

Abstract
The ecological significance of rare microorganisms within microbial communities remains an important, unanswered question. Microorganisms of extremely low abundance (the ‘rare biosphere’) are believed to be largely inaccessible and unknown. To understand the structure of complex environmental microbial communities, including the representation of rare and prevalent community members, we coupled traditional cultivation with pyrosequencing. We compared cultured and uncultured bacterial members of the same agricultural soil, including eight locations within one apple orchard and four time points. Our analysis revealed that soil bacteria captured by culturing were in very low abundance or absent in the culture-independent community, demonstrating unexpected accessibility of the rare biosphere by culturing.
Microorganisms are the most abundant organisms on Earth and represent an unfathomably high level of diversity (Whitman et al., 1998; Rappé and Giovannoni, 2003; Schloss and Handelsman, 2007). Traditional culturing techniques expose a small subset of this vast environmental microbial diversity (Lane et al., 1985; Hugenholtz and Pace, 1996; Rondon et al., 2000). To reveal greater diversity, culturing was replaced first by Sanger sequencing of 16S rRNA genes (Olsen et al., 1986; Rappé and Giovannoni, 2003) and then by high-throughput sequencing (Liu et al., 2007). High-throughput sequencing revealed that detection of microorganisms of extremely low abundance was inadequate by either sequencing method (Sogin et al., 2006).
Microorganisms of extremely low abundance have been designated ‘the rare biosphere’ (Sogin et al., 2006). The ecological significance of rare microorganisms is just beginning to be understood (Pedrós-Alió, 2012). One hypothesis is that rare members represent a dormant seed bank. Members of this seed bank may become active at random (Epstein, 2009), or in direct response to changes in the environment, for instance, to initiate community recovery after disturbance (Epstein, 2009; Lennon and Jones, 2011). This hypothesis is supported by a recent investigation of Baltic Sea bacterioplankton responses to organic carbon additions, wherein rare members increased in abundance from less than 10 sequences to as many as thousands after carbon amendment (Sjöstedt et al., 2012). Similarly, a study in the Western English Channel showed that community members in low abundance were persistent over time, and that, in a few cases, populations of rare members occasionally bloomed (Caporaso et al., 2011). However, there also are situations in which rare members are hypothesized to be less important for the community, such as when populations are becoming extinct or are between favourable environments (Pedrós-Alió, 2012). Despite advances in sequencing depth that reveal rare organisms and a few intriguing observations using these sequencing technologies, the significance of the rare biosphere remains obscure (Hugenholtz and Pace, 1996; Sogin et al., 2006; Huse et al., 2010). However, because members of the rare biosphere may provide novel products and processes, bioprospecting for these organisms has been made a priority (Reid and Buckley, 2011).
Soil harbours a complex microbial consortium that contains a preponderance of low-abundance microorganisms (e.g. Roesch et al., 2007), representing perhaps the most significant habitat of the rare biosphere. To estimate the contribution of rare members to soil microbial communities, a clone library of 16S rRNA genes, including 13 000nearly full-length sequences, was analysed (Elshahed et al., 2008). The results suggested that many rare members represented either phylogenetically divergent or generally uncommon lineages for the soil environment (Elshahed et al., 2008). The goal of the present study was to understand the contributions of cultured and as-yet-uncultured members of soil bacterial communities to community structure. To directly compare culture-based and culture-independent communities, it was important to have both assessed using the same technology. Thus, we compared taxa identified by pyrosequencing of the 16S rRNA genes isolated by culture-based and culture-independent methods.
Soil samples from eight locations within one apple orchard were collected twice per year over 2 years. DNA was extracted directly from soil on the day of collection (culture-independent), and from bacteria cultured from soil on rhizosphere isolation medium (RIM) modified by replacing nystatin with cycloheximide (100 µg ml−1) to inhibit fungal growth (Buyer, 1995). Plates were incubated for 6 days at room temperature. Pyrosequencing of the 16S rRNA gene V3-V4 variable region was performed with primers 515/806, and sequences were analysed using default QIIME v. 1.2.1 workflow (see Supporting information, Caporaso et al., 2010). Pyrosequencing generated 83 992 quality sequences across 58 samples, representing 5822 operational taxonomic units (OTUs) containing at least 97% sequence identity (Table S1, Fig. 1A). These sequences are available (MG-RAST ID 4487654.3). To ensure a conservative estimate of richness, singleton OTUs (i.e. OTUs observed once throughout the entire dataset) were omitted (Fig. 1B). There were no global differences in the composition of bacterial communities across locations in the orchard (P = 0.87, 0.89) or time (P = 0.33, 0.21), as assessed by analysis of similarity from ranked Bray–Curtis distances and from Morista–Horn similarity respectively.
Fig. 1.
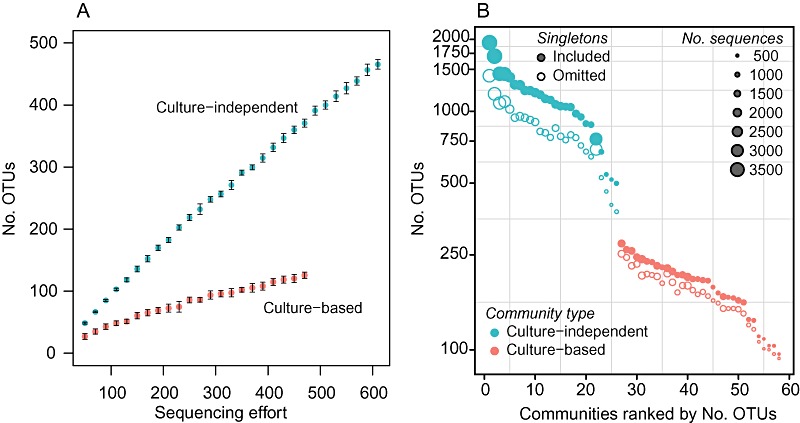
Community structure varies between culture-based and culture-independent assessments.
A. To determine the completeness of the sequencing effort, rarefaction was performed from a minimum of 50 sequences to the median number of sequences observed across all samples (610 sequences, as per QIIME default parameters for the script alpha_rarefaction.py). Error bars are standard error around the mean of 10 subsamples at each level of sequencing effort.
B. Omitting singletons has minimal effect on community structure. Communities are ranked by number of OTUs. Closed symbols represent communities including singleton OTUs, and open symbols represent communities without singleton OTUs. Thus, the difference on the y-axis between the closed and open symbols depicts the change in the number of OTUs within a community after omitting singletons. Symbols are scaled in size to the total number of sequences observed within a community before rarefaction.
Most OTUs in the collection of cultured microorganisms (61%) were not detected in the communities described by culture-independent means (Fig. 2A). We identified the most abundant OTUs as those positioned before the beginning of the ‘tail’ of the log-scale rank abundance curve (Fig. S1), as a way of partitioning abundant taxa from rare taxa in a community. For the top 26 OTUs, we observed a range of 300 to 7102 total sequences. Of the 26 most-prevalent OTUs across the entire dataset, 22 were abundant only among the cultured organisms and were in low abundance or below detection among the culture-independent sequences (Fig. 2B). The four that were prevalent in both groups were OTUs 4789, 6271, 27 and 5980. Moreover, 23 of 26 dominant OTUs among the cultured members were rare among the uncultured members (Fig. S2A), and 23 of 26 dominant OTUs among the uncultured were rare among the cultured organisms (Fig. S2B). These observations indicate that most OTUs obtained by culturing under standard conditions (see Appendix S1) were rare among community members represented by pyrosequencing DNA directly isolated from soil. These cultured rare members likely represent a fraction of the total rare biosphere, but a subset that would have been undiscovered by pyrosequencing at the given sequencing depth (a depth suitable for uncovering patterns of microbial beta diversity, e.g. Kuczynski et al., 2010). Thus, a combination of culture-dependent and culture-independent analysis described the soil microbial community more comprehensively than either approach alone and uncovered members of the rare biosphere.
Fig. 2.
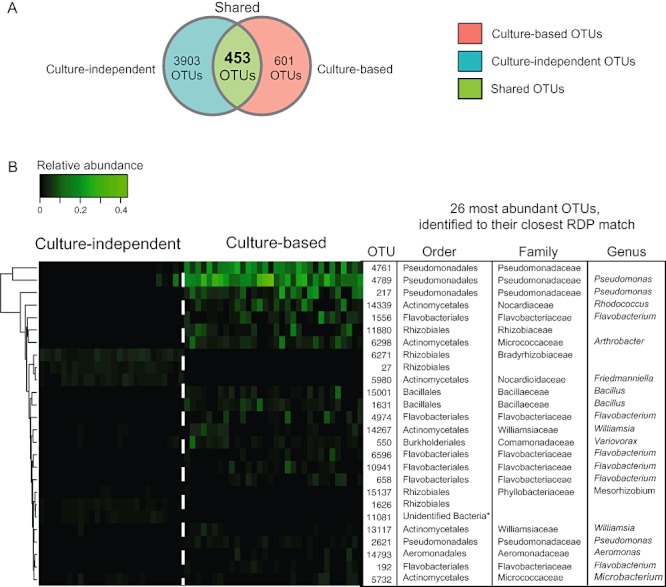
Culture-based and culture-independent analyses of soil bacterial communities.
A. Number of OTUs shared between culture-based and culture-independent analyses, and number of OTUs unique to each analysis.
B. Heat map of 26 most abundant OTUs in the dataset, organized by response patterns (clusters shown by dendrogram). Columns are community samples from the culture-independent method (n = 26, left side of the dashed white line) or the cultured-based method (n = 32, right side of the dashed white line). Each OTU is in a row and colour intensity indicates its relative abundance, with brighter green indicating higher abundance. Asterisk indicates that the OTU could not be identified to the Order level.
Although culture-based analysis of microbial communities has been largely replaced by sequence-based studies such as 16S rRNA gene analysis and metagenomics, our study demonstrates an advantage of including culturing in characterization of the microbial communities. Additionally, cultivation provides access to diverse characteristics of each microorganism, offering a rich platform for physiological analyses, which could advance bioprospecting, one of the motivations for pursuing the rare biosphere (Reid and Buckley, 2011). However, not all rare community members will be captured readily by culturing. Culturing from the murine gut, for example, revealed many abundant members of the microbial community (Goodman et al., 2011), and thus in that habitat, deep sequencing of culture-independent samples will likely provide better access to rare members. Therefore, success of cultivating rare members will depend, first, on the life strategies of microorganisms inhabiting a niche (e.g. copiotrophic or oligotrophic, Dethlefsen and Schmidt, 2007; Fierer et al., 2007) and, second, on the precise conditions of cultivation.
Life strategies of microorganisms are important for understanding microbial community ecology (Dethlefsen and Schmidt, 2007; Fierer et al., 2007), and perhaps also for understanding the roles of rare members in the community. Cultivation is thought to select for opportunistic ‘weed’ species of bacteria, or copiotrophs, that grow quickly in resource-rich conditions common to many culture media (e.g. Garland et al., 2001). Some members of the soil rare biosphere may be copiotrophs waiting for favourable resources to flourish. This mechanism for maintenance of copiotrophic rare taxa would be in agreement with the ‘storage effect’ (Warner and Chesson, 1985), which posits that strategies of temporal bet-hedging (e.g. dormancy) promote persistence of populations.
Furthermore, some soil bacterial phyla are associated with copiotrophic or oligotrophic life strategies. For example, in one study, soil Acidobacteria had a negative relationship with carbon concentration and were classified as oligotrophs, while Betaproteobacteria and Bacteroidetes had a positive relationship with carbon concentration and were classified as copiotrophs (Fierer et al., 2007). Although not all phyla could be assigned to a copiotrophic or oligotrophic life strategy, the authors observed that Acidobacteria were prevalent in communities assessed using culture-independent techniques but underrepresented in isolate collections, whereas Betaproteobacteria and Bacteroidetes were common in isolate collections (Fierer et al., 2007). Similarly, in our soil analysis we detected no Acidobacteria in the culture-based collection, but detected many in the culture-independent collection (Fig. S3). By proportion, Bacteroidetes-affiliated taxa comprised most of the culture-based members, and were also prevalent among culture-independent OTUs. There was a similar pattern for Betaproteobacteria.
The precise conditions of cultivation, including choice of medium, also determine which community members are detected using culture-based methods. Our results show that even standard cultivation conditions can uncover members of the rare biospehere. We used a modified RIM, a medium developed specifically to inhibit growth of Bacillus mycoides, a bacterium with a filamentous colony morphology that obscures smaller bacterial colonies. Although RIM lacks five common amino acids, it supported growth of a similar number of bacterial colonies and more bacterial genera than a medium containing Casamino Acids (Buyer, 1995). Furthermore, we identified a similar number of bacterial taxa on RIM and another medium, 0.1× tryptic soy agar, which is commonly used for isolation of soil bacteria (Fig. S4). Both media revealed taxa representing four phyla (Actinobacteria, Bacteroidetes, Firmicutes and Proteobacteria) and three Proteobacteria classes (Alpha-, Beta- and Gammaproteobacteria; Fig. S4). Thus, while RIM is a selective medium, it does not appear to inhibit soil bacteria other than B. mycoides.
Members of the rare biosphere of soil were accessible using standard cultivation conditions, suggesting that we may not be as ignorant about the soil rare biosphere as previously suspected. Our study also supports the prediction that members of the rare biosphere may mediate functions that are essential for community viability, potentially acting as keystone species. For example, readily cultured soil microorganisms that were in low abundance based on culture-independent pyrosequencing, such as some Rhizobiales taxa and Streptomyces (e.g. Janssen, 2006), are extremely important in nitrogen fixation and production of secondary metabolites respectively, which are critical functions in soil ecology. As microorganisms in low abundance will likely prove important in other habitats, understanding the rare biosphere is therefore a key feature of global microbial ecology. The results presented here show that organisms known to play important ecological roles are members of the rare biosphere, that combining culture-based methods with next generation sequencing can provide a richer portrait of microbial diversity than does either approach alone, and that the diminishing use of culturing in modern microbiology may obscure critical dimensions of community structure and function.
In summary, our findings demonstrate that through culturing of soil samples, microbiologists have been studying members of the rare biosphere intensively since the 19th Century. Moreover, the ecological importance of many readily cultured members of the soil community is well established, validating that members of the rare biosphere contribute vital functions to the soil community.
Acknowledgments
USDA NIFA Microbial Observatories 2006-35319-17466 funded this work. A. S. is a Gordon and Betty Moore Foundation Fellow of the Life Sciences Research Foundation. We thank R. Losick, T. Schmidt and E. Yashiro for comments on an earlier version of the work.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Rank abundance distribution of OTUs from singleton-omitted, rarefied dataset from soil.
Figure S2. Heatmap visualizations of the 26 most abundant OTUs within the (A) culture-based and (B)culture-independent communities, and organized within each map by similar response patterns. Columns are communities, and rows are OTUs. Please note differences in scale of relative abundances for A and B. Variovorax (OTU 550) was the only abundant OTU shared across both cultured-based and culture-independent communities. OTUs marked with an asterisk could not be resolved to the Order level.
Figure S3. Frequency of OTUs across Bacteria phyla and Proteobacteria class for (A) culture-independent and (B) culture-based communities.
Figure S4. Comparison of taxa detected in rhizosphere isolation medium (RIM) and 0.1× tryptic soy agar (TSA) at the (A) phylum level and (B) Proteobacteria class. (C) Venn analysis of OTUs detected on each type of medium. OTUassignments were made with Ribosomal Database Project assignment of full-length 16S RNA gene sequences from clone libraries of metagenomic DNA extracted from a pool of isolates for each culturing media. RIM was ultimately selected for cultivation because it inhibited the growth of filamentous Bacillus mycoides, which often obscured quantification of smaller, non-filamentous colonies on TSA.
Table S1. Summary of sequencing effort and number of OTUs detected in culture-based, culture-independent, and the total dataset. Note that the total is not necessarily the sum of the culture-based and culture-independent (e.g. some OTUs were shared across both communities).
Appendix S1. Supplemental methods.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Buyer JS. A soil and rhizosphere microorganism isolation and enumeration medium that inhibits Bacillus mycoides. Appl Environ Microbiol. 1995;61:1839–1842. doi: 10.1128/aem.61.5.1839-1842.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high- throughput community sequencing data. Nature. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Paszkiewicz K, Field D, Knight R, Gilbert JA. The Western English Channel contains a persistent microbial seed bank. ISME J. 2011;6:1089–1093. doi: 10.1038/ismej.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, Schmidt TM. Performance of the translational apparatus varies with the ecological strategies of bacteria. J Bacteriol. 2007;189:3237–3245. doi: 10.1128/JB.01686-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshahed MS, Youssef NH, Spain AM, Sheik C, Najar FZ, Sukharnikov LO, et al. Novelty and uniqueness patterns of rare members of the soil biosphere. Appl Environ Microbiol. 2008;74:5422–5428. doi: 10.1128/AEM.00410-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein SS. Microbial awakenings. Nature. 2009;457:1083–1083. doi: 10.1038/4571083a. [DOI] [PubMed] [Google Scholar]
- Fierer N, Bradford MA, Jackson RB. Toward an ecological classification of soil bacteria. Ecology. 2007;88:1354–1364. doi: 10.1890/05-1839. [DOI] [PubMed] [Google Scholar]
- Garland J, Cook K, Adams J, Kerkhof L. Culturability as an indicator of succession in microbial communities. Microb Ecol. 2001;42:150–158. doi: 10.1007/s00248-001-0002-3. [DOI] [PubMed] [Google Scholar]
- Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, Gordon JI. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci USA. 2011;108:6252–6257. doi: 10.1073/pnas.1102938108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugenholtz P, Pace NR. Identifying microbial diversity in the natural environment: a molecular phylogenetic approach. Trends Biotechnol. 1996;14:190–197. doi: 10.1016/0167-7799(96)10025-1. [DOI] [PubMed] [Google Scholar]
- Huse SM, Welch DM, Morrison HG, Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol. 2010;12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen PH. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ Microbiol. 2006;72:1719–1728. doi: 10.1128/AEM.72.3.1719-1728.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczynski J, Costello EK, Nemergut DR, Zaneveld J, Lauber CL, Knights D, et al. Direct sequencing of the human microbiome readily reveals community differences. Genome Biol. 2010;11:210. doi: 10.1186/gb-2010-11-5-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DJ, Pace B, Olsen GJ, Stahl DA, Sogin ML, Pace NR. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci USA. 1985;82:6955–6959. doi: 10.1073/pnas.82.20.6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon JT, Jones SE. Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat Rev Microbiol. 2011;9:119–130. doi: 10.1038/nrmicro2504. [DOI] [PubMed] [Google Scholar]
- Liu Z, Lozupone C, Hamady M, Bushman FD, Knight R. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res. 2007;35:e120. doi: 10.1093/nar/gkm541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen GJ, Lane DJ, Giovannoni SJ, Pace NR, Stahl DA. Microbial ecology and evolution: a ribosomal RNA approach. Annu Rev Microbiol. 1986;40:337–365. doi: 10.1146/annurev.mi.40.100186.002005. [DOI] [PubMed] [Google Scholar]
- Pedrós-Alió C. The rare bacterial biosphere. Ann Rev Mar Sci. 2012;4:449–466. doi: 10.1146/annurev-marine-120710-100948. [DOI] [PubMed] [Google Scholar]
- Rappé MS, Giovannoni SJ. The uncultured microbial majority. Annu Rev Microbiol. 2003;57:369–394. doi: 10.1146/annurev.micro.57.030502.090759. [DOI] [PubMed] [Google Scholar]
- Reid A, Buckley M. Washington, DC: American Academy of Microbiology; 2011. The Rare Biosphere: A report from the American Academy of Microbiology. [Google Scholar]
- Roesch LFW, Fulthorpe RR, Riva A, Casella G, Hadwin AKM, Kent AD, et al. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 2007;1:283–290. doi: 10.1038/ismej.2007.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondon MR, August PR, Bettermann D, Brady SF, Grossman TH, Liles MR, et al. Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl Environ Microbiol. 2000;66:2541–2547. doi: 10.1128/aem.66.6.2541-2547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Handelsman J. The last word: books as a statistical metaphor for microbial communities. Annu Rev Microbiol. 2007;61:23–34. doi: 10.1146/annurev.micro.61.011507.151712. [DOI] [PubMed] [Google Scholar]
- Sjöstedt J, Koch-Schmidt P, Pontarp M, Canbäck B, Tunlid A, Lundberg P, et al. Recruitment of members from the rare biosphere of marine bacterioplankton communities after an environmental disturbance. Appl Environ Microbiol. 2012;78:1361–1369. doi: 10.1128/AEM.05542-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, et al. Microbial diversity in the deep sea and the underexplored ‘rare biosphere. Proc Natl Acad Sci USA. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner RR, Chesson PL. Coexistence mediated by recruitment fluctuations: a field guide to the storage effect. Am Nat. 1985;125:769–787. [Google Scholar]
- Whitman WB, Coleman DC, Wiebe WJ. Prokaryotes: the unseen majority. Proc Natl Acad Sci USA. 1998;95:6578–6583. doi: 10.1073/pnas.95.12.6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.