

Abstract
Background
The major concept behind augmentation therapy with human α1-antitrypsin (AAT) is to raise the levels of AAT in patients with protease inhibitor phenotype ZZ (Glu342Lys)-inherited AAT deficiency and to protect lung tissues from proteolysis and progression of emphysema.
Objective
To evaluate the short-term effects of augmentation therapy (Prolastin®) on plasma levels of AAT, C-reactive protein, and chemokines/cytokines.
Materials and methods
Serum and exhaled breath condensate were collected from individuals with protease inhibitor phenotype ZZ AAT deficiency-related emphysema (n = 12) on the first, third, and seventh day after the infusion of intravenous Prolastin. Concentrations of total and polymeric AAT, interleukin-8 (IL-8), monocyte chemotactic protein-1, IL-6, tumor necrosis factor-α, vascular endothelial growth factor, and C-reactive protein were determined. Blood neutrophils and primary epithelial cells were also exposed to Prolastin (1 mg/mL).
Results
There were significant fluctuations in serum (but not in exhaled breath condensate) levels of AAT polymers, IL-8, monocyte chemotactic protein-1, IL-6, tumor necrosis factor-α, and vascular endothelial growth factor within a week of augmentation therapy. In general, augmented individuals had higher AAT and lower serum levels of IL-8 than nonaugmented subjects. Prolastin added for 3 hours to neutrophils from protease inhibitor phenotype ZZ individuals in vitro reduced IL-8 release but showed no effect on cytokine/chemokine release from human bronchial epithelial cells.
Conclusion
Within a week, augmentation with Prolastin induced fluctuations in serum levels of AAT polymers and cytokine/chemokines but specifically lowered IL-8 levels. It remains to be determined whether these effects are related to the Prolastin preparation per se or to the therapeutic efficacy of augmentation with AAT.
Keywords: Prolastin, augmentation therapy, cytokines, IL-8, exhaled breath condensate, neutrophils
Introduction
α1-antitrypsin (AAT), also referred to as α1-proteinase inhibitor or SERPINA1, is an acute phase protein and one of the most abundant serine protease inhibitors (Pi) in human plasma. The AAT protein is encoded by the Pi locus on chromosome 14q32.1 as part of a gene cluster called SERPIN supergene. The level of plasma AAT is controlled by a pair of codominant alleles. The nomenclature used to identify the AAT variants of the allelic system called “Pi system” was developed during the 1970s, based on the migration velocity of the AAT variants in an electric field.
Severe Pi phenotype ZZ (PiZZ) AAT deficiency (AATD) is one of the proven genetic risk factors that is associated with a predisposition to early onset, rapidly progressive chronic obstructive pulmonary disease where emphysema is a major component.1 It is well established that a single amino acid mutation (Glu342Lys) in Z variant of AAT perturbs the folding and tertiary structure of protein leading to its spontaneous polymerization and retention in the endoplasmic reticulum of the hepatocytes.2 This results in a dramatic reduction (up to 90%) in circulating levels of monomeric AAT protein, an important inhibitor of neutrophil derived proteases and a modulator of host inflammatory responses.3 Extrahepatic Z AAT polymerization also occurs, and Z AAT polymers have been inferred to have chemotactic and proinflammatory activities, particularly if oxidized.4,5
Augmentation therapy with purified, pooled human AAT was introduced for emphysema patients with AATD during the 1980s.6 This specific therapy is typically considered for those AATD patients with forced expiratory volume in 1 second between 30%–65%.7 The approved regimen of 3–5 g AAT (60 mg/kg body weight) weekly intravenous infusions has been the most studied therapy, although biweekly and monthly regimens are sometimes used. The major concept behind the therapy is that raising the levels of blood and tissue AAT would protect the lungs from continuous destruction by proteases, particularly neutrophil elastase.8 Whether this biochemical normalization of AAT levels influences the pathogenic processes of lung disease is still debated, although existing results do suggest that augmentation therapy has beneficial effects in reducing the frequency of lung infections and reducing the rate of decline of lung function.9–13 On the other hand, the fundamental mechanisms of human lung injury are difficult to investigate, as clinical data are descriptive regarding the evolution of inflammatory changes inside pulmonary tissue. It is also noteworthy that different commercial preparations of human purified plasma AAT are used for augmentation therapy. There is no definitive evidence to suggest superiority of any one formulation; however, the biological activity of AAT has not been consistent across preparations.14
The aim of the current study was to investigate the short-term (within 1 week) effects of augmentation therapy with Prolastin® (Grifols Biotherapeutics, Barcelona, Spain) on the levels of AAT and inflammatory cytokines/chemokines in serum and exhaled breath condensate (EBC) from individuals with AATD.
Material and methods
Prolastin was used for intravenous infusions and for cell experiments in vitro.
Study participants
This observational study enrolled 24 subjects with PiZZ AATD-related emphysema and has not been registered as a clinical trial. Individuals who’d had recent acute exacerbations (within the last 3 months) or were taking oral steroids were excluded from the study. Exacerbation was defined according to the Gold Report: “An exacerbation of chronic obstructive pulmonary disease is an acute event characterized by a worsening of the patient’s respiratory symptoms that is beyond normal day-to-day variations and leads to a change in medication”.15 All participants had signed an informed consent and answered a questionnaire regarding the symptoms, smoking history, health status, and medical history. The study was approved by the Philipps University of Marburg local ethics committee (number 59/06).
Out of 24 patients, twelve were under weekly (60 mg/kg body weight) augmentation therapy with Prolastin and twelve had not received augmentation therapy. At the time of the study all participants were nonsmokers; however, there were former smokers among the augmented (n = 8) and nonaugmented (n = 7) patients. The mean forced expiratory volume in 1 second was 44.4% and 64.8% in augmented and nonaugmented patients, respectively (Table 1).
Table 1.
Patient characteristics
| Augmentation therapy with Prolastin® (60 mg/kg body weight) | ||
|---|---|---|
|
|
||
| Augmented | Nonaugmented | |
| Age, years (SD) | 58 (9.9) | 53 (13.3) |
| Sex, F/M | 3/9 | 5/7 |
| FEV1, % (SD) | 44.4 (15.7) | 64.8 (24.0) |
| Smokers, current/previous | 0/8 | 0/7 |
| Pack-years smoked, years (SD) | 9.7 (7.7) | 13.4 (16.6) |
Abbreviations: F, females; FEV1, forced expiratory volume in 1 second; M, males; SD, standard deviation.
Study design
EBC and blood samples were obtained from patients receiving weekly intravenous Prolastin therapy just before therapy, and 2 hours and 3 days after therapy. EBC and serum samples were obtained from control individuals who were not on augmentation with Prolastin.
EBC collection and analysis
EBC from all participants was obtained according to the American Thoracic Society/European Respiratory Society guidelines using RTube™ (Respiratory Research, Inc, Austin, TX) by breathing tidally into the device precooled to −20°C for 10 minutes.16 Samples were aliquoted for direct assessment of pH as described previously.17 The rest of the samples were stored at −80°C for subsequent analysis. The levels of EBC interleukin-1β (IL-1β), IL-6, IL-8, tumor necrosis factor-α (TNFα), vascular endothelial growth factor (VEGF), and monocyte chemotactic protein-1 (MCP-1) were measured using a multiplex fluorescent bead immunoassay (Cytometric Bead Array; BD Biosciences, San Jose, CA) as described previously.18 Detection limits were: IL-1β: 2.3 pg/mL; IL-6: 1.6 pg/mL; IL-8: 1.2 pg/mL; MCP-1: 1.3 pg/mL; TNFα: 1.2 pg/mL, and VEGF: 4.5 pg/mL. C-reactive protein (CRP) analysis was performed using a salivary CRP enzyme-linked immunosorbent assay (ELISA; Salimetrics LLC, State College, PA). The sensitivity for this assay was 10 pg/mL and the detection limit was 90 pg/mL.
Serum assays
Routine blood samples were obtained from all participants during the clinical routine visits and analyzed for differential blood cell counts and levels of AAT, CRP, IL-6, IL-8, and TNFα. Additional blood samples were taken for IL-6, VEGF, and MCP-1 analysis by the multiplex arrays (Cytometric Bead Array; BD Biosciences). The sensitivity of the assays was 1.2 pg/mL. AAT polymer ELISAs were performed from serum samples as described previously.19 Due to a lack of sample, this assay was performed only in ten study participants of the Prolastin-treated subgroup. Serum CRP was analyzed using an ELISA kit ( R&D Systems, Minneapolis, MN). Samples were diluted 1:100 with Calibrator Diluent Calibrator Diluent RD5P (R&D Systems, Minneapolis, MN). The minimum detection concentration of CRP ranged from 0.005–0.022 ng/mL.
Cell experiments
Epithelial cells
For all cell experiments, Prolastin was first prepared according to manufacturer’s recommendations and then diluted in phosphate buffered saline until the intended concentration was reached. Primary human bronchial epithelial cells were cultured in air liquid interface culture systems as described previously.20 Cells were stimulated with 1 mg/mL (19.2 μM) of Prolastin for 4 hours and 24 hours at 37°C 5% volume/volume carbon dioxide. The concentrations of IL-6, IL-8, TNFα, and MCP-1 were measured in cell culture supernatants by ELISA kits (R&D Systems). The sensitivity of the ELISAs were: IL-6: 10 pg/mL, IL-8: 30 pg/mL, TNFα: 15 pg/mL, and MCP-1: 15 pg/mL.
Neutrophil isolation and assays
Neutrophils from healthy donors (n = 3), PiZZ AATD patients (n = 12) before and after Prolastin infusion, and AATD patients without augmentation therapy (n = 3) were isolated from the peripheral blood using Polymorphprep™ (Axis-Shield PoC AS, Oslo, Norway) according to the manufacturer’s recommendations as previously described.21 The neutrophil purity and cell viability exceeded 95%. Purified neutrophils (1 × 106 cells/mL) were resuspended in Roswell Park Memorial Institute medium supplemented with 10% volume/volume fetal calf serum and stimulated with 1 mg/mL (19.2 μM) Prolastin or 0.1 μg/mL lipopolysaccharide (LPS) separately or in combination at 37°C 5% volume/volume carbon dioxide for the determined time points. The concentrations of IL-8, IL-6, and MCP-1 in cell culture supernatants were measured by ELISA kits (R&D Systems).
Statistics
Statistical analysis was performed using SigmaStat® 3.1. (Systat Software, Inc, San Jose, CA) and GraphPad Prism® 5.0 software (GraphPad Software, Inc, La Jolla, CA). Data are presented as mean ± standard deviation. The D’Agostino– Pearson omnibus normality test was performed.22 Normally distributed values were compared using the paired t-test. If the D’Agostino–Pearson omnibus normality test failed, the Wilcoxon matched-pairs signed-ranks test was employed. Differences between values of groups were explored by one-way analysis of variance followed by post hoc multiple comparisons according to Tukey’s test.
Results
Serum levels of total and polymeric AAT
As expected, mean basal serum concentrations of AAT were lower in AATD patients without augmentation therapy than those with augmentation therapy (n = 12; 0.26 ± 0.04 mg/mL versus 0.79 ± 0.28 mg/mL, respectively; P < 0.001). Directly after augmentation therapy levels of serum AAT rose from 0.79 ± 0.28 mg/mL to 2.678 ± 0.74 mg/mL (P < 0.0001). On day three, the serum AAT concentrations decreased to 1.19 ± 0.24 mg/mL, but were still significantly higher relative to day seven after therapy (P = 0.0014).
Determination of serum AAT polymer levels revealed significantly higher mean polymer concentration at day one postaugmentation therapy relative to the baseline (day seven) (n = 10; 6.94 ± 2.2 μg/mL versus 4.74 ± 1.6 μg/mL; P = 0.002). However, AAT polymer concentrations dropped to 4.98 ± 1.3 μg/mL on the third day after augmentation therapy and did not differ significantly from the baseline. Notably, AAT polymer levels correlated with total AAT levels (r = 0.55, P = 0.0017). Due to the lack of samples, polymer concentrations were not able to be measured in nonaugmented patients.
Serum levels of CRP
There was no significant difference between the levels of CRP in the augmented and nonaugmented individuals (n = 12; 3.17 ± 2.7 μg/mL versus 2.92 ± 2.9 μg/mL). Moreover, no change was found in CRP levels during augmentation therapy (2.76 ± 2.9 μg/mL on the first day after therapy and 2.29 ± 2.7 μg/mL at day three).
Serum levels of cytokines and chemokines
As shown in Table 2, lower serum levels of IL-8 (P = 0.02) but higher levels of MCP-1 (P = 0.029) were found the day before augmentation therapy compared to nonaugmented patients. On the other hand, serum levels of IL-6, TNFα, and VEGF did not differ significantly between augmented and nonaugmented patients (Table 2).
Table 2.
Serum markers in Prolastin® augmented and nonaugmented patients
| Analyte | Patient, n | A | N | A vs N | ||
|---|---|---|---|---|---|---|
|
|
|
|
|
|||
| A/N | Mean ± SD (pg/mL) | Mean ± SD (pg/mL) | P value | |||
| IL-8 | 12/12 | 4.3 | 3.9 | 8.4 | 4.2 | 0.02 |
| MCP-1 | 12/12 | 85.0 | 65.1 | 38.3 | 23.6 | 0.029 |
| IL-6 | 12/11 | 1.6 | 1.8 | 1.45 | 3.3 | NS |
| TNFα | 12/12 | 9.5 | 2.6 | 11.4 | 2.4 | NS |
| VEGF | 12/11 | 35.3 | 38.9 | 30.0 | 57.7 | NS |
Abbreviations: A, augmented patients; IL-6, interleukin 6; IL-8, interleukin-8; MCP-1, monocyte chemotactic protein-1; N, nonaugmented patients; n, number; NS, not significant; SD, standard deviation; TNFα, tumor necrosis factor-α; vs, versus; VEGF, vascular endothelial growth factor.
Further analysis of the effects of weekly augmentation therapy on cytokine, chemokine, and VEGF serum levels revealed that serum levels of IL-8 increased on the first day after augmentation therapy, whereas IL-6 and VEGF levels decreased relative to those on day seven, ie, before the weekly infusion. Interestingly, at day three after Prolastin infusion, the levels of MCP-1 were lower, whereas TNFα levels were higher relative to day seven after augmentation (Table 3).
Table 3.
Changes in serum analyte concentrations (pg/mL) within a week after augmentation therapy
| Analyte | Day 1 | Day 3 | P value | Day 7 | P value | |||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|||||
| Mean ± SD (pg/mL) | Mean ± SD (pg/mL) | 1 vs 3 | Mean ± SD (pg/mL) | |||||
| IL-8 | 6.5 | 3.5 | 7.92 | 3.3 | 0.032 | 4.26 | 3.9 | 1 vs 7, 0.008 |
| MCP-1 | 67.0 | 22.3 | 40.2 | 22.3 | NS | 85.1 | 65.1 | 3 vs 7, 0.035 |
| IL-6 | 0.42 | 0.99 | 1.17 | 1.85 | NS | 1.58 | 1.78 | 1 vs 7, 0.012 |
| TNFα | 9.86 | 2.9 | 11.7 | 2.6 | NS | 9.51 | 2.6 | 3 vs 7, 0.030 |
| VEGF | 15.4 | 16.3 | 20.0 | 34.4 | NS | 35.3 | 38.8 | 1 vs 7, 0.023 |
Abbreviations: IL-6, interleukin-6; IL-8, interleukin-8; MCP-1, monocyte chemotactic protein-1; NS, not significant; SD, standard deviation; TNFα, tumor necrosis factor-α; vs, versus; VEGF, vascular endothelial growth factor.
Biomarker analysis in EBC
CRP levels in EBC were found to be higher in nonaugmented (220.2 ± 57.4 pg/mL) patients compared to augmented patients (P < 0.0001; Figure 1). Remarkably, CRP levels rose directly after augmentation therapy (59.5 ± 16.6 pg/mL to 84.8 ± 27.2 pg/mL; P = 0.013) and remained higher on day three (98.4 ± 19.4 pg/mL; P < 0.0001). However, despite significant fluctuations due to augmentation therapy, CRP levels remained within a normal range and were just above the detection limit of the assay. None of the other EBC markers (IL-1β, IL-6, IL-8, TNFα, MCP-1, and VEGF) were significantly changed during augmentation therapy (data not shown).
Figure 1.

C-reactive protein levels in exhaled breath condensate. Exhaled breath condensate samples were obtained according to the American Thoracic Society/European Respiratory Society guidelines from augmented patients at different time points and from nonaugmented patients at least once.
Notes: Measurements were performed using an enzyme-linked immunosorbent assay. C-reactive levels in exhaled breath condensate were found to be higher in nonaugmented patients as compared to augmented patients (***P < 0.0001). C-reactive protein levels rose directly after augmentation therapy (*P < 0.05) and remained higher on day three (***P < 0.0001).
Abbreviation: CRP, C-reactive protein.
Electrophoretic characterization of Prolastin preparation
Prolastin vials contain 1059 mg of AAT, as determined by its capacity to inhibit porcine pancreatic elastase. Prolastin was dissolved in sterile water for injections provided by the manufacturer. Electrophoretically separated samples of Prolastin were immunoblotted with a polyclonal antibody against AAT. Figure 2 illustrates the typical profile of the preparations of Prolastin on nondenaturing and sodium dodecyl sulfate polyacrylamide gel electrophoresis. Prolastin migrates as several different bands on nondenaturing polyacrylamide gel electrophoresis. In contrast, electrophoretic separation of Prolastin under reducing conditions manifests a single protein with the molecular size of native AAT. These data demonstrate that Prolastin contains high molecular mass polymers of AAT.
Figure 2.
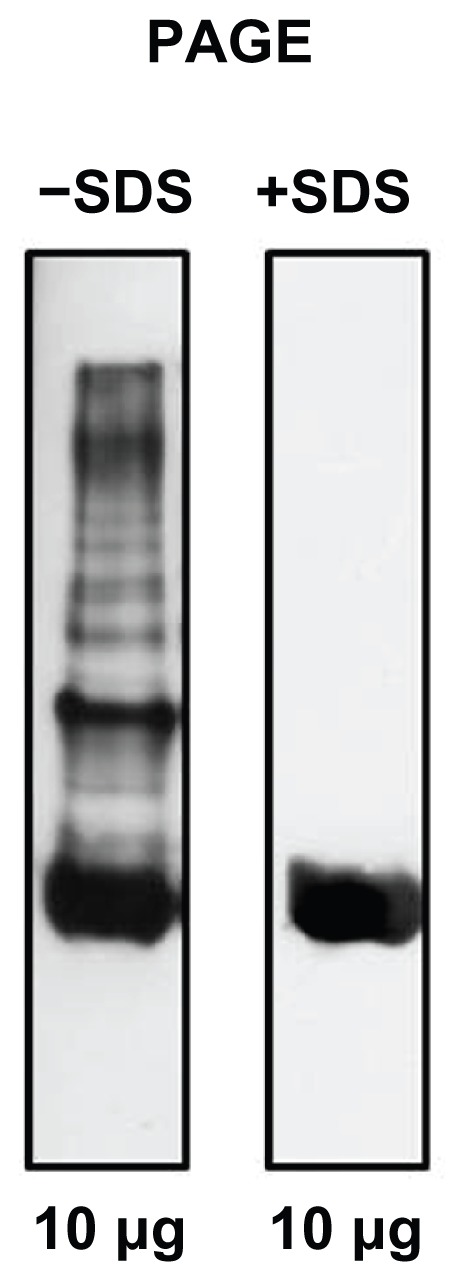
A representative western blot of Prolastin®.
Notes: Equal amounts of Prolastin were analyzed by 7% weight/volume acrylamide nondenaturing polyacrylamide gel electrophoresis without and with sodium dodecyl sulfate, blotted and probed with antihuman α1-antitrypsin antibodies (goat antihuman α1-antitrypsin antibody, dilution 1:10000 and rabbit antigoat immunoglobulin G peroxidase-conjugated secondary antibody, dilution 1:20000). Blots were developed with Amersham® ECL Western Blot analysis system (GE Healthcare, Little Chalfont, United Kingdom).
Abbreviations: PAGE, polyacrylamide gel electrophoresis; SDS, sodium dodecyl sulfate.
Biological activities of Prolastin in cell culture models in vitro
Neutrophils from healthy donors
Blood neutrophils were stimulated either with LPS or with Prolastin for 3 hours in media supplemented with 10% volume/volume fetal calf serum. The release of IL-8 was determined in cell culture supernatants. As illustrated in Figure 3, Prolastin induced the release of IL-8 as compared to nontreated neutrophil controls. However, when compared to LPS, the effect of Prolastin was significantly lower. Additional investigation revealed that after 6 hours, Prolastin (0.1 mg/mL and 1 mg/mL) showed no effect on IL-8 release relative to controls but inhibited LPS-induced IL-8 and MCP-1 release (Figure 4).
Figure 3.

Effects of Prolastin® interleukin-8 release from neutrophils isolated from healthy donors after 3 hours incubation time.
Notes: Isolated blood neutrophils (1 × 106 cells/mL) were suspended in medium containing 10% volume/volume fetal calf serum and treated with lipopolysaccharide or Prolastin for 3 hours. The release of interleukin-8 was determined in cell culture supernatants. Each bar represents mean ± standard deviation from four separate repeats of two independent experiments; *P < 0.05; **P < 0.01.
Abbreviations: IL-8, interleukin-8; LPS, lipopolysaccharide.
Figure 4.

Effects of Prolastin® on lipopolysaccharide-induced interleukin-8 and monocyte chemotactic protein-1 release from neutrophils isolated from healthy donor blood after 6 hours incubation time.
Notes: Isolated blood neutrophils (1 × 106 cells/mL) were suspended in medium containing 10% volume/volume fetal calf serum and treated with lipopolysaccharide (0.1 μg/mL) and Prolastin (0.1 mg/mL and 1 mg/mL) for 6 hours separately or in combination. The release of interleukin-8 and monocyte chemotactic protein-1 was determined in cell culture supernatants. Each bar represents mean ± standard deviation from three separate repeats of three independent experiments; *P < 0.05; **P < 0.01; ***P < 0.001.
Abbreviations: IL-8, interleukin-8; LPS, lipopolysaccharide; MCP-1, monocyte chemotactic protein-1.
Neutrophils from PiZZ patients before and after augmentation therapy
Prolastin (1 mg/mL) was added to blood neutrophils isolated before and 1 day after augmentation therapy. Cells stimulated with LPS served as a positive control, while phosphate buffered saline-stimulated neutrophils served as negative controls. As illustrated in Figure 5A and B, incubation of neutrophils in media supplemented with 10% volume/volume fetal calf serum for 3 hours in the presence of LPS resulted in a significant induction in IL-8 release compared to the control cells. Under these experimental conditions, Prolastin reduced IL-8 levels in neutrophil culture supernatants relative to nontreated control cells. Notably, the effects of Prolastin on IL-8 release did not differ significantly when added to neutrophils isolated before or after augmentation therapy.
Figure 5.

Effects of an interleukin-8 release from neutrophils isolated from protease inhibitor phenotype ZZ emphysema patients after 3 hours incubation time. Isolated blood neutrophils (A) before and the (B) first day after augmentation therapy were suspended in medium containing 10% volume/volume fetal calf serum (1 × 106 cells/mL) and treated with lipopolysaccharide (0.1 μg/mL), Prolastin®, or phosphate buffered saline (0.1 mg/mL and 1 mg/mL) for 3 hours.
Notes: The release of interleukin-8 was determined in cell culture supernatants. **P < 0.01; ***P < 0.001.
Abbreviations: IL-8, interleukin-8; LPS, lipopolysaccharide.
Epithelial cells
To assess the effect of Prolastin on human bronchial epithelial cells, the levels of IL-8, MCP-1, and IL-6 were measured in cell culture supernatants after 4 hours and 24 hours of cell culture. Under the same experimental conditions, cells were exposed to LPS, TNFα, and IL-8 as the positive controls. As shown in Figure 6A–C, independently of the proinflammatory stimuli, levels of IL-6, IL-8, and MCP-1 in cell culture supernatants increased with time. Prolastin showed no significant effect on cytokine/chemokine release.
Figure 6.

The effects of Prolastin® on bronchial epithelial cells. Primary human bronchial epithelial cells isolated from protease inhibitor phenotype MM controls and cultured in air liquid interface culture systems were treated for 4 hours and 24 hours with lipopolysaccharide (0.1 μg/mL), interleukin-8 (0.1 μg/mL), tumor necrosis factor-α (0.1 μg/mL), and Prolastin. Stimulation was performed in triplicate. Levels of (A) interleukin-8, (B) interleukin-6, and (C) monocyte chemotactic protein-1 in cell culture supernatants.
Abbreviations: h, hours; IL-6, interleukin-6; IL-8, interleukin-8; LPS, lipopoly-saccharide; MCP-1, monocyte chemotactic protein-1; TNF, tumor necrosis factor.
Discussion
A major focus of augmentation therapy is to correct the AATD state by increasing serum AAT levels with intravenous infusions of human plasma-derived AAT. Although AAT augmentation therapy was introduced 30 years ago, clinical trials have produced conflicting results and to date there remains no widely accepted clinical evidence of the efficacy of therapy.23 Stockley et al have shown that augmentation therapy (60 mg/kg weekly for 4 weeks) favorably changes sputum inflammatory milieu, ie, reduces sputum elastase activity and levels of the leukotriene-B4, IL-8, and myeloperoxidase.24 Furthermore, Prolastin (2.5 mg per nostril) has been found to inhibit nasal IL-8 release in response to LPS challenge and in a concentration-dependent manner to inhibit LPS-induced TNFα and IL-1β release from monocytes and IL-8 release from neutrophils, in vitro.25 Thus, experience with AAT therapy suggests that it is safe, with few and usually well-tolerated side effects, and that it expresses multiply antiinflammatory effects. Nevertheless, still not much is known about the direct systemic and local effects of augmentation therapy on the immune-inflammatory markers. In the current study, cytokines (IL-1β, IL-6, and TNFα), chemokines (IL-8 and MCP-1), and VEGF were measured in patients undergoing weekly augmentation therapy just before intravenous AAT infusion, 1 day and 3 days after therapy. These markers were chosen because they are well studied and have been previously linked to the AATD-related emphysema.24,26
As expected, basal serum concentrations of AAT were significantly higher in AATD patients with augmentation therapy when compared to nonaugmented individuals. The augmentation therapy within 1–3 days restored serum levels of AAT within a physiological range in keeping with previous findings.6,24,27 All patients had normal serum CRP levels during augmentation therapy, which did not differ from nonaugmented AATD patients. Remarkably, the levels of CRP in EBC were much higher in the nonaugmented patients compared to augmented patients. This finding provides further support for the local antiinflammatory effect of augmentation therapy.11,24 It is of particular interest that CRP levels in EBC, but not in serum, increased directly after augmentation therapy and remained higher on day three. The reason for the observed temporary rise in EBC levels of CRP is not known. Moreover, it is not clear why these changes in CRP levels can only be detected in EBC, but not in serum. It is possible that, in general, much lower CRP levels in EBC than in serum is too variable to give a meaningful result. Previous work has shown that Prolastin contains a small fraction of a latent conformation.28 This finding provided an important demonstration that the commercial preparation process of AAT protein may induce conformational changes and alter both physicochemical and functional properties of the fraction of AAT protein with potential unforeseen physiological consequences. Characterization of the molecular forms of Prolastin was outside the scope of the current study but it was further confirmed that Prolastin preparations contain other forms of AAT protein. By using ELISA to detect Z as well as M polymers,19 it was found that serum levels of AAT polymers are significantly increased on the first day after augmentation therapy but decrease again at days three and seven. Moreover, there was a direct correlation between serum levels of total AAT and polymers of AAT. This finding implies that Prolastin preparations may contain some fraction of polymeric AAT. Polymers of Pi phenotype MM (PiMM) and PiZZ AAT have been shown to be proinflammatory and therefore it is possible that they induce the temporary increase in CRP levels in EBC.
Further analysis revealed that the augmentation therapy induces serum (but not EBC) fluctuations of IL-8 and MCP-1 in particular. For example, 7 days after augmentation therapy, serum levels of IL-8 were found to be significantly lower while levels of MCP-1 were higher, whereas TNFα, IL-6, and VEGF did not differ from nonaugmented patients. The reason behind these diverse effects of Prolastin on serum IL-8 and MCP-1 levels is not known but it is most likely linked either to the fast rise of AAT serum levels or to the Prolastin preparation per se. IL-8 is a well-known regulator of the migration of neutrophils and the reduction of serum levels of IL-8 has a beneficial effect.29,30 MCP-1 is an important mediator of monocyte recruitment into the vascular wall and therefore the enhancement of MCP-1 during augmentation therapy may have a beneficial effect. Indeed, short-term enhancement of MCP-1 has been shown to improve reperfusion in a hindlimb ischemia model.31 In support of this latter report, another study demonstrated that cardiac MCP-1 overexpression induces macrophage infiltration, neovascularization, and the accumulation of cardiac myofibroblasts, thereby resulting in the prevention of cardiac dysfunction and remodeling after myocardial infarction.32
AAT is a broad regulator of neutrophil functions such as chemotaxis, adhesion, and release of free radical and cytokines.36–38 In this way, AAT is an important controller of neutrophilic inflammation that plays a pivotal role in the pathogenesis of chronic obstructive pulmonary disease. Particularly in AATD-related chronic obstructive pulmonary disease, the increased neutrophil recruitment to the lung is thought to have a major role in the rapidly progressive lung destruction.33 Therefore, in the next set of experiments, isolated blood neutrophils from healthy PiMM donors and PiZZ patients were used. It is important to stress, that to mimic the in vivo environment as closely as possible, 10% serum (which also contains AAT) was introduced into the cell culture medium. Hence, in PiZZ neutrophils within the short term (3 hours), Prolastin inhibited IL-8 release. Remarkably, under the same experimental conditions, Prolastin added to PiMM neutrophils enhanced IL-8 release relative to nontreated controls, whereas Prolastin had no effect on IL-8 by itself after 6 hours but inhibited LPS-induced IL-8 and IL-6 release. These findings confirm that Prolastin exhibits time-dependent effects on IL-8 release that are also linked to the unique properties of PiMM and PiZZ neutrophils. Additionally, the current study revealed unique differences in the functional activities of neutrophils from patients with AATD-related emphysema compared with other non-AATD groups.34 Notably, IL-8 concentrations released from PiZZ patients’ neutrophils were 1000-fold higher than from PiMM neutrophils, which further supports the notion that neutrophils from AATD patients are activated.
In epithelial cells isolated from PiMM controls, Prolastin added for 3 hours did not change basal levels of IL-8, MCP-1, and IL-6. Thus, it can be speculated that the beneficial effects of Prolastin therapy might be primarily linked to the regulation of functional activities of innate immune cells such as neutrophils and monocytes. Experiments with monocytes were not performed in this study; however, existing data show that Prolastin reduces LPS-induced IL-8 production from monocytes.35
Conclusion
In summary, the results support the notion that weekly intravenous Prolastin therapy provides protective levels of circulating AAT protein and it is a well-tolerated and safe therapy. However, within 1 week of augmentation therapy, fluctuations of certain cytokines were observed, pointing out that potentially unacknowledged immunologic properties are carried out by weekly infusion of AAT. Furthermore, Prolastin preparations contain fractions of modified molecular forms of AAT generated during the manufacturing process that may potentially express unrevealed biological activities and/or lower therapeutic efficacy. Therefore, the quality of Prolastin and other commercial preparations of AAT and the application regimen are critically important factors for treatment efficacy.
Acknowledgments
The authors would like to thank Mrs Elssner, Mrs Patzke, Mrs Hoffmann, and Mrs Liebmann for performing the spirometries. The study was partially funded and supported by Grifols grant and SFB A18 (S Janciauskiene), SFB A19 (AR Koczulla), and Behring Rontgen grant 56-0036 (AR Koczulla).
Footnotes
Disclosure
S Schmid, A Hattesohl, J Koepke, E Frenzel, J Perez, E Miranda, H Teschler, and M Dresel have nothing to declare. S Janciauskiene received honoraria, research, and travel support from Grifols. DA Lomas has received honoraria and grant support, and acts as a consultant for GSK. He is a member of the Talecris (now Grifols)-funded European Alpha-1 Laurell Training Award panel. S Noeske has received speaker fees and travel support from Talecris (now Grifols). M Wencker is a former employee of Talecris. AR Koczulla has received speaker fees and travel support from Talecris (now Grifols). C Vogelmeier has received speaker fees and travel support from Talecris (now Grifols).
References
- 1.Seersholm N, Kok-Jensen A. Clinical features and prognosis of life time non-smokers with severe alpha 1-antitrypsin deficiency. Thorax. 1998;53(4):265–268. doi: 10.1136/thx.53.4.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Janciauskiene S, Eriksson S, Callea F, et al. Differential detection of PAS-positive inclusions formed by the Z, Siiyama, and Mmalton variants of alpha1-antitrypsin. Hepatology. 2004;40(5):1203–1210. doi: 10.1002/hep.20451. [DOI] [PubMed] [Google Scholar]
- 3.Janciauskiene SM, Bals R, Koczulla R, Vogelmeier C, Kohnlein T, Welte T. The discovery of ?1-antitrypsin and its role in health and disease. Respir Med. 2011;105(8):1129–1139. doi: 10.1016/j.rmed.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Mahadeva R, Atkinson C, Li Z, et al. Polymers of Z alpha1-antitrypsin co-localize with neutrophils in emphysematous alveoli and are chemotactic in vivo. Am J Pathol. 2005;166(2):377–386. doi: 10.1016/s0002-9440(10)62261-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parmar JS, Mahadeva R, Reed BJ, et al. Polymers of alpha(1)-antitrypsin are chemotactic for human neutrophils: a new paradigm for the pathogenesis of emphysema. Am J Respir Cell Mol Biol. 2002;26(6):723–730. doi: 10.1165/ajrcmb.26.6.4739. [DOI] [PubMed] [Google Scholar]
- 6.Hubbard RC, Sellers S, Czerski D, Stephens L, Crystal RG. Biochemical efficacy and safety of monthly augmentation therapy for alpha 1-antitrypsin deficiency. JAMA. 1988;260(9):1259–1264. [PubMed] [Google Scholar]
- 7.American Thoracic Society; European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168(7):818–900. doi: 10.1164/rccm.168.7.818. [DOI] [PubMed] [Google Scholar]
- 8.Silverman EK, Sandhaus RA. Clinical practice. Alpha1-antitrypsin deficiency. N Engl J Med. 2009;360(26):2749–2757. doi: 10.1056/NEJMcp0900449. [DOI] [PubMed] [Google Scholar]
- 9.Seersholm N, Wencker M, Banik N, et al. Does alpha1-antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary alpha1-antitrypsin deficiency? Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL) alpha1-AT study group. Eur Respir J. 1997;10(10):2260–2263. doi: 10.1183/09031936.97.10102260. [DOI] [PubMed] [Google Scholar]
- 10.Wencker M, Fuhrmann B, Banik N, Konietzko N. Longitudinal follow-up of patients with alpha(1)-protease inhibitor deficiency before and during therapy with IV alpha(1)-protease inhibitor. Chest. 2001;119(3):737–744. doi: 10.1378/chest.119.3.737. [DOI] [PubMed] [Google Scholar]
- 11.Lieberman J. Augmentation therapy reduces frequency of lung infections in antitrypsin deficiency: a new hypothesis with supporting data. Chest. 2000;118(5):1480–1485. doi: 10.1378/chest.118.5.1480. [DOI] [PubMed] [Google Scholar]
- 12.Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med. 1999;160(5 Pt 1):1468–1472. doi: 10.1164/ajrccm.160.5.9901055. [DOI] [PubMed] [Google Scholar]
- 13.Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J. 2009;33(6):1345–1353. doi: 10.1183/09031936.00159408. [DOI] [PubMed] [Google Scholar]
- 14.Kueppers F. The role of augmentation therapy in alpha-1 antitrypsin deficiency. Curr Med Res Opin. 2011;27(3):579–588. doi: 10.1185/03007995.2010.548750. [DOI] [PubMed] [Google Scholar]
- 15.Cazzola M, MacNee W, Martinez FJ, et al. Outcomes for COPD pharmacological trials: from lung function to biomarkers. Eur Respir J. 2008;31(2):416e69. doi: 10.1183/09031936.00099306. [DOI] [PubMed] [Google Scholar]
- 16.Horvath I, Hunt J, Barnes PJ, et al. Exhaled breath condensate: methodological recommendations and unresolved questions. Eur Respir J. 2005;26(3):523–548. doi: 10.1183/09031936.05.00029705. [DOI] [PubMed] [Google Scholar]
- 17.Koczulla AR, Noeske S, Herr C, et al. Ambient temperature impacts on pH of exhaled breath condensate. Respirology. 2010;15(1):155–159. doi: 10.1111/j.1440-1843.2009.01664.x. [DOI] [PubMed] [Google Scholar]
- 18.Sack U, Scheibe R, Wotzel M, et al. Multiplex analysis of cytokines in exhaled breath condensate. Cytometry A. 2006;69(3):169–172. doi: 10.1002/cyto.a.20231. [DOI] [PubMed] [Google Scholar]
- 19.Miranda E, Perez J, Ekeowa UI, et al. A novel monoclonal antibody to characterize pathogenic polymers in liver disease associated with alpha1-antitrypsin deficiency. Hepatology. 2010;52(3):1078–1088. doi: 10.1002/hep.23760. [DOI] [PubMed] [Google Scholar]
- 20.Bals R, Beisswenger C, Blouquit S, Chinet T. Isolation and air-liquid interface culture of human large airway and bronchiolar epithelial cells. J Cyst Fibros. 2004;3(Suppl 2):49–51. doi: 10.1016/j.jcf.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 21.Janciauskiene S, Zelvyte I, Jansson L, Stevens T. Divergent effects of alpha1-antitrypsin on neutrophil activation, in vitro. Biochem Biophys Res Commun. 2004;315(2):288–296. doi: 10.1016/j.bbrc.2004.01.055. [DOI] [PubMed] [Google Scholar]
- 22.Surhone LM, Timpledon MT, Marseken SF, editors. Normality Test: Data Set, Normal Distribution, Random Variable, Model Selection, Interpretations of Probability. Betascript Publishing; 2010. [Google Scholar]
- 23.Dickens JA, Lomas DA. Why has it been so difficult to prove the efficacy of alpha-1-antitrypsin replacement therapy? Insights from the study of disease pathogenesis. Drug Des Devel Ther. 2011;5:391–405. doi: 10.2147/DDDT.S14018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stockley RA, Bayley DL, Unsal I, Dowson LJ. The effect of augmentation therapy on bronchial inflammation in alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2002;165(11):1494–1498. doi: 10.1164/rccm.2109013. [DOI] [PubMed] [Google Scholar]
- 25.Kasahara Y, Tuder RM, Taraseviciene-Stewart L, et al. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest. 2000;106(11):1311–1319. doi: 10.1172/JCI10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nita I, Hollander C, Westin U, Janciauskiene SM. Prolastin, a pharmaceutical preparation of purified human alpha1-antitrypsin, blocks endotoxin-mediated cytokine release. Respir Res. 2005;6:12. doi: 10.1186/1465-9921-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gadek JE, Klein HG, Holland PV, Crystal RG. Replacement therapy of alpha 1-antitrypsin deficiency. Reversal of protease-antiprotease imbalance within the alveolar structures of PiZ subjects. J Clin Invest. 1981;68(5):1158–1165. doi: 10.1172/JCI110360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lomas DA, Elliott PR, Carrell RW. Commercial plasma alpha1-antitrypsin (Prolastin) contains a conformationally inactive, latent component. Eur Respir J. 1997;10(3):672–675. [PubMed] [Google Scholar]
- 29.Boring L, Gosling J, Chensue SW, et al. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest. 1997;100(10):2552–2561. doi: 10.1172/JCI119798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baggiolini M, Clark-Lewis I. Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett. 1992;307(1):97–101. doi: 10.1016/0014-5793(92)80909-z. [DOI] [PubMed] [Google Scholar]
- 31.Capoccia BJ, Gregory AD, Link DC. Recruitment of the inflammatory subset of monocytes to sites of ischemia induces angiogenesis in a monocyte chemoattractant protein-1-dependent fashion. J Leukoc Biol. 2008;84(3):760–768. doi: 10.1189/jlb.1107756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morimoto H, Hirose M, Takahashi M, et al. MCP-1 induces cardioprotection against ischaemia/reperfusion injury: role of reactive oxygen species. Cardiovasc Res. 2008;78(3):554–562. doi: 10.1093/cvr/cvn035. [DOI] [PubMed] [Google Scholar]
- 33.Rouhani F, Paone G, Smith NK, Krein P, Barnes P, Brantly ML. Lung neutrophil burden correlates with increased pro-inflammatory cytokines and decreased lung function in individuals with alpha(1)-antitrypsin deficiency. Chest. 2000;117(5 Suppl 1):250S–251S. doi: 10.1378/chest.117.5_suppl_1.250s. [DOI] [PubMed] [Google Scholar]
- 34.Boutten A, Dehoux MS, Seta N, et al. Compartmentalized IL-8 and elastase release within the human lung in unilateral pneumonia. Am J Respir Crit Care Med. 1996;153(1):336–342. doi: 10.1164/ajrccm.153.1.8542140. [DOI] [PubMed] [Google Scholar]
- 35.Janciauskiene S, Larsson S, Larsson P, Virtala R, Jansson L, Stevens T. Inhibition of lipopolysaccharide-mediated human monocyte activation, in vitro, by alpha1-antitrypsin. Biochem Biophys Res Commun. 2004;321(3):592–600. doi: 10.1016/j.bbrc.2004.06.123. [DOI] [PubMed] [Google Scholar]
- 36.Pott GB, Chan ED, Dinarello CA, Shapiro L. Alpha-1-antitrypsin is an endogenous inhibitor of proinflammatory cytokine production in whole blood. J Leukoc Biol. 2009;85(5):886–895. doi: 10.1189/jlb.0208145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bergin DA, Reeves EP, Meleady P, et al. ?-1 antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J Clin Invest. 2010;120(12):4236–4250. doi: 10.1172/JCI41196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sapey E, Stockley JA, Greenwood H, et al. Behavioral and structural differences in migrating peripheral neutrophils from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183(9):1176–1186. doi: 10.1164/rccm.201008-1285OC. [DOI] [PubMed] [Google Scholar]