


Abstract
Although most deoxyribonucleic acid (DNA) lesions are accurately repaired before replication, replication across unrepaired lesions is the main source of point mutations. The lesion tolerance processes, which allow damaged DNA to be replicated, entail two branches, error-prone translesion synthesis (TLS) and error-free damage avoidance (DA). While TLS pathways are reasonably well established, DA pathways are poorly understood. The fate of a replication-blocking lesion is generally explored by means of plasmid-based assays. Although such assays represent efficient tools to analyse TLS, we show here that plasmid-borne lesions are inappropriate models to study DA pathways due to extensive replication fork uncoupling. This observation prompted us to develop a method to graft, site-specifically, a single lesion in the genome of a living cell. With this novel assay, we show that in Escherichia coli DA events massively outweigh TLS events and that in contrast to plasmid, chromosome-borne lesions partially require RecA for tolerance.
INTRODUCTION
Replication of damaged deoxyribonucleic acid (DNA) is a universal problem faced by all organisms. DNA lesions arise continuously due to endogenous or environmental agents. Despite the efficient action of numerous repair systems, some lesions that escape these repair mechanisms are present when the genome is being replicated. To overcome the challenge of replicating damaged DNA, cells have developed several lesion tolerance mechanisms that enable the replication machinery to bypass sites of damaged DNA. The conceptually simplest procedure of bypassing lesions encountered during DNA replication is translesion synthesis (TLS), whereby the replicative polymerase is replaced by a specialized polymerase that can synthesize the new DNA strand across the site of damage. This process, although not always mutagenic, is inherently error-prone [for a review, see (1)]. On the other hand, error-free bypass of DNA lesions is possible by using the information present in the undamaged sister chromatid (2–6). These processes, collectively referred as damage avoidance (DA), embrace several pathways related to homologous recombination and are still poorly defined to date. Failure to achieve TLS or DA will lead to incomplete replication of the genome and therefore to the cell death.
Until today, studies on the consequences of lesions in DNA in vivo are usually limited to the analysis of induced mutations. Indeed, mutations that result from TLS events represent the major biological consequence of the presence of lesions in DNA because DA events are considered to be error-free. However, the respective proportion of TLS versus DA events within DNA damage tolerance events is presently not known. Moreover, most induced-mutagenesis studies involve treatment of cells with damaging agents that introduce a variety of different lesions randomly distributed all over the genome. Consequently, the DNA lesion that causes a given mutation can only be guessed. Over the past recent years, many studies aimed at uncovering the mechanism of TLS by introducing single site-specific lesions within plasmids. This approach has been instrumental to unravel the complexity of TLS pathways and the genetics of mutagenesis in Escherichia coli, Saccharomyces cerevisiae and recently in human cells (7–9). In the present paper, we show that, during plasmid replication, a single blocking lesion triggers replication fork uncoupling (10) accompanied by full unwinding of the two sister chromatids. As DA events require the two sisters to be maintained in close proximity, plasmid systems are not suited for the analysis of DA events. To overcome the limitations inherent to plasmids, we developed a methodology that allows single lesions to be site-specifically introduced into the E. coli chromosome. Under these assay conditions we show that, in contrast to plasmid-borne lesions, tolerance of chromosomal DNA lesions by DA is partially dependent on a functional recA gene. Until now, it was essentially impossible to monitor the rescue of a blocked replication fork in vivo by lack of an appropriate methodology. Indeed, current assays involve lesions randomly distributed over the chromosome, making it impossible to determine the structure and the dynamic of a fork blocked at a specific lesion site.
MATERIALS AND METHODS
Plasmids construction
pVP135 expresses the integrase and excisionase (int–xis) genes from lambda under control of a trc promoter that has been weakened by mutations in the −35 and the −10 region (11). Transcription from Ptrc is regulated by the lac repressor, supplied by a copy of lacIq on the plasmid. The vector is derived from pDSW206 (11) by replacing chloramphenicol resistance by a kanamycin resistance cassette. A polymerase chain reaction (PCR) fragment containing the int–xis operon from pTSC29cxi (12) was cloned into the NcoI-PstI restriction sites.
pVP146 is derived from pACYC184 plasmid where the chloramphenicol resistance gene has been deleted by BsaAI digestion and re-ligation. This vector, which carries only the tetracycline resistance gene, serves as an internal control for transformation efficiency.
pVP141-144 and pGP1-2 are derived from pLDR9-attL-lacZ plasmid (12). These plasmid vectors contain the following genes characteristics: the ampicillin resistance gene, the R6K replication origin that allows plasmid replication only if the recipient strain carries the pir gene (13), and the 5′ end of lacZ gene in fusion with the attL site-specific recombination site of phage lambda. The P′3 site of attL has been mutated (AATCATTAT to AATTATTAT) to avoid the excision of the plasmid once integrated (14). pVP141-144 and pGP1-2 are produced in strain EC100D pir-116 (from Epicentre Biotechnologies—cat# EC6P0950H) in which the pir-116 allele supports higher copy number of R6K origin plasmids.
Construction of vectors carrying a single lesion
Vectors for integration
Duplex plasmids carrying a single lesion were constructed following the gap-duplex method previously described (15). A 13-mer oligonucleotide, 5′-GCAAGTTAACACG, containing no lesion, a cyclobutane pyrimidine dimer (TT-CPD) or a thymine-thymine pyrimidine(6-4)pyrimidone photoproduct [TT(6-4)] lesion (underlined) was inserted into the gapped-duplex pGP1/2 leading to an in frame lacZ gene. Because the G-AAF (N-2-acetylaminofluorene) lesion can be bypassed by two distinct pathways (7), two vectors were constructed to monitor all TLS events. A 15-mer oligonucleotide containing or not a single G-AAF adduct (underlined) in the NarI site (ATCACCGGCGCCACA) was inserted into a gapped duplex pVP141/142 or pVP143/144, leading respectively to an in frame lacZ gene, and a +2 frameshift lacZ. Therefore, the construct pVP141/142 Nar3AAF/Nar+3 monitors TLS0 events, whereas pVP143/144 Nar3AAF/Nar+3 monitors TLS−2 events.
Replicating vectors
pCUL− series and pCUL+ series contain the G-AAF lesion, respectively, in the lagging and leading strand. pCUL−Nar0/Nar+3, pCUL−Nar3AAF/Nar+3, pCUL+Nar0/Nar+3 and pCUL+Nar3AAF/Nar+3 were constructed as previously described (10).
Strains
All strains used in the present study for site-specific recombination are derivative of strain FBG152 (16). Strain FBG152 is derived from strain MG1655 in which the original λ attB site was replaced by an artificial promoterless operon carrying attR fused to the 3′ part of lacZ upstream of the aadA gene and between ybhC and ybhB (at ∼ 17th min) (16). Gene disruptions of recA, mutS, uvrA and phrB were achieved by the one-step PCR method (17). The following FBG152-derived strains were constructed by P1 transduction: EVP23 (FBG152 uvrA::frt mutS::frt), EVP123 (FBG152 uvrA::frt mutS::frt recA::frt), EVP184 (FBG152 uvrA::frt mutS::frt phrB::frt) and EVP206 (FBG152 uvrA::frt mutS::frt phrB::frt recA::frt). All strains carry plasmid pVP135 that allows the expression of the int–xis under the control of IPTG. Following the site-specific recombination reaction, the lesion [G-AAF, TT-CPD or TT(6-4)] is located in the leading strand of strain FBG152.
Strand segregation assay protocol
After transformation of plasmid pCUL-Nar3AAF/Nar+3 or pCUL+Nar3AAF/Nar+3 in strain JM103uvrAmutS, and plating on X-gal indicator plates, individual blue colonies (i.e. colonies in which TLS has occurred) are collected and grown in Luria Broth (LB) media containing ampicillin. Plasmids extracted from a single colony are re-transformed into JM103 and plated on X-gal indicator plates to score blue and white clones.
Integration protocol—measurement of lesion tolerance pathways: TLS and DA
LB (50 ml) containing kanamycin to maintain plasmid pVP135 that expresses the int–xis operon and 200 µM of IPTG (to induce the expression of int–xis) are inoculated with 500 µl of an overnight starter. When the culture reaches OD600 ≈ 0.5, cells are washed twice in water, once in 10% glycerol and finally re-suspended in 200 µl of 10% glycerol and frozen in 40 µl aliquots.
To the 40 µl aliquot of cells, 1 ng of the lesion carrying vector mixed with 1 ng of the internal standard (pVP146) were added, and electroporated (in a GenePulser Xcell from BioRad, 2.5 kV, 25 µF, 200 Ω). 1 ml of SOC medium containing 200 µM IPTG is then added, and cells are incubated 1 h at 37°C. Part of the cells are plated on LB + 10 µg/ml tetracycline to measure the transformation efficiency of plasmid pVP146 (internal transformation standard), and the rest is plated on LB + 50 µg/ml Ampicillin and 80 µg/ml X-gal to select for integrants (Amp®) and TLS events (blue colonies). The integration rate is about 2000 clones per picogram of non-damaged vector for a wild-type (WT) strain.
Following the integration of the damaged vector, blue colonies represent TLS events, whereas white colonies represent DA events. The relative integration efficiencies of damaged vectors compared with their non-damaged homologues, and normalized by the transformation efficiency of pVP146 plasmid in the same electroporation experiment, allow the overall rate of tolerance of the lesion to be measured. DA events are estimated by subtracting TLS events from the total lesion tolerance events.
RESULTS AND DISCUSSION
Evidence for strand uncoupling during replication of a plasmid containing a single replication-blocking lesion
We developed an assay to analyse the products that are generated during replication of a plasmid vector that contains a single replication-blocking lesion. For this purpose, we used an adduct formed by covalent binding of the chemical carcinogen N-2-acetylaminofluorene (AAF) to the C-8 position of guanine (G-AAF) (18). This adduct blocks replication similarly to the ultraviolet (UV)-induced lesions such as TT-CPDs or 6-4 photoproducts (19). The plasmid construct also contains a short sequence heterology in the complementary strand across from the single adduct (18). The lesion is located in the lacZ gene such that TLS events give rise to lac+ plasmids, while the complementary strand is out of frame and thus gives rise to lac− plasmids. These constructs are transformed into a recipient cell (primary transformation) and plated on X-gal indicator plates to identify colonies that have experienced TLS (TLS colonies) as blue colonies (lacZ+) (Figure 1A). Individual ‘TLS colonies’ are further analysed to determine the average proportion of blue and white plasmids within the cells by extracting plasmid DNA and retransforming it into an indicator strain to determine the proportion of white and blue plasmids (secondary transformation) (Figure 1A). When a lesion-free construct is analysed, essentially all primary transformants are blue because the blue phenotype is dominant. Analysis of the plasmid content of individual blue colonies by secondary transformation shows that, as expected, the progeny of the two strands are about equally represented in the majority of colonies analysed (although there is a slight, as yet unexplained, bias towards <50% of blue colonies, Figure 1B). When a construct that monitors TLS events across a single G-AAF adduct is introduced in a non-SOS induced WT strain, only 2–3% of colonies are blue (TLS colonies) in the primary transformation, showing that TLS across G-AAF is a rare event. Analysis of individual blue colonies shows that most ‘TLS colonies’ contain <20% of blue plasmids (and thus >80% white plasmids) (Figure 1B). The same distribution is seen whether the lesion is located in the leading or in the lagging strand. These results illustrate the ‘replisome uncoupling’ phenomenon as initially identified by molecular analysis of replication intermediates (10). It was concluded from these experiments that when a replisome encounters a replication-blocking lesion, synthesis in the lesion strand is transiently arrested, whereas replication of the undamaged strand continues (10) (Figure 1C). It is estimated that following a leading strand block the replicative helicase can unwind the parental strands beyond the blocking lesion over a distance of at least 2–3 kb (10). Therefore, the commonly used single-adducted plasmid constructs become fully unwound leading to unbalanced amplification of the non-damaged strand with respect to the damaged strand. Replication of the damaged strand lags behind due to the time required to perform TLS (Figure 1C) (10).
Figure 1.

Strand segregation analysis using plasmids with a single replication-blocking lesion. (A) Outline of the strand segregation analysis assay (see the text). (B) Histogram showing the distribution, within individual primary blue colonies, of the proportion of lacZ+ versus lacZ− plasmids. For a lesion-free construct, most primary colonies are blue and carry a similar proportion of blue and white plasmids (although there is a slight, as yet unexplained, bias towards <50% of blue colonies). In contrast, most primary blue TLS colonies obtained with a plasmid carrying a single G-AAF adduct contain a small proportion of lacZ+ plasmids: 80–100% of primary TLS colonies contain <20% of lacZ+ plasmids. (C) Scheme representing full replication fork uncoupling in plasmids (sister-chromatid separation) following the encounter by the fork of a single replication-blocking lesion in one of the template strands: while replication is blocked at the damage site awaiting for TLS to occur, the non-damaged strand is fully replicated and amplified, leading to a high proportion of lacZ− (white) over lacZ+ (blue) plasmid progeny as revealed in the secondary transformation experiment.
In conclusion, these data suggest that due to their limited size, plasmids are inappropriate tools for the analysis of DA pathways that require the two sister chromatids to be maintained in close proximity to undergo a recombination-based transfer of information that characterize DA events (20). When the lesion is located in a larger genome such as a chromosome, even if the helicase travels over a certain distance beyond the replication blocking lesion, the fork will eventually come to an arrest as observed by electron microscopy in S. cerevisiae after UV irradiation (21). In an attempt to investigate DA pathways, we developed a method that allows a single lesion to be located site-specifically in a genome.
Site-specific insertion of a single lesion in the E. coli chromosome
To overcome the intrinsic limitations of plasmid systems we developed a methodology to insert a single lesion at a precise chromosomal location in E. coli. The experimental system (Figure 2A), based on phage lambda site-specific recombination, entails the following two major components: a recipient E. coli strain with a single attR site and a non-replicating plasmid construct containing the single lesion of interest and an attL site and an ampicillin resistance gene. The recombination reaction between attL and attR is controlled by ectopic expression of phage lambda int–xis proteins (12), and leads to the integration of the lesion-containing vector into the chromosome. Integrants are selected on the basis of their resistance to ampicillin. The chromosomal integration region carries the 3′-end of the lacZ gene fused to attR, whereas the remaining 5′-end is located on the incoming fragment in fusion with attL, so that precise integration restores a functional ß-galactosidase gene (lacZ). The non-damaged opposite strand contains a short sequence heterology that inactivates the lacZ gene and serves as a genetic marker that allows strand discrimination (22). Precise integration of the vector at nucleotide resolution was shown by the presence of only LacZ+ colonies following the integration of a lesion-free construct. Individual integration events were analysed by PCR and found to be precisely located at the expected chromosomal position. The present experimental approach allows a single replication-blocking lesion to be located in the leading strand template at min 17. The way this block is tolerated either by TLS or via DA pathways can thus be studied genetically and at the molecular level.
Figure 2.
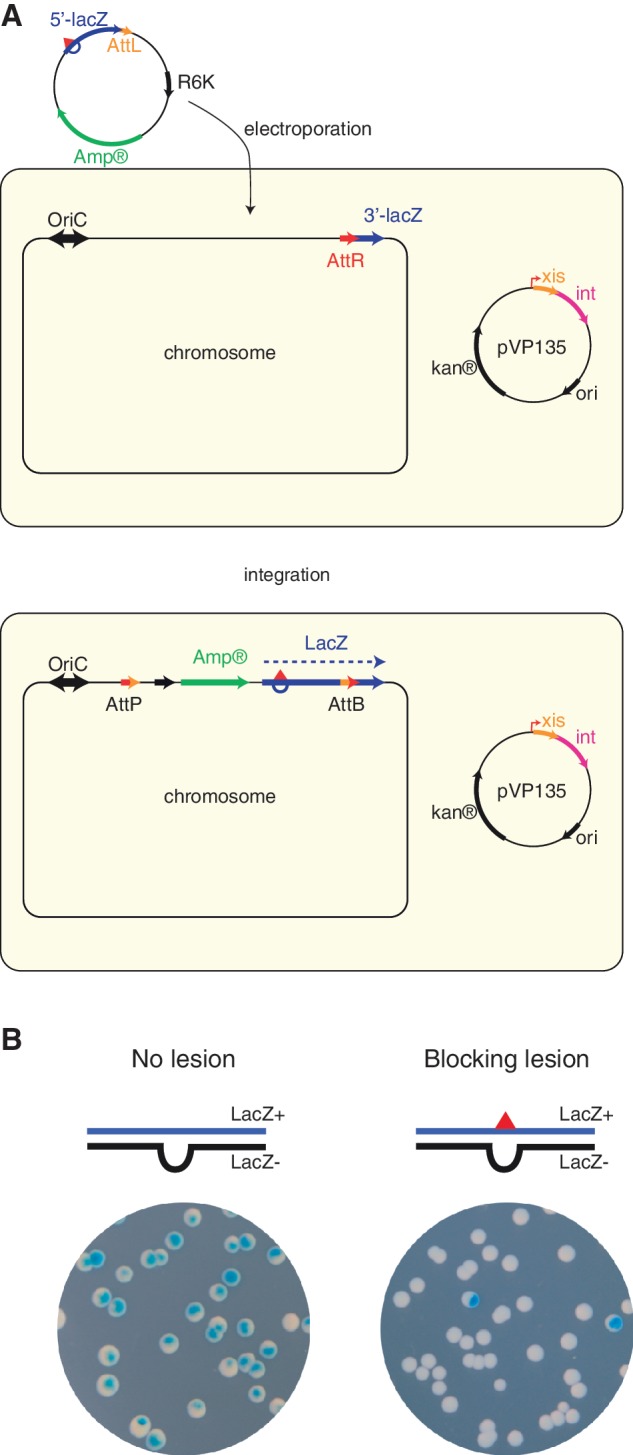
Grafting a single lesion into the chromosome. (A) The recipient strain contains a single attR integration site in fusion with the 3′ end of lacZ gene at min 17 in the E. coli chromosome. Following ectopic expression of phage lambda int–xis, the lesion-carrying construct is introduced by electroporation. Its attL site will recombine with the chromosomal attR, leading to integration of the entire lesion-containing construct. Integration events are selected on the basis of their resistance to ampicillin. Integration at nucleotide level resolution restores a functional lacZ gene allowing these events to be monitored on X-gal indicator plates. (B) Chromosomal integration of lac−/lac+ heteroduplex constructs. As expected, integration of a lesion-free heteroduplex gives rise to essentially only sectored colonies on X-gal plates. Replication of the G-AAF heteroduplex yields a lac+ event when the lesion is bypassed by TLS, whereas complementary strand replication yields a lac− event. TLS events are characterized as sectored colonies illustrating the coupling during replication of both strands even in the presence of a replication-blocking lesion.
Proper strand segregation is maintained during replication of a single chromosomal replication block
To observe what happens when a lesion escapes the repair mechanisms and is encountered by the replication fork, we conducted the following experiments in a nucleotide excision repair (uvrA) deficient strain. For experiments involving UV lesions, the photolyase gene (phrB) was additionally inactivated. Also, the recipient strain carried mutations in mismatch repair (mutS) to prevent correction of the heteroduplex. To assess normal chromosomal strand segregation we first integrated into the chromosome a lesion-free heteroduplex in which one strand carries a lacZ+ allele and the other strand a lacZ− allele. Selection of individual integration events on lac indicator plates (X-gal plates) shows mostly blue:white sectored colonies (Figure 2B). This is expected because chromosomal replication is semi-conservative and daughter chromosomes segregate in a one to one ratio. Molecular analysis of the sectored colonies reveals that blue and white sectors contain the bacterial progeny that stems from the replication and subsequent segregation of the lac+ and lac− strands, respectively. Occasional non-sectored colonies are observed probably reflecting a low extent of heteroduplex correction before replication despite inactivation of mismatch repair.
Next, we integrated a lac+/lac− heteroduplex containing a single G-AAF adduct in the lac+ strand. A majority of integration events form non-sectored pure white colonies on the X-gal indicator plate. Molecular analysis of these events suggests that they result from the processing of the single adduct via a DA pathway. Indeed, these colonies only contain the specific lac− allele carried in the non-damaged strand in the initial construct. However, it is presently not possible to eliminate the possibility that the damaged strand is degraded and that some white colonies originate from the sole replication of the non-damaged strand. A small amount of colonies are sectored blue:white colonies (Figure 2B). Molecular analysis of the sectored colonies reveals that the blue and white sectors contain the bacterial progeny that stems from the replication and subsequent segregation of the damaged and undamaged strands, respectively. The blue sectors contain the sequence of the lesion-containing strand and thus represent TLS events. The white sectors carry the lac− allele initially present in the complementary strand (Figure 3). This observation illustrates ‘coupled replication’ of both strands even in the presence of a replication-blocking lesion in contrast to the situation of strand uncoupling encountered with plasmid probes (see previous text). These observations fully validate the use of the present system to study the different lesion tolerance pathways including the yet to be defined DA pathways.
Figure 3.

Molecular characterization of blue and white sectors taken from lesion-containing sectored colonies. The sequence around the G-AAF adduct site shows the local heteroduplex that allows strand discrimination following replication. The non-damaged strand carries a 3 nt insertion that will generate an additional NlaIII restriction site during replication. Two PCR primers were designed to amplify a 610-bp fragment around the lesion site. The figure represents the analysis of individual sectors from seven sectored clones resulting from the integration of an AAF damaged vector (Figure 2B). Analysis of the white sectors (bottom gel) by NlaIII digestion shows the presence of an additional NlaIII restriction site corresponding to the replication of the non-damaged strand of the chromosome. Analysis of the blue sectors (top gel) lacks the additional NlaIII site; sequencing of these PCR fragments showed the expected sequence resulting from TLS events on the damaged strand (i.e. frameshift-2 in the NarI hotspot where the lesion is located (23).
DA events require the function of recA
DA pathways are defined as representing the error-free branch of post recombinational repair. It is supposed that replication-blocking lesions are non-informative and that the missing information is accurately retrieved from the sister chromatid by means of mechanisms akin to homologous recombination. Consequently, we wanted to investigate the tolerance of single chromosomal lesions in recA− and recA+ strains. For this purpose, we compared three common replication blocking lesions, namely G-AAF and the two major UV-induced photoproducts, the TT-CPD and the TT(6-4). In a recA+ strain, there is no detectable loss in the integration efficiency of any of these lesions relative to the respective lesion-free construct (Figure 4A). In contrast, in a recA−deficient strain, the relative integration efficiency, hereafter referred to as the ‘survival’, dropped to about 40–50% for all three lesions (Figure 4A). This loss in survival reflects a severe impairment to process a single lesion by DA in the absence of RecA protein. Although the involvement of RecA or its homologues in DA was expected from existing models of DA (24,25), the present results offer, for the first time, an experimental quantification of the phenomenon at the level of a single chromosomal lesion. Meanwhile, the results also suggest the existence of a recA-independent DA pathway that accounts for ∼50% of the lesion tolerance events. Classical models of DA entail either template switching at regressed forks or daughter strand gap formation that are subsequently filled in by homologous recombination (26). It is likely that the recA-dependent DA pathway corresponds to the classical daughter strand gap-filling model (27), whereas the recA-independent component may relate to DA via template switching. It should be stressed that under our conditions, where SOS is not induced, TLS events represent a minor fraction (0.5–3%) with respect to DA events (Figure 4A). Moreover, the small amount of TLS events is further suppressed in the recA− background (Figure 4A) because Pol V requires RecA for its activation (28,29).
Figure 4.
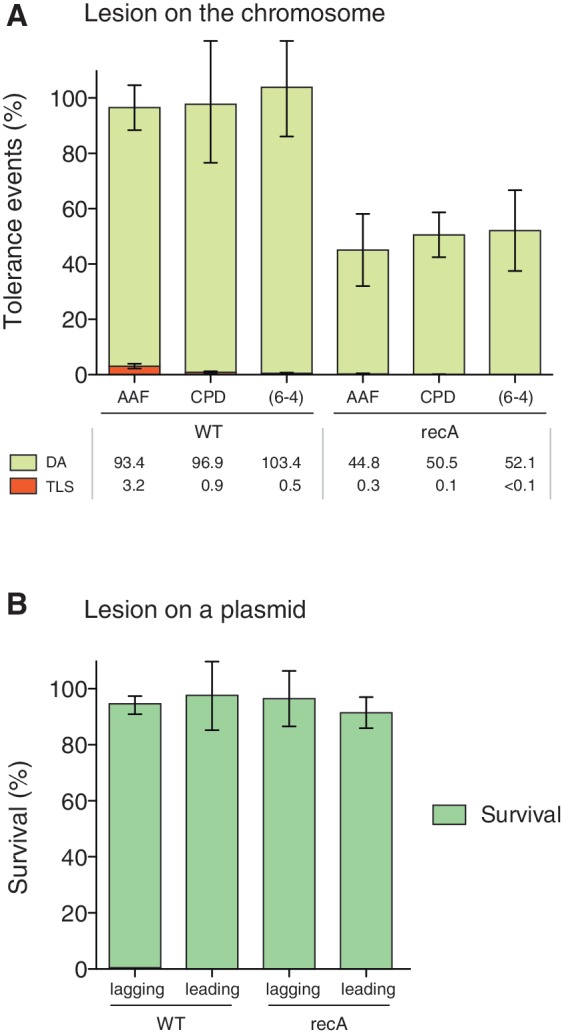
Survival of chromosomal and plasmid-borne replication-blocking lesions in WT and recA strains. (A) Relative integration efficiency (survival) and TLS events of a damage-containing construct relative to the corresponding damage-free construct, is measured in WT and recA strains. Three lesions, namely G-AAF, TT-CPD and TT(6-4), are introduced in the leading strand at position 17 min in the chromosome by site-specific recombination. The data represent the average value and standard deviation of at least three independent experiments. (B) Relative transformation efficiency (survival) of a ColE1-based plasmid containing a single G-AAF adduct in the leading or lagging strand. In both orientations, for a G-AAF adduct located in a plasmid there is no loss of survival in a recA versus rec+ strain. In contrast to plasmid-borne lesions, tolerance of chromosomal DNA lesions by DA is partially dependent on a functional recA gene.
Similarly, we measured the survival of a plasmid-borne G-AAF adduct in a WT and recA mutant strain. Low levels of TLS, similar to those measured for this adduct in the chromosome, are seen in a non-SOS induced WT strain (7,30). With a G-AAF adduct located in a plasmid rather than in the chromosome, no loss of survival in a recA− versus rec+ strain was observed (Figure 4B). As discussed earlier, damaged plasmid replication exhibits uncoupled amplification of the two strands, a process that appears not to be impeded in a recA− strain in contrast to the observation made for the chromosome-borne lesions. This observation reinforces the notion that plasmids are not suitable tools for the analysis of genuine DA events as discussed in the previous section.
CONCLUSION
The present work provides the first experimental system to study TLS and DA pathways in vivo, in a quantitative manner. Depending on the nature of the replication block, tolerance pathways occur either directly at the fork (on the fly) or at gaps left opposite lesion, i.e. behind the fork (26). Indeed, when encountering a strong replication block in the leading strand, the fork can restart by means of a priming event that occurs downstream of the lesion, leaving a gap that is subsequently processed by TLS or DA (31,32). We show that for common replication-blocking lesions, such as UV-induced photoproducts or G-AAF adducts, DA events massively outweigh TLS events (97% versus 1–3%) in non-SOS induced cells. The present methodology opens new ways to explore the metabolism of defined replication fork blocks at the chromosomal level. We highlight a major drawback of plasmid-borne lesion assays by showing that the extensive uncoupling between sister chromatids that occurs during plasmid replication precludes DA events to be monitored. This point is illustrated by the fact that, in contrast to plasmid-borne lesions, tolerance of chromosomal DNA lesions by DA is partially dependent on a functional recA gene. Until now, it was essentially impossible to monitor the repair of blocked replication forks in vivo by lack of an appropriate methodology.
This approach offers an experimental system able to open up the field of replication fork recovery in a way comparable with the development of double-strand break repair by the introduction of single double-strand breaks by HO and I-SceI endonucleases (33,34). In the present article, lesion tolerance pathways have been monitored using only genetic read-outs. However, given the efficiency of integration, i.e. 5–10% of all cells, we intend to develop molecular methods able to detect early replication intermediates that should allow us to monitor replication fork rescue in real time.
FUNDING
LIGUE Contre le Cancer (labellization 2011, in part); ANR grant ForkRepair [ANR 11 BSV8 017 01, in part]; fellowship from the Fondation ARC pour la recherche sur le cancer (to V.P. and K.N., in part). Funding for open access charge: LIGUE Contre le Cancer.
Conflict of interest statement. None declared.
REFERENCES
- 1.Pagès V, Fuchs RPP. How DNA lesions are turned into mutations within cells? Oncogene. 2002;21:8957–8966. doi: 10.1038/sj.onc.1206006. [DOI] [PubMed] [Google Scholar]
- 2.Prakash L. The structure and function of RAD6 and RAD18 DNA repair genes of Saccharomyces cerevisiae. Genome. 1989;31:597–600. doi: 10.1139/g89-111. [DOI] [PubMed] [Google Scholar]
- 3.Frampton J, Irmisch A, Green CM, et al. Postreplication repair and PCNA modification in Schizosaccharomyces pombe. Mol. Biol. Cell. 2006;17:2976–2985. doi: 10.1091/mbc.E05-11-1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rudolph CJ, Upton AL, Lloyd RG. Replication fork stalling and cell cycle arrest in UV-irradiated Escherichia coli. Genes Dev. 2007;21:668–681. doi: 10.1101/gad.417607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karras GI, Jentsch S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell. 2010;141:255–267. doi: 10.1016/j.cell.2010.02.028. [DOI] [PubMed] [Google Scholar]
- 6.Daigaku Y, Davies AA, Ulrich HD. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature. 2010;465:951–955. doi: 10.1038/nature09097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Napolitano R, Janel-Bintz R, Wagner J, Fuchs RP. All three SOS-inducible DNA polymerases (Pol II, Pol IV and Pol V) are involved in induced mutagenesis. EMBO J. 2000;19:6259–6265. doi: 10.1093/emboj/19.22.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pagès V, Johnson RE, Prakash L, Prakash S. Mutational specificity and genetic control of replicative bypass of an abasic site in yeast. Proc. Natl Acad. Sci. USA. 2008;105:1170–1175. doi: 10.1073/pnas.0711227105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoon J-H, Prakash L, Prakash S. Error-free replicative bypass of (6-4) photoproducts by DNA polymerase zeta in mouse and human cells. Genes Dev. 2010;24:123–128. doi: 10.1101/gad.1872810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pagès V, Fuchs RP. Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo. Science. 2003;300:1300–1303. doi: 10.1126/science.1083964. [DOI] [PubMed] [Google Scholar]
- 11.Weiss DS, Chen JC, Ghigo JM, Boyd D, Beckwith J. Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J. Bacteriol. 1999;181:508–520. doi: 10.1128/jb.181.2.508-520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valens M, Penaud S, Rossignol M, Cornet F, Boccard F. Macrodomain organization of the Escherichia coli chromosome. EMBO J. 2004;23:4330–4341. doi: 10.1038/sj.emboj.7600434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inuzuka M. Plasmid-encoded initiation protein is required for activity at all three origins of plasmid R6K DNA replication in vitro. FEBS Lett. 1985;181:236–240. doi: 10.1016/0014-5793(85)80266-0. [DOI] [PubMed] [Google Scholar]
- 14.Bauer CE, Hesse SD, Gumport RI, Gardner JF. Mutational analysis of integrase arm-type binding sites of bacteriophage lambda. Integration and excision involve distinct interactions of integrase with arm-type sites. J. Mol. Biol. 1986;192:513–527. doi: 10.1016/0022-2836(86)90273-1. [DOI] [PubMed] [Google Scholar]
- 15.Koehl P, Burnouf D, Fuchs RP. Construction of plasmids containing a unique acetylaminofluorene adduct located within a mutation hot spot: a new probe for frameshift mutagenesis. J. Mol. Biol. 1989;207:355–364. doi: 10.1016/0022-2836(89)90259-3. [DOI] [PubMed] [Google Scholar]
- 16.Esnault E, Valens M, Espéli O, Boccard F. Chromosome structuring limits genome plasticity in Escherichia coli. PLoS Genet. 2007;3:e226. doi: 10.1371/journal.pgen.0030226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl Acad. Sci. USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koffel-Schwartz N, Coin F, Veaute X, Fuchs RP. Cellular strategies for accommodating replication-hindering adducts in DNA: control by the SOS response in Escherichia coli. Proc. Natl. Acad. Sci. USA. 1996;93:7805–7810. doi: 10.1073/pnas.93.15.7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujii S, Fuchs RP. Defining the position of the switches between replicative and bypass DNA polymerases. EMBO J. 2004;23:4342–4352. doi: 10.1038/sj.emboj.7600438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010;11:208–219. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- 21.Lopes M, Foiani M, Sogo JM. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell. 2006;21:15–27. doi: 10.1016/j.molcel.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 22.Veaute X, Fuchs RP. Greater susceptibility to mutations in lagging strand of DNA replication in Escherichia coli than in leading strand. Science. 1993;261:598–600. doi: 10.1126/science.8342022. [DOI] [PubMed] [Google Scholar]
- 23.Burnouf D, Koehl P, Fuchs RP. Single adduct mutagenesis: strong effect of the position of a single acetylaminofluorene adduct within a mutation hot spot. Proc. Natl Acad. Sci. USA. 1989;86:4147–4151. doi: 10.1073/pnas.86.11.4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Courcelle J, Hanawalt PC. RecA-dependent recovery of arrested DNA replication forks. Annu. Rev. Genet. 2003;37:611–646. doi: 10.1146/annurev.genet.37.110801.142616. [DOI] [PubMed] [Google Scholar]
- 25.Zhang H, Lawrence CW. The error-free component of the RAD6/RAD18 DNA damage tolerance pathway of budding yeast employs sister-strand recombination. Proc. Natl Acad. Sci. USA. 2005;102:15954–15959. doi: 10.1073/pnas.0504586102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann AR, Fuchs RP. Gaps and forks in DNA replication: rediscovering old models. DNA Repair. 2006;5:1495–1498. doi: 10.1016/j.dnarep.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 27.Rupp WD, Howard-Flanders P. Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation. J. Mol. Biol. 1968;31:291–304. doi: 10.1016/0022-2836(68)90445-2. [DOI] [PubMed] [Google Scholar]
- 28.Dutreix M, Moreau PL, Bailone A, et al. New recA mutations that dissociate the various RecA protein activities in Escherichia coli provide evidence for an additional role for RecA protein in UV mutagenesis. J. Bacteriol. 1989;171:2415–2423. doi: 10.1128/jb.171.5.2415-2423.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sweasy JB, Witkin EM, Sinha N, Roegner-Maniscalco V. RecA protein of Escherichia coli has a third essential role in SOS mutator activity. J. Bacteriol. 1990;172:3030–3036. doi: 10.1128/jb.172.6.3030-3036.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Napolitano RL, Fuchs RP. New strategy for the construction of single-stranded plasmids with single mutagenic lesions. Chem. Res. Toxicol. 1997;10:667–671. doi: 10.1021/tx970018w. [DOI] [PubMed] [Google Scholar]
- 31.Michel B, Grompone G, Florès M-J, Bidnenko V. Multiple pathways process stalled replication forks. Proc. Natl Acad. Sci. USA. 2004;101:12783–12788. doi: 10.1073/pnas.0401586101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heller RC, Marians KJ. Replication fork reactivation downstream of a blocked nascent leading strand. Nature. 2006;439:557–562. doi: 10.1038/nature04329. [DOI] [PubMed] [Google Scholar]
- 33.Plessis A, Perrin A, Haber JE, Dujon B. Site-specific recombination determined by I-SceI, a mitochondrial group I intron-encoded endonuclease expressed in the yeast nucleus. Genetics. 1992;130:451–460. doi: 10.1093/genetics/130.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pâques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]