse serum, 1% l-glutamine, and antibiotics supplemented with 7S-nerve growth factor (BD Biosciences Discovery Labware, Bedford, MA). For each experiment, the culture medium was aspirated, and cells were washed twice with Na Ringer (140 mM NaCl, 5 mM KCl, 10 mM HEPES, 1 mM MgCl2, 2 mM glucose, and 2 mM CaCl2), then incubated with Alda-1 [N-(1,3-benzodioxol-5-ylmethyl)-2,6-dichlorobenzamide], an ALDH2 activator (100 μM) (Chen et al., 2008), 2-chloro-N-cyclopentyl-2′-methyladenosine (2′-MeCCPA), an adenosine A1-receptor agonist (10 nM) (Franchetti et al., 1998), 1-deoxy-1-[6-[[(3-iodophenyl)methyl]amino]-9H-purin-9-yl]-N-methyl-β-d-ribofuranuronamide (IB-MECA), an adenosine A3-receptor agonist (50 nM) (Gallo-Rodriguez et al., 1994), or methimepip [4-(1H-imidazol-4-ylmethyl)-1-methylpiperidine], a histamine H3-receptor agonist (1 nM) (Kitbunnadaj et al., 2005) for 20 min in an incubator at 37°C in either the absence or presence of glyceryl trinitrate (GTN; 2 μM), which causes desensitization of ALDH2 (Chen et al., 2008), 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), an adenosine A1-receptor antagonist (300 nM) (von der Leyen et al., 1989), 3-propyl-6-ethyl-5- [(ethylthio)carbonyl]-2phenyl-4-propyl-3-pyridine carboxylate (MRS1523), an adenosine A3-receptor antagonist (100 nM) (Li et al., 1998), 1-{3-[4-(piperidin-1-ylmethyl)phenoxy]propyl}piperidine (JNJ5207852), a histamine H3-receptor antagonist (30 nM) (Barbier et al., 2004), peptide εV1–2 (EAVSLKPT), a PKCε inhibitor (1 μM) (Johnson et al., 1996), or peptide ΨεRACK [HDAPIGYD; PKCε (85–92)], a PKCε activator (Dorn et al., 1999). When these drugs were used, PC12-H3 cells were preincubated with them for 30 min. Controls were incubated for an equivalent length of time without drugs. At the end of each experiment, cell lysates were analyzed for ALDH2 activity.
ALDH2 Enzymatic Activity Assay.
Enzymatic activity of ALDH2 in PC12-H3 cells was determined spectrophotometrically by monitoring the reductive reaction of NAD+ to NADH at 340 nm as described previously (Koda et al., 2010). The assays were carried out in 50 mM sodium pyrophosphate buffer, pH 9.0, at 25°C, and 300 μg of cell lysates and 2.5 mM NAD were added to the buffer. To start the reaction, 10 mM acetaldehyde was added, and the accumulation of NADH was recorded for 3 min with measurements taken every 15 s. ALDH2 reaction rates were calculated as μmole NADH per minute per milligram of protein and compared with control cells (i.e., treated with Na Ringer) and expressed as percentage increase from control.
Statistics.
Values are expressed as means ± S.E.M. Unpaired t test and one-way and two-way ANOVA were performed where appropriate, as indicated in the figure legends. A value of P < 0.05 was considered statistically significant.
Drugs and Chemicals.
Acetaldehyde, desipramine hydrochloride (DMI), ω-conotoxin GVIA (ω-CTX), IB-MECA, DPCPX, and MRS1523 were purchased from Sigma-Aldrich. Methimepip dihydrobromide, JNJ5207852, and 2′-MeCCPA were purchased from Tocris Bioscience (Ellisville, MO). GTN was purchased from Hospira, Inc. (Lake Forest, IL). Peptide ΨεRACK, εV1–2, and Alda-1 were synthesized and kindly provided by the Mochly-Rosen laboratory (Stanford University School of Medicine, Palo Alto, CA). IB-MECA, Alda-1, MRS1523, JNJ5207852, ω-conotoxin GVIA, and 2′-MeCCPA were dissolved in dimethyl sulfoxide. Further dilutions were made with HBS; at the concentration used, dimethyl sulfoxide did not affect mediator release.
Results
ALDH2 Activation Reduces Hypoxic NE Release from Cardiac Sympathetic Nerve Endings: Involvement of Adenosine A1 and A3 Receptors and PKCε.
Incubation of cardiac synaptosomes in hypoxic conditions for 30 min caused a ≥50% increase in NE release above basal conditions. In the presence of the ALDH2 activator Alda-1 (20 μM; 12 min) (Chen et al., 2008) the hypoxic increase in NE release was reduced by ∼60%. Preincubation with GTN (2 μM; 30 min), which causes ALDH2 desensitization (Chen et al., 2008), prevented the effect of Alda-1 (Fig. 1A). Selective PKCε activation with ΨεRACK (500 nM; 12 min) (Inagaki et al., 2003) attenuated the hypoxia-induced NE release by ∼45%. ALDH2 desensitization with GTN (2 μM; 30 min) prevented the effect of ΨεRACK (Fig. 1B). Selective activation of adenosine A1 receptors with 2′-MeCCPA (10 nM; 12 min) (Franchetti et al., 1998) reduced the hypoxia-induced increase in NE release by ∼50%. ALDH2 desensitization with GTN (2 μM; 30 min) and PKCε blockade with εV1–2 (1 μM; 20 min) (Johnson et al., 1996) prevented the effects of 2′-MeCCPA (Fig. 1C). Blockade of adenosine A1 receptors with DPCPX (300 nM; 12 min) (von der Leyen et al., 1989) enhanced hypoxia-induced NE release by ≥30% (Fig. 1D). Selective activation of adenosine A3 receptors with IB-MECA (50 nM; 12 min) (Gallo-Rodriguez et al., 1994) reduced the hypoxia-induced increase in NE release by ∼35%. ALDH2 desensitization with GTN (2 μM; 30 min) and PKCε blockade with εV1–2 (1 μM; 20 min) prevented the effects of IB-MECA (Fig. 1E). Blockade of adenosine A3 receptors with MRS1523 (100 nM; 12 min) (Li et al., 1998) enhanced hypoxia-induced NE release by ∼35% (Fig. 1F).
Fig. 1.
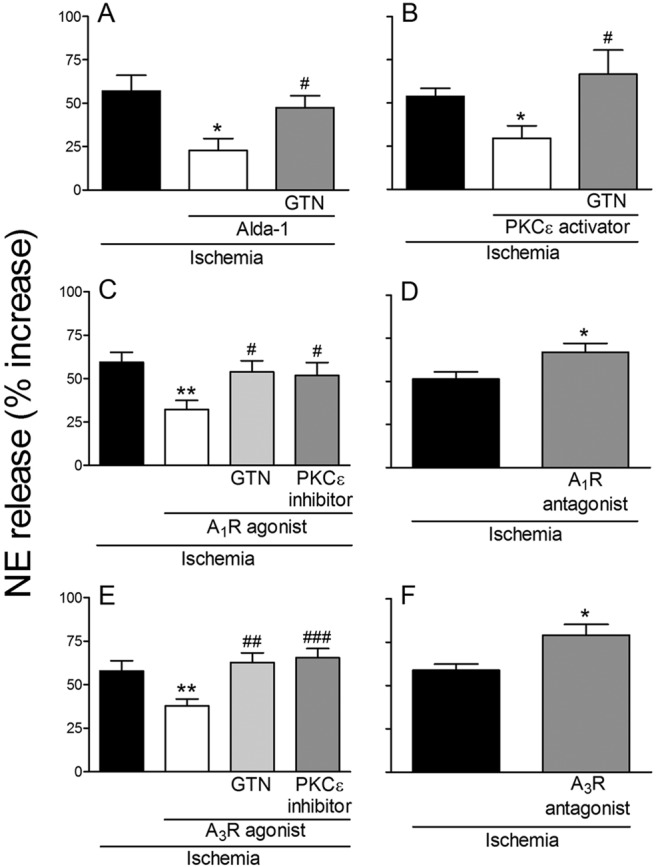
Release of NE from cardiac synaptosomes during 30-min hypoxia. Bars (means ± S.E.M.) represent the hypoxia-induced increase in NE release above the normoxic basal level of 273 ± 11.5 pmol/mg protein (n = 49). A, activation of ALDH2 with Alda-1 (20 μM; 12 min) reduces hypoxia-induced NE release. Desensitization of ALDH2 with GTN (2 μM; 30 min) prevents the effect of Alda-1 (n = 7–12). *, P < 0.05 versus hypoxia. #, P < 0.05 versus Alda-1 by unpaired t test. B, PKCε activation with ΨεRACK (PKCε activator; 500 nM; 12 min) reduces hypoxia-induced NE release. ALDH2 desensitization with GTN (2 μM; 30 min) prevents the effect of ΨεRACK (n = 7–8). *, P < 0.05 versus hypoxia. #, P < 0.05 versus ΨεRACK by unpaired t test. C, activation of adenosine A1 receptor with 2′-MeCCPA (A1R agonist; 10 nM; 12 min) diminishes hypoxia-induced NE release. ALDH2 desensitization with GTN (2 μM; 30 min) and PKCε blockade with εV1–2 (PKCε inhibitor; 1 μM; 20 min) each prevents the effect of 2′-MeCCPA (n = 7–10). **, P < 0.005 versus hypoxia. #, P < 0.05 versus 2′-MeCCPA by unpaired t test. D, blockade of adenosine A1-receptor with DPCPX (A1R antagonist; 300 nM; 12 min) enhances hypoxia-induced NE release (n = 15–17). *, P < 0.05 versus hypoxia by unpaired t test. E, selective activation of adenosine A3 receptor with IB-MECA (A3R agonist; 50 nM; 12 min) reduces hypoxia-induced NE release. ALDH2 desensitization with GTN (2 μM; 30 min) and PKCε blockade with εV1–2 (1 μM; 20 min) each prevents the effect of IB-MECA (n = 7–11). **, P < 0.01 versus hypoxia. ##, P < 0.005 versus IB-MECA. ###, P < 0.001 versus IB-MECA by unpaired t test. F, blockade of adenosine A3 receptor with MRS1523 (A3R antagonist; 100 nM; 12 min) enhances hypoxia-induced NE (n = 7–9). *, P < 0.05 versus hypoxia by unpaired t test.
These results suggested that activation of ALDH2 in cardiac sympathetic neurons reduces hypoxic NE release by removing toxic aldehydes, which are known to be formed in ischemic conditions, most likely acetaldehyde (Esterbauer et al., 1991; Cordis et al., 1993). This process likely involves adenosine release, activation of A1 and A3 receptors, and activation/translocation of PKCε.
Administration of Exogenous Acetaldehyde to Cardiac Sympathetic Nerve Endings Elicits NE Release: Inhibition by Activation of ALDH2 and PKCε.
Incubation of cardiac synaptosomes with acetaldehyde (100–1000 μM; 10 min) elicited a concentration-dependent increase in NE release that was abolished by ALDH2 activation with Alda-1 (20 μM; 10 min). ALDH2 desensitization with GTN (2 μM; 30 min) prevented the effect of Alda-1 (Fig. 2A). PKCε activation with ΨεRACK (500 nM; 10 min) (Inagaki et al., 2003) attenuated the acetaldehyde-induced release of NE. ALDH2 desensitization with GTN (2 μM; 30 min) prevented the effect of ΨεRACK (Fig. 2B).
Fig. 2.

Incubation of isolated cardiac synaptosomes with acetaldehyde elicits a concentration-dependent increase in NE release, which is inhibited by the activation of either ALDH2 or PKCε. Points (means ± S.E.M.; n = 4–46 and 12–24 in A and B, respectively) represent percentage increases in NE release above a mean basal control level of 205 ± 7.4 pmol/mg protein (n = 92). A, ALDH2 activation with Alda-1 (20 μM; 10 min) attenuates the acetaldehyde-induced release of NE. ALDH2 desensitization with GTN (2 μM; 30 min) prevents the effect of Alda-1. B, PKCε activation with ΨεRACK (500 nM, 10 min) attenuates the release of NE. ALDH2 desensitization with GTN (2 μM; 30 min) prevents the effect of ΨεRACK. ***, P < 0.0001 from control and GTN + Alda-1 in A and from control and GTN + ΨεRACK in B, by unpaired t test.
Thus, exogenous acetaldehyde elicits NE release from cardiac sympathetic nerve endings, and activation of ALDH2 abolishes the effect of acetaldehyde. Activation/translocation of PKCε mimics the effect of Alda-1; this action is prevented by prior desensitization of ALDH2. This suggests that activation/translocation of PKCε in turn activates ALDH2, which then attenuates the NE-releasing effect of acetaldehyde.
Activation of Adenosine A1 and A3 Receptors Inhibits the Acetaldehyde-Induced NE Release from Isolated Cardiac Synaptosomes: Prevention by Either PKCε Inhibition or ALDH2 Desensitization.
Activation of adenosine A1 receptors with 2′-MeCCPA (10 nM; 10 min) shifted downward and to the right of the concentration-response curve for the NE-releasing effect of acetaldehyde in isolated sympathetic nerve endings (cardiac synaptosomes) (Fig. 3A). The effect of 2′-MeCCPA was prevented by the selective A1-receptor antagonist DPCPX (300 nM; 10 min) (Fig. 3A), the PKCε inhibitor εV1–2 (1 μM; 10 min) (Fig. 3B), and ALDH2 desensitization with GTN (2 μM; 30 min) (Fig. 3C).
Fig. 3.

Activation of adenosine A1 and A3 receptors inhibits the acetaldehyde-induced NE release from isolated cardiac synaptosomes: prevention by either PKCε inhibition or ALDH2 desensitization. Points (means ± S.E.M.; n = 8–42) represent percentage increases in NE release above a mean basal control level of 267 ± 6.6 pmol/mg protein (n = 124). A, selective activation of A1 receptors with 2′-MeCCPA (10 nM; 10 min) attenuates NE release by acetaldehyde, an action that is prevented by A1-receptor blockade with DPCPX (300 nM; 10 min). B and C, the A1 receptor-induced attenuation of NE release is prevented either by PKCε inhibition with εV1–2 (1 μM; 10 min) (B) or ALDH2 desensitization with GTN (2 μM; 30 min) (C). D, selective activation of A3 receptors with IB-MECA (50 nM; 10 min) attenuates NE release by acetaldehyde, an action that is prevented by A3-receptor blockade with MRS1523 (100 nM; 10 min). E and F, the A3 receptor-induced attenuation of NE release is prevented either by PKCε inhibition with εV1–2 (1 μM; 10 min) (E) or ALDH2 desensitization with GTN (2 μM; 30 min) (F). **, P < 0.01 and ***, P < 0.001 from control and A1- and A3-receptor agonists in combination with respective antagonists or in combination with εV1–2 or GTN, by unpaired t test.
Activation of adenosine A3 receptors with IB-MECA (50 nM; 10 min) also shifted downward and to the right of the concentration-response curve for the NE-releasing effect of acetaldehyde (Fig. 3D). The effect of IB-MECA was prevented by the selective A3-receptor antagonist MRS1523 (100 nM; 10 min) (Fig. 3D), the PKC inhibitor εV1–2 (1 μM; 10 min) (Fig. 3E), and ALDH2 desensitization with GTN (2 μM; 30 min) (Fig. 3F).
Thus, activation of adenosine A1 and A3 receptors attenuates the NE-releasing effect of acetaldehyde in cardiac sympathetic nerve terminals, an action that depends on PKCε and ALDH2 activation.
ALDH2 Activation Prevents the Acetaldehyde-Induced Release of NE and Associated Tachycardia in the Ex Vivo Guinea Pig Heart.
Perfusion of guinea pig hearts ex vivo with acetaldehyde (500 μM; 20 min) in a Langendorff apparatus elicited a ∼6-fold increase in NE overflow that was associated with a ∼30 beats/min increase in heart rate. Both of these changes subsided at the end of the acetaldehyde perfusion. When acetaldehyde was preceded by a 10-min perfusion with the ALDH2 activator Alda-1 (20 μM) the increases in NE overflow and heart rate both were markedly reduced (Fig. 4). This indicated that acetaldehyde provokes the release of NE from cardiac sympathetic neurons, and NE probably is responsible for the associated tachycardia, because ALDH2 activation diminished both NE release and tachycardia.
Fig. 4.

ALDH2 activation inhibits the positive chronotropic effect of acetaldehyde and associated increase in NE overflow in guinea pig hearts ex vivo. Top, time course of the increase in spontaneous heart rate during perfusion with acetaldehyde (500 μM). Pretreatment with the ALDH2 activator Alda-1 (20 μM; 10 min) inhibits the acetaldehyde-induced tachycardia and attenuates the associated increase in NE overflow. Points are heart rates recorded at the corresponding times on the abscissa (means ± S.E.M.; n = 5 and 6 for control and Alda-1, respectively). †, P < 0.05 and ††, P <0.01 from control by one-way ANOVA + Bonferroni's test. **, P < 0.01 and ***, P < 0.001 from acetaldehyde, by two-way ANOVA + Bonferroni's test. Bottom, time course of the increase in NE overflow (measured in 5-min intervals) in the same hearts as in Top. Bars are means (means ± S.E.M.; n = 5 and 6 for control and Alda-1, respectively). †, P < 0.05 and ††, P < 0.01 from control by one-way ANOVA + Bonferroni's test. **, P < 0.01 from acetaldehyde, by two-way ANOVA + Bonferroni's test.
Mechanism of Acetaldehyde-Induced NE Release.
Blockade of N-type Ca2+ channels with ω-conotoxin GVIA (100 nM; 10 min) markedly reduced the acetaldehyde-induced NE release; in fact, the acetaldehyde concentration-response curve for the NE-releasing effect of acetaldehyde in isolated sympathetic nerve endings was significantly shifted to the right and downward (Fig. 5). In contrast, preincubation of cardiac synaptosomes with the NE transporter inhibitor DMI (300 nM; 10 min) significantly potentiated the NE-releasing effect of acetaldehyde, resulting in a marked upward and leftward shift of the concentration-response curve for the NE-releasing effect of acetaldehyde (Fig. 5). This indicated that acetaldehyde most likely promotes NE exocytosis.
Fig. 5.
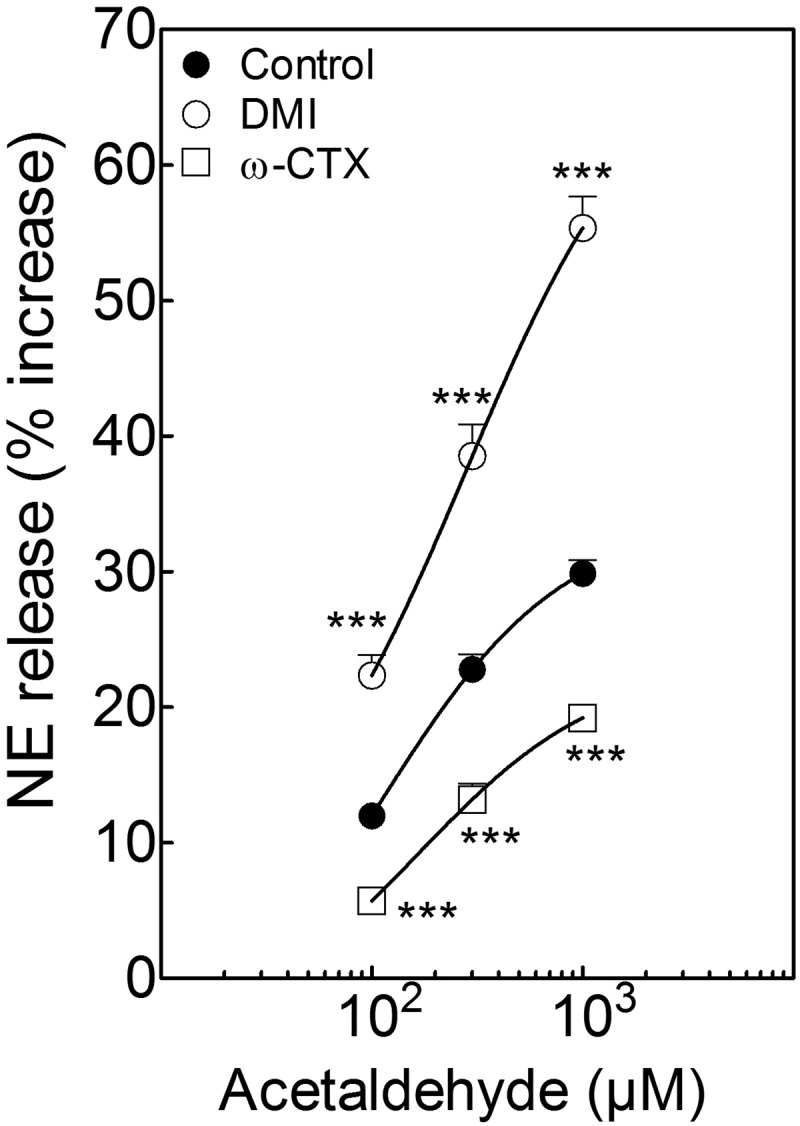
Incubation of isolated cardiac synaptosomes with acetaldehyde elicits a concentration-dependent increase in NE release that is potentiated by the NE transporter inhibitor DMI (300 nM; 10 min). A selective N-type Ca2+ channel blocker, ω-CTX (100 nM; 10 min), markedly reduces the acetaldehyde-induced NE release. Points (means ± S.E.M.; n = 8–32) represent percentage increases in NE release above a mean basal control level of 246 ± 6.80 pmol/mg protein (n = 32). ***, P < 0.001 from acetaldehyde control by unpaired t test.
Histamine H3-Receptor Activation Attenuates Hypoxic NE Release from Cardiac Synaptosomes: Prevention by PKCε Inhibition or ALDH2 Desensitization.
Incubation of cardiac synaptosomes in hypoxic conditions for 30 min caused a ≥50% increase in NE release above basal conditions (Figs. 1 and 6). In the presence of the selective histamine H3-receptor agonist methimepip (1 nM; 12 min) (Kitbunnadaj et al., 2005) the hypoxic increase in NE release was reduced by ∼50%, an effect that was prevented by the addition of the selective H3-receptor antagonist JNJ5207852 (30 nM; 12 min) (Barbier et al., 2004). Addition of the PKCε inhibitor εV1–2 (1 μM; 20 min) or GTN (2 μM; 30 min), which causes ALDH2 desensitization, prevented the effect of methimepip (Fig. 6). These results suggested that, similar to A1 and A3 receptors, the activation of H3 receptors in cardiac sympathetic neurons also reduces hypoxic NE release by an action that involves PKCε and ALDH2.
Fig. 6.
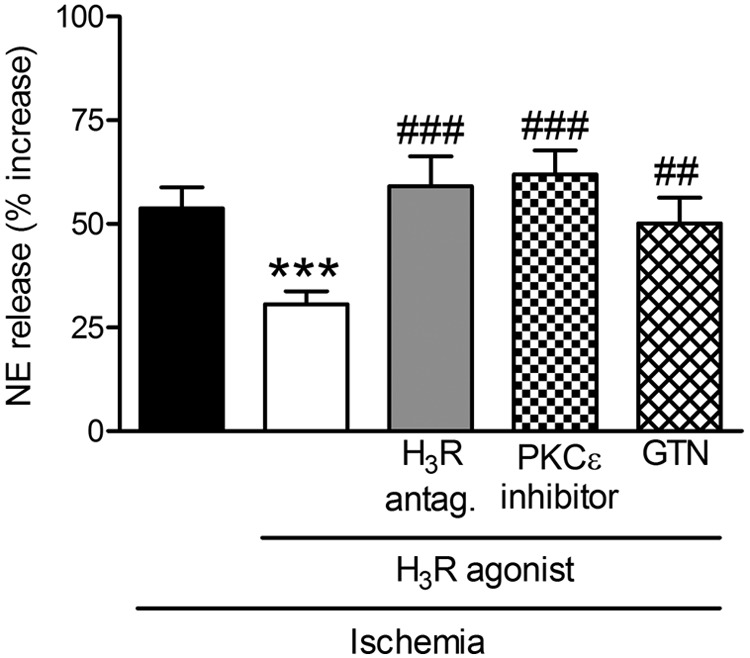
Release of NE from cardiac synaptosomes during 30-min hypoxia. Bars (means ± S.E.M.) represent the hypoxia-induced increase in NE release above the normoxic basal level of 291 ± 24 pmol/mg protein (n = 14). Treatment with the selective histamine H3-receptor agonist methimepip (1 nM; 12 min) reduces hypoxia-induced NE release, and this effect is blocked by a previous addition of the selective histamine H3-receptor antagonist JNJ5207852 (30 nM; 12 min). PKCε blockade with εV1–2 (1 μM; 20 min) and ALDH2 desensitization with GTN (2 μM; 30 min) each prevents the effect of methimepip (n = 5–16). ***, P < 0.001 versus hypoxia. ##, P < 0.005 versus methimepip. ###, P < 0.0005 versus methimepip by unpaired t test.
Activation of Histamine H3 Receptors Attenuates Acetaldehyde-Induced NE Release from Cardiac Sympathetic Nerve Endings: Inhibition by Blockade of PKCε or Desensitization of ALDH2.
Incubation of cardiac synaptosomes with acetaldehyde (100–1000 μM; 10 min) elicited a concentration-dependent increase in NE release that was markedly attenuated by H3-receptor activation with methimepip (1 nM; 10 min). This effect was prevented by H3-receptor blockade with JNJ5207852 (30 nM; 10 min) (Fig. 7A). PKCε blockade with εV1–2 (1 μM; 10 min) or ALDH2 desensitization with GTN (2 μM; 30 min) prevented the effect of methimepip (Fig. 7, B and C, respectively). Thus, exogenous acetaldehyde elicits NE release from cardiac sympathetic nerve endings; similar to A1 and A3 receptors, H3-receptor activation reduces acetaldehyde-induced NE release by an action that involves PKCε and ALDH2.
Fig. 7.

Activation of histamine H3 receptors inhibits the acetaldehyde-induced NE release from isolated cardiac synaptosomes: prevention by either PKCε inhibition or ALDH2 desensitization. Points (means ± S.E.M.; n = 12–28) represent percentage increases in NE release above a mean basal control level of 268 ± 9.2 pmol/mg protein (n = 96). A, selective activation of H3 receptors with methimepip (1 nM; 10 min) attenuates NE release by acetaldehyde, an action that is prevented by H3-receptor blockade with JNJ5207852 (30 nM; 10 min). B and C, the H3-receptor-induced attenuation of NE release is prevented either by PKCε inhibition with εV1–2 (1 μM; 10 min) (B) or ALDH2 desensitization with GTN (2 μM; 30 min) (C). ***, P < 0.0001 from control and H3-receptor agonists with respective antagonists or εV1–2 and GTN, by unpaired t test.
Activation of Adenosine A1 and A3 Receptors and Histamine H3 Receptors Increases ALDH2 Activity in PC12-H3 Cells: Prevention by ALDH2 Desensitization or PKCε Inhibition.
As described above, activation of adenosine A1 and A3 receptors as well as histamine H3 receptors in cardiac synaptosomes markedly reduced hypoxia- and acetaldehyde-induced NE release, and all of these effects were abolished by PKCε inhibition or ALDH2 desensitization. This suggested that the stimulation of A1, A3, and H3 receptors may sequentially increase PKCε and ALDH2 activity. We verified this hypothesis in nerve growth factor-differentiated pheochromocytoma PC12 cells stably transfected with H3 receptors, a recognized model of peripheral sympathetic neuron (Morrey et al., 2008; Corti et al., 2011). We found that Alda-1 (100 μM; 20 min) elicited a ≥60% increase in ALDH2 activity (Fig. 8). Similar to Alda-1, activation of adenosine A1 and A3 receptors as well as histamine H3 receptors with the respective selective agonists 2′-MeCCPA (10 nM; 20 min; Fig. 8A), IB-MECA (50 nM; 20 min; Fig. 8B), and methimepip (1 nM; 20 min; Fig. 8C) caused a ∼40 to 55% increase in ALDH2 activity, which was abolished by the respective antagonists DPCPX (300 nM; 30 min), MRS1523 (100 nM; 30 min), and JNJ5207852 (30 nM; 30 min) (Fig. 8). Similar to Alda-1, the effects of 2′-MeCCPA, IB-MECA, and methimepip were abolished by prior ALDH2 desensitization with GTN (2 μM; 30 min) (Fig. 8). Prior blockade of PKCε with εV1–2 (1 μM; 30 min) also prevented the ALDH2-stimulating effect of 2′-MeCCPA, IB-MECA, and methimepip (Fig. 8). Thus, the prevention of the NE-releasing effects of hypoxia or acetaldehyde by A1-, A3-, and H3-receptor activation probably results from an increase in ALDH2 activity in sympathetic nerve endings via PKCε activation/translocation.
Fig. 8.

Activation of adenosine A1 receptors, A3 receptors, or histamine H3 receptors increases ALDH2 activity in PC12-H3 cells. Incubation of PC12-H3 cells with the selective ALDH2 activator Alda-1 (100 μM; 20 min) increases ALDH2 activity (measured by the rate of NADH production at 340 nm). A, incubation of PC12-H3 cells with the A1-receptor agonist 2′-MeCCPA (10 nM; 20 min) increases ALDH2 activity. Selective desensitization of ALDH2 with GTN (2 μM; 30 min) prevents the effects of Alda-1 and A1-receptor agonist. Pretreatment of PC12-H3 cells with the selective A1-receptor antagonist DPCPX (300 nM; 30 min) or the selective PKCε inhibitor εV1–2 (1 μM; 30 min) prevents the effects of A1-receptor activation. B, incubation of PC12-H3 cells with the A3-receptor agonist IB-MECA (50 nM; 20 min) increases ALDH2 activity. Selective desensitization of ALDH2 with GTN (2 μM; 30 min) prevents the effects of Alda-1 and IB-MECA. Pretreatment of PC12-H3 cells with the selective A3-receptor antagonist MRS1523 (100 nM; 30 min) or the selective PKCε inhibitor εV1–2 (1 μM; 30 min) prevents the effects of A3-receptor activation. C, incubation of PC12-H3 cells with the H3-receptor agonist methimepip (1 nM; 20 min) increases ALDH2 activity. Selective desensitization of ALDH2 with GTN (2 μM; 30 min) prevents the effects of Alda-1 and methimepip. Pretreatment of PC12-H3 cells with the selective H3-receptor antagonist JNJ5207852 (30 nM; 30 min) or the selective PKCε inhibitor εV1–2 (1 μM; 30 min) prevents the effects of H3-receptor activation. Bars are mean percentage increases from control (± S.E.M.; n = 4–16). Basal NADH production was 7.47 ± 0.13 μmol/min/mg protein. ***, P < 0.0001 from control. †††, P < 0.0001 from Alda-1. ♦♦♦, P < 0.0001 from A1R agonist. ‡‡‡, P < 0.001 from A3R agonist. ###, P < 0.0001 from methimepip by unpaired t test.
Discussion
We hypothesized that toxic aldehydes formed in the heart during ischemia and reperfusion may elicit the release of NE from sympathetic nerve terminals, and aldehyde detoxification by ALDH2 might inhibit this process. Our findings suggest that in myocardial ischemia locally released endogenous mediators, such as adenosine and histamine, promote a receptor-initiated sequential activation of neuronal PKCε and ALDH2, culminating in an attenuation of NE release.
We had observed previously that incubation of cardiac sympathetic nerve endings in hypoxic conditions in vitro elicits a marked increase in NE release (Sesti et al., 2003). Here, we uncovered that selective pharmacologic activation of neuronal ALDH2 with Alda-1 (Chen et al., 2008) markedly diminishes hypoxic NE release, an action that is reversed by ALDH2 desensitization with GTN. This suggests that ALDH2 activation eliminates NE-releasing effect in ischemic conditions.
We had found that PKCε translocation/activation leads to ALDH2 activation in mast cell mitochondria (Koda et al., 2010). PKCε activation now seems to also be an essential precursor of ALDH2 activation in the ischemic sympathetic nerve terminal. Indeed, PKCε and ALDH2 activation each inhibited hypoxic NE release, and these actions were prevented by ALDH2 desensitization with GTN. Because activation of Gi/o-coupled receptors leads to PKCε translocation/activation (Inagaki et al., 2006), and adenosine is formed copiously in the ischemic myocardium where it inhibits NE release (Endou et al., 1994; Imamura et al., 1996), we hypothesized that activation of Gi/o-coupled adenosine receptors might initiate the PKCε-ALDH2 cascade, culminating in the inhibition of NE release. Indeed, we found that blockade of adenosine A1 and A3 receptors enhanced NE release in hypoxic sympathetic nerve terminals, indicating that these receptors were already activated by an endogenous ligand, most likely adenosine. Alternatively, it is conceivable that hypoxia caused an increase of constitutive activity of these receptors and the antagonists were acting as inverse agonists. In any event, activation of adenosine A1 or A3 receptors with selective exogenous ligands, 2′-MeCCPA (Franchetti et al., 1998) and IB-MECA (Gallo-Rodriguez et al., 1994), respectively, inhibited hypoxic NE release, an action that was prevented by blockade of PKCε or ALDH2. This suggested a protective chain of events initiated by the sequential activation of A1 and A3 receptors, PKCε and ALDH2. It is noteworthy that activation of the Gs-coupled adenosine A2b receptors with 2-amino-4-(3-hydroxyphenyl)-6-(1H-imidazol-2-ylmethylsulfanyl)pyridine-3,5-dicarbonitrile (LUF5835) (Baraldi et al., 2008) failed to modify ischemic NE release (data not shown), confirming that Gi/o coupling is a prerequisite for the initiation of the PKCε/ALDH2-mediated inhibition of ischemic NE release. To strengthen this postulate, we resolved to activate another Gi/o-coupled receptor, i.e., the histamine H3 receptor that we had reported previously to be present in cardiac sympathetic nerve endings where it negatively modulates ischemic NE release (Levi and Smith, 2000). Indeed, the selective H3-receptor agonist methimepip (Kitbunnadaj et al., 2005) significantly attenuated hypoxic NE release by an action involving both PKCε and ALDH2, because it was prevented by PKCε blockade and ALDH2 desensitization.
That acetaldehyde, produced in the heart in ischemic conditions, is at least partially responsible for the characteristic increase in NE release, is substantiated by our finding that the administration of exogenous acetaldehyde to isolated cardiac sympathetic nerve terminals elicits NE release. It is noteworthy that this NE release increases within a range of acetaldehyde concentrations known to be reached in the ischemic heart (Cordis et al., 1993; Eaton et al., 1999). Most important, in cardiac synaptosomes, acetaldehyde-induced NE release was inhibited by the activation of PKCε and ALDH2, revealing that the same mechanisms modulate hypoxic and acetaldehyde-induced NE release from cardiac sympathetic nerve endings. Similar to what was observed in sympathetic nerve endings subjected to hypoxia, selective activation of neuronal adenosine A1 and A3 receptors inhibited acetaldehyde-induced NE release, and both of these effects were prevented by the inhibition of PKCε and ALDH2. Analogous to A1 and A3 receptors, selective activation of neuronal histamine H3 receptors also decreased acetaldehyde-induced NE release, which again was prevented by PKCε and ALDH2 inhibition. It seems likely, therefore, that the inhibition of acetaldehyde-induced NE release initiated by the activation of Gi/o-coupled receptors entails a pathway in which PKCε and ALDH2 play a pivotal role. Indeed, we found that activation of these Gi/o-coupled receptors enhanced in each case the activity of ALDH2 in differentiated pheochromocytoma cells expressing a sympathetic phenotype. This enhancement in ALDH2 activity by adenosine A1 and A3 receptors and histamine H3 receptors mimicked the effect of the prototypical ALDH2 activator Alda-1 (Chen et al., 2008) and was abolished by the inhibition of PKCε. This confirmed that translocation/activation of neuronal PKCε is essential for the phosphorylation/activation of ALDH2 initiated by the stimulation of Gi/o-coupled receptors.
As to the mechanism by which acetaldehyde enhances NE release from cardiac sympathetic terminals, it probably involves a promotion of NE exocytosis. Indeed, we found that pharmacological inhibition of neuronal N-type Ca2+ channels with ω-conotoxin GVIA (Seyedi et al., 2005) markedly attenuated acetaldehyde-induced NE release. Moreover, pharmacological inhibition of the NE transporter at the level of the neuronal membrane with DMI (Aloyo et al., 1991) potentiated the acetaldehyde-induced NE release. Both of these findings suggest an acetaldehyde-induced enhancement of NE exocytosis. Indeed, were acetaldehyde to enhance a nonvesicular carrier-mediated mechanism of NE release, one would have expected an inhibition, rather than a potentiation, of NE release upon blockade of the NE transporter (Levi and Smith, 2000). Moreover, perfusion of isolated Langendorff hearts with acetaldehyde elicited sinus tachycardia that was attenuated ALDH2 activation; arrhythmias, which typically arise with carrier-mediated NE release (Levi and Smith, 2000), did not occur. In protracted ischemic conditions, various locally produced endogenous factors enhance Na+/H+ exchanger activity, leading to nonvesicular carrier-mediated NE release and associated arrhythmic dysfunction (Levi and Smith, 2000). It is plausible that acetaldehyde formed in severe ischemia might contribute to Na+/H+ exchanger activation, carrier-mediated NE release, and arrhythmias, because acetaldehyde was implied to stimulate Na+/H+ exchanger activity and increase intracellular Na+ in liver cells (Carini et al., 2000).
In conclusion, our findings reveal a novel mechanism of inhibition of hypoxic NE release in cardiac sympathetic nerve terminals (Fig. 9). This involves the pivotal phosphorylation/activation of mitochondrial ALDH2 by PKCε, which is translocated/activated when Gi/o-coupled receptors, such as A1, A3, and H3 receptors, are activated by adenosine and histamine produced in the close proximity of sympathetic nerve endings. Because excessive NE release is a major cause of arrhythmic cardiac dysfunction, cardiac failure, and hypertension (Julius, 1993; Esler and Kaye, 2000; Selwyn and Braunwald, 2001), activation of ALDH2 may constitute an important new therapeutic target in these conditions.
Fig. 9.

Proposed pathway for the inhibition of NE release from cardiac sympathetic neurons upon activation of mitochondrial ALDH2. This involves the pivotal phosphorylation/activation of mitochondrial ALDH2 by PKCε, which is translocated/activated when Gi/o-coupled receptors, such as A1, A3, and H3 receptors, are activated by adenosine and histamine released in ischemic conditions in close proximity to sympathetic nerve endings.
Acknowledgments
We thank Dr. Daria Mochly-Rosen (Stanford University School of Medicine, Palo Alto, CA) for the compounds Alda-1, ΨεRACK, and εV1–2.
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute [Grant HL034215]; an American Heart Association Grant-in-Aid; the Caja Madrid Foundation; and a Pharmaceutical Research Manufacturers Association of America Foundation predoctoral fellowship.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
- NE
- norepinephrine
- ALDH2
- aldehyde dehydrogenase type 2
- ANOVA
- analysis of variance
- A1R
- A1 receptor
- A3R
- A3 receptor
- DMI
- desipramine hydrochloride
- DPCPX
- 8-cyclopentyl-1,3-dipropylxanthine
- GTN
- glyceryl trinitrate
- H3R
- H3 receptor
- HBS
- HEPES-buffered saline solution
- IB-MECA
- 1-deoxy-1-[6-[[(3-iodophenyl)methyl]amino]-9H-purin-9-yl]-N-methyl-β-d-ribofuranuronamide
- JNJ5207852
- 1-{3-[4-(piperidin-1-ylmethyl)phenoxy]propyl}piperidine
- 2′-MeCCPA
- 2-chloro-N-cyclopentyl-2′-methyladenosine
- MRS1523
- 3-propyl-6-ethyl-5-[(ethylthio)carbonyl]-2phenyl-4-propyl-3-pyridine carboxylate
- PKCε
- protein kinase Cε
- ω-CTX
- ω-conotoxin GVIA
- LUF5835
- 2-amino-4-(3-hydroxyphenyl)-6-(1H-imidazol-2-ylmethylsulfanyl)pyridine-3,5-dicarbonitrile.
Authorship Contributions
Participated in research design: Robador, Chan, Koda, and Levi.
Conducted experiments: Robador, Seyedi, Chan, and Koda.
Performed data analysis: Robador, Seyedi, Chan, and Koda.
Wrote or contributed to the writing of the manuscript: Robador, Chan, and Levi.
References
- Aloyo VJ, McIlvain HB, Bhavsar VH, Roberts J. (1991) Characterization of norepinephrine accumulation by a crude synaptosomal-mitochondrial fraction isolated from rat heart. Life Sci 48:1317–1324 [DOI] [PubMed] [Google Scholar]
- Baraldi PG, Tabrizi MA, Fruttarolo F, Romagnoli R, Preti D. (2008) Recent improvements in the development of A2B adenosine receptor agonists. Purinergic Signal 4:287–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbier AJ, Berridge C, Dugovic C, Laposky AD, Wilson SJ, Boggs J, Aluisio L, Lord B, Mazur C, Pudiak CM, et al. (2004) Acute wake-promoting actions of JNJ-5207852, a novel, diamine-based H3 antagonist. Br J Pharmacol 143:649–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carini R, De Cesaris MG, Spendore R, Albano E. (2000) Ethanol potentiates hypoxic liver injury: role of hepatocyte Na+ overload. Biochim Biophys Acta 1502:508–514 [DOI] [PubMed] [Google Scholar]
- Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. (2008) Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 321:1493–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordis GA, Maulik N, Bagchi D, Engelman RM, Das DK. (1993) Estimation of the extent of lipid peroxidation in the ischemic and reperfused heart by monitoring lipid metabolic products with the aid of high-performance liquid chromatography. J Chromatogr 632:97–103 [DOI] [PubMed] [Google Scholar]
- Corti F, Olson KE, Marcus AJ, Levi R. (2011) The expression level of ecto-NTP diphosphohydrolase1/CD39 modulates exocytotic and ischemic release of neurotransmitters in a cellular model of sympathetic neurons. J Pharmacol Exp Ther 337:524–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW, 2nd, Souroujon MC, Liron T, Chen CH, Gray MO, Zhou HZ, Csukai M, Wu G, Lorenz JN, Mochly-Rosen D. (1999) Sustained in vivo cardiac protection by a rationally designed peptide that causes ε protein kinase C translocation. Proc Natl Acad Sci U S A 96:12798–12803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eade NR. (1959) Mechanism of sympathomimetic action of aldehydes. J Pharmacol Exp Ther 127:29–34 [PubMed] [Google Scholar]
- Eaton P, Li JM, Hearse DJ, Shattock MJ. (1999) Formation of 4-hydroxy-2-nonenal-modified proteins in ischemic rat heart. Am J Physiol Heart Circ Physiol 276:H935–H943 [DOI] [PubMed] [Google Scholar]
- Endou M, Poli E, Levi R. (1994) Histamine H3-receptor signaling in the heart: possible involvement of Gi/Go proteins and N-type Ca2+ channels. J Pharmacol Exp Ther 269:221–229 [PubMed] [Google Scholar]
- Esler M, Kaye D. (2000) Measurement of sympathetic nervous system activity in heart failure: the role of norepinephrine kinetics. Heart Fail Rev 5:17–25 [DOI] [PubMed] [Google Scholar]
- Esterbauer H, Schaur RJ, Zollner H. (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 11:81–128 [DOI] [PubMed] [Google Scholar]
- Franchetti P, Cappellacci L, Marchetti S, Trincavelli L, Martini C, Mazzoni MR, Lucacchini A, Grifantini M. (1998) 2′-C-Methyl analogues of selective adenosine receptor agonists: synthesis and binding studies. J Med Chem 41:1708–1715 [DOI] [PubMed] [Google Scholar]
- Gallo-Rodriguez C, Ji XD, Melman N, Siegman BD, Sanders LH, Orlina J, Fischer B, Pu Q, Olah ME, van Galen PJ. (1994) Structure-activity relationships of N6-benzyladenosine- 5′-uronamides as A3-selective adenosine agonists. J Med Chem 37:636–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellström E, Tottmar O. (1982) Acute effects of ethanol and acetaldehyde on blood pressure and heart rate in disulfiram-treated and control rats. Pharmacol Biochem Behav 17:1103–1109 [DOI] [PubMed] [Google Scholar]
- Imamura M, Lander HM, Levi R. (1996) Activation of histamine H3-receptors inhibits carrier-mediated norepinephrine release during protracted myocardial ischemia. Comparison with adenosine A1-receptors and α2-adrenoceptors. Circ Res 78:475–481 [DOI] [PubMed] [Google Scholar]
- Imamura M, Poli E, Omoniyi AT, Levi R. (1994) Unmasking of activated histamine H3-receptors in myocardial ischemia: their role as regulators of exocytotic norepinephrine release. J Pharmacol Exp Ther 271:1259–1266 [PubMed] [Google Scholar]
- Inagaki K, Churchill E, Mochly-Rosen D. (2006) Epsilon protein kinase C as a potential therapeutic target for the ischemic heart. Cardiovasc Res 70:222–230 [DOI] [PubMed] [Google Scholar]
- Inagaki K, Hahn HS, Dorn GW, 2nd, Mochly-Rosen D. (2003) Additive protection of the ischemic heart ex vivo by combined treatment with δ-protein kinase C inhibitor and ε-protein kinase C activator. Circulation 108:869–875 [DOI] [PubMed] [Google Scholar]
- James TN, Bear ES. (1967) Effects of ethanol and acetaldehyde on the heart. Am Heart J 74:243–255 [DOI] [PubMed] [Google Scholar]
- Johnson JA, Gray MO, Chen CH, Mochly-Rosen D. (1996) A protein kinase C translocation inhibitor as an isozyme-selective antagonist of cardiac function. J Biol Chem 271:24962–24966 [DOI] [PubMed] [Google Scholar]
- Julius S. (1993) Corcoran Lecture. Sympathetic hyperactivity and coronary risk in hypertension. Hypertension 21:886–893 [DOI] [PubMed] [Google Scholar]
- Kitbunnadaj R, Hashimoto T, Poli E, Zuiderveld OP, Menozzi A, Hidaka R, de Esch IJ, Bakker RA, Menge WM, Yamatodani A, et al. (2005) N-substituted piperidinyl alkyl imidazoles: discovery of methimepip as a potent and selective histamine H3 receptor agonist. J Med Chem 48:2100–2107 [DOI] [PubMed] [Google Scholar]
- Koda K, Salazar-Rodriguez M, Corti F, Chan NY, Estephan R, Silver RB, Mochly-Rosen D, Levi R. (2010) Aldehyde dehydrogenase activation prevents reperfusion arrhythmias by inhibiting local renin release from cardiac mast cells. Circulation 122:771–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi R, Smith NC. (2000) Histamine H3-receptors: a new frontier in myocardial ischemia. J Pharmacol Exp Ther 292:825–830 [PubMed] [Google Scholar]
- Li AH, Moro S, Melman N, Ji XD, Jacobson KA. (1998) Structure-activity relationships and molecular modeling of 3, 5-diacyl-2,4-dialkylpyridine derivatives as selective A3 adenosine receptor antagonists. J Med Chem 41:3186–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrey C, Brazin J, Seyedi N, Corti F, Silver RB, Levi R. (2010) Interaction between sensory C-fibers and cardiac mast cells in ischemia/reperfusion: activation of a local renin-angiotensin system culminating in severe arrhythmic dysfunction. J Pharmacol Exp Ther 335:76–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrey C, Estephan R, Abbott GW, Levi R. (2008) Cardioprotective effect of histamine H3-receptor activation: pivotal role of Gβγ-dependent inhibition of voltage-operated Ca2+ channels. J Pharmacol Exp Ther 326:871–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider FH. (1971) Acetaldehyde-induced catecholamine secretion from the cow adrenal medulla. J Pharmacol Exp Ther 177:109–118 [PubMed] [Google Scholar]
- Selwyn AP, Braunwald E. (2001) Ischemic heart disease, in Harrison's Principles of Internal Medicine (Braunwald E, Fauci AS, Kasper DL, Hauser SL, Longo DL, Jameson JL. eds) pp 1399–1410, McGraw-Hill, New York [Google Scholar]
- Sesti C, Koyama M, Broekman MJ, Marcus AJ, Levi R. (2003) Ectonucleotidase in sympathetic nerve endings modulates ATP and norepinephrine exocytosis in myocardial ischemia. J Pharmacol Exp Ther 306:238–244 [DOI] [PubMed] [Google Scholar]
- Seyedi N, Mackins CJ, Machida T, Reid AC, Silver RB, Levi R. (2005) Histamine H3-receptor-induced attenuation of norepinephrine exocytosis: a decreased protein kinase A activity mediates a reduction in intracellular calcium. J Pharmacol Exp Ther 312:272–280 [DOI] [PubMed] [Google Scholar]
- Seyedi N, Win T, Lander HM, Levi R. (1997) Bradykinin B2-receptor activation augments norepinephrine exocytosis from cardiac sympathetic nerve endings. Mediation by autocrine/paracrine mechanisms. Circ Res 81:774–784 [DOI] [PubMed] [Google Scholar]
- Thadani PV, Truitt EB., Jr (1977) Effect of acute ethanol or acetaldehyde administration on the uptake, release, metabolism and turnover rate of norepinephrine in rat brain. Biochem Pharmacol 26:1147–1150 [DOI] [PubMed] [Google Scholar]
- von der Leyen H, Schmitz W, Scholz H, Scholz J, Lohse MJ, Schwabe U. (1989) Effects of 1,3-dipropyl-8-cyclopentylxanthine (DPCPX), a highly selective adenosine receptor antagonist, on force of contraction in guinea-pig atrial and ventricular cardiac preparations. Naunyn Schmiedebergs Arch Pharmacol 340:204–209 [DOI] [PubMed] [Google Scholar]