

Abstract
Natural killer (NK) cells are specialized innate lymphocytes important in the early defense against tumor and virus bearing cells. Many factors influence the immune system’s effectiveness against pathogens, including stress. Social disruption (SDR) “primes” macrophages/monocytes and dendritic cells thereby enhancing their antimicrobial function. What remains unclear is whether similar responses are evident in NK cells. Current studies investigated the cellular distribution and activation/inhibitory phenotypes of NK cells in the spleen, lung, and blood of C57BL/6 male mice following SDR. Furthermore, cytolytic activity and anti-viral cytokine production of splenic NK cells were determined. Lastly, β-adrenergic receptor (β-AR) signaling was investigated to determine possible mechanisms behind the SDR-induced NK cell alterations. Results indicated NK cells from SDR mice have increased expression of CD16 and CD69 and reduced NKG2a and Ly49a expression on splenic CD3-/DX5+ NK cells indicative of an activated phenotype, both immediately and 14hrs post-SDR. Administration of propranolol (10mg/kg; non-selective β-adrenergic receptor antagonist) was shown to block these “priming” effects at the 14hr time-point. In the lung, SDR had similar effects on activation and inhibitory receptors 14hr post-SDR, however no alterations were evident in the blood besides increased NK cells directly after SDR. Additionally, splenic NK cells from SDR mice had increased CD107a surface expression, cytolytic activity, and IFN-γ production was increased upon costimulation with IgG and IL-2 ex vivo. Collectively, these data suggest that social stress “primes” NK cells in the spleen and lung to be more proficient in their cytolytic and antiviral/tumor effecter functions through β-adrenergic receptor dependent signaling.
Keywords: NK cells, Inhibitory, Activation, Degranulation, Social Disruption, Stress, Interferon Gamma
1. INTRODUCTION
There are many factors that affect innate immune cell functioning, stress being one of them. Our laboratory as well as others have shown repeated social disruption (SDR) stress causes biological responses similar to other stressors, such as hypothalamic-pituitary-adrenal (HPA) axis activation, increased corticosterone, and increased sympathetic nervous system (SNS) activity (Bailey et al., 2004; Hanke et al., under review; Wohleb et al., 2011). However, in addition to responses similar to other stressors, SDR results in a unique set of microenvironmental alterations that have a profound effect on peripheral immunity. For example, despite increased levels in circulating corticosterone which normally act to suppress immune cell function, SDR causes splenomegaly, functional immune cell glucocorticoid (GC) resistance, increased percentages in splenic CD11b+ myeloid cells and granulocytes, increased trafficking capabilities, and increased pro-inflammatory cytokine production (i.e., IL-6 and TNF-α) (Avitsur et al., 2001; Avitsur et al., 2005; Engler et al., 2004a; Powell et al., 2009; Stark et al., 2002; Wohleb et al., 2011). In addition to CD11b+ cells, CD11c+ dendritic cells are also susceptible to the microenvironmental changes induced by repeated social disruption. Specifically, SDR causes CD11c+ cells to acquire an activated phenotype by increasing the surface expression of CD80, MHC I, and CD44. Furthermore, CD11c+ dendritic cells produce a greater cytokine response (i.e., TNF-α, IL-6, and IL-10) upon TLR stimulation (Powell et al., 2009). Phenotypic alterations in CD11b+ and CD11c+ cells are attributed to the acquisition of functional GC insensitivity, thus reducing the normal suppressive effect that GCs have on immunity (Powell et al., 2009; Stark et al., 2001). Recently, additional mechanisms for SDR-induced immune “priming” have been revealed. For example, Wohleb et al. showed SDR-induced anxiety-like behavior, cFos expression in areas associated with threat appraisal, and inflammatory changes in CNS microglia/macrophages are ameliorated by the pre-administration of the non-selective β-adrenergic receptor (β-AR) antagonist propranolol (Wohleb et al., 2011). Additional studies from our lab show that SDR propranolol-treated mice show reduced IL-6 plasma levels, reduced splenocyte “priming”, and reduced GC insensitivity compared to animals that received SDR treatment alone (Hanke et al., under review).
Overall, SDR “primes” the immune system to respond to infectious insults in a more robust, timelier manner. A report by Bailey et al. used an intravenous E. coli infection model to show that socially disrupted animals effectively clear bacteria from the spleen and blood quicker than control animals that were not stressed (Bailey et al., 2007). Furthermore, splenic CD11b+ monocytes and macrophages cultured ex vivo from SDR mice had a greater propensity to kill E. coli compared to cells from control animals. In addition to increases in immunity to bacterial challenges, SDR enhances immunity in a viral mouse model of influenza A. Mice infected with influenza A/PR/8/34 after an adoptive transfer of dendritic cells from SDR mice showed an increased number of influenza specific CD8+ T-cells, increased IFN-γ and IFN-α mRNA expression, and decreased influenza M1 gene expression in lung tissue (Powell et al., 2010). In a primary HSV-1 infection model, SDR increased the percentage of CD11b+ macrophages in trigeminal (TG) nerve tissue. Furthermore, SDR increased gene expression of the anti-viral genes IFN-α and TNF-α and reduced viral-associated genes in the TG (Dong-Newsom et al., 2010). These data indicate that repeated social stress is capable of “priming” the immune system to enhance anti-microbial immunity.
Natural killer (NK) cells are bone marrow-derived lymphocytes that are important in the early immune response to viral infection and tumor suppression. During the initial stages of a viral infection NK cells mediate perforin and granzyme-dependent lysis, and secrete a number of anti-viral cytokines (e.g., TNF-α and IFN-γ) when coming in contact with infected cells or as a result of stimulation by cytokines (e.g., IL-2, IL-12, and IL-15) produced by other innate immune cells (Andoniou et al., 2005; Feau et al., 2005; Granucci et al., 2004; Henney et al., 1981; Krug et al., 2004; Liu et al., 1996; Orange et al., 1996). Additionally, NK cells can also be stimulated through CD16 (FcγRIII) ligation with IgG, determining the absence of MHC type I-like molecules, and/or CD69 activation, in turn mediating antibody-dependent cellular cytotoxicity (ADCC) and antiviral cytokine production (Trinchieri, 1989).
Within infected tissue, NK cell activation occurs upon differential ligation of activating and inhibitory NK cell receptors. Recent reports have suggested a role for inhibitory receptor (e.g., Ly49a and CD94/NKG2a) dysfunction on an NK cell’s ability to modulate the cytolytic activity toward virally infected cells. C-type lectin superfamily inhibitory receptors, including Ly49a and NKG2a, are able to recognize MHC type I-like receptors on the surface of cells much like their activation receptor counter parts. Inhibitory receptors are essential for the inhibition of the immunoreceptor tyrosine-based activating motif (ITAM) pathway, through the signaling of the immunoreceptor tyrosine-based inhibitory motif (ITIM) pathway, ultimately keeping NK cytolytic activity in close regulation. In addition to activating and inhibitory receptors, NK cells express high levels of β-ARs. Following catecholamine receptor ligation, NK cell cytolytic functionality becomes altered, although it remains under debate if it becomes enhanced or suppressed in animals. Changes in catecholamine-induced NK cell cytotoxicity is dependent on a variety of variables such as tissue type (e.g., blood, spleen, liver), species (e.g., humans vs. rodents), experimental stressor (e.g., social stressor, restraint, shock), etc. (Sanders et al., 2002).
Knowing SDR enhances innate immunity to become more effective, the objective of the current experiments was to test whether SDR causes similar changes in NK cells in the absence of infection. To date, NK cell phenotype and functionality has not been examined in the SDR model. Additionally, due to the importance of SNS signaling in the SDR-induced “priming” of innate immune cells, potential β-AR dependent signaling mechanisms behind SDR-induced NK cell phenotype and functional changes were examined. Alterations in SDR-induced “priming” of NK cells, through SNS stimulatory pathways, may prove to be an important and effective mediator in viral/tumor challenges and could possibly lead to advances in numerous novel anti-viral/tumor therapies.
2. METHODS
2.1. Subjects
Subjects were 6 to 8 week-old male C57BL/6 mice purchased from Charles River Laboratories (Wilmington, MA). Animals were allowed to acclimate in the animal facility for ~1 week prior to experimental procedures. Mice were group housed 3/cage in standard polycarbonate mouse cages (aggressors were singly housed in a separate room). Animal housing rooms were maintained on a 12 hr light/dark cycle with lights being turned on at 0600 in an AAALAC (American Association of Accreditation of Laboratory Animal Care) facility. Food and water was available ad libitum. Animals were treated in compliance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996) and experiments were conducted in accordance with a protocol approved by the Institutional Laboratory Animal Care and Use Committee (ILACUC) at The Ohio State University.
2.2. Drug Treatment: Propranolol a Non-selective β-adrenergic Receptor Antagonist
Experiments conducted to examine the mechanisms behind SDR-induced NK cell alterations employed the use of the non-selective β-adrenergic receptor antagonist propranolol. One hour prior to each of the 6 cycles of SDR, both HCC and SDR mice were injected with either 10 mg/kg of propranolol (Sigma-Aldrich; St. Louis, MO) or vehicle (0.02% ETOH in PBS). Fourteen hours after the last cycle of propranolol/SDR treatment, animals were humanely euthanized and tissue was analyzed accordingly to address each studies specific experimental hypotheses.
2.3. Social Disruption Stress
The SDR paradigm has been previously described to a greater extent in previous publications from our laboratory (Avitsur et al., 2001; Bailey et al., 2007). Briefly, experimental mice underwent 6 consecutive cycles of SDR over 6 days where aggressive intruders were placed into established cohorts of 3 naïve mice between the hours of 1700 and 1900. Intruder mice were outbred CD-1 retired breeders that were individually housed upon arrival to the animal facility and had previously shown aggressive behavior prior to experimentation. If the aggressor mouse did not attack and defeat the resident mice, or if the resident mice attacked the intruder within the first 5 min of the SDR cycle, the aggressor was removed and another aggressor was introduced into the cage. During SDR, resident mice were observed for submissive behaviors such as upright posture that exposed the under flank, fleeing, and cowering in the corners of the cage (Avitsur et al., 2001; Bailey et al., 2007). At the end of each SDR cycle, intruder mice were removed and placed into their home cage until the next SDR cycle began. Of note, no intruder mice were used more than once per cage over the course of SDR. Home cage control (HCC) animals were housed in a separate room isolated from stressor cues (i.e., visual, olfactory, and auditory) throughout SDR until tissue harvesting occurred. Immediately following or 14hrs after the final cycle of SDR (depending on the experimental design), socially disrupted and control animals were humanely euthanized by CO2 asphyxiation and tissues were harvested for further analyses. During and post-SDR, animals were monitored for excessive cutaneous wounding and/or signs of infection. During SDR experimentation, no apparent infection was observed.
2.4. Tissue Processing and Cell Isolation
After euthanization by CO2 asphyxiation, spleens and lungs from experimental and control mice were excised aseptically. In addition, blood was collected by cardiac puncture and placed in heparin lined tubes. Spleens were put directly in 5 mls of Hanks’ balanced salt solution (HBSS) while whole lungs were placed in 5 mls of HBSS containing 1.58 mg/ml of type-I collagenase (Worthington, Lakewood, NJ). Tissues were then mechanically disrupted using a Model 80 Biomaster Stomacher (Seward, Riverview, FL) to obtain single cell suspensions. Red blood cells from spleens, lungs, and whole blood were lysed in 1 ml/sample of room temperature RBC lysis buffer (0.16 M NH4Cl, 10 mM KHCO3, and 0.13 mM EDTA) for 2 min followed by the addition of HBSS+10% heat-inactivated fetal bovine serum (FBS) to stop active lysis. Cell pellets were then washed, filtered through a 70 μm nylon cell strainer, and resuspended in HBSS. Final cell counts were obtained using a Beckman Z2 Coulter Counter (Corixa, Seattle, WA). All solutions and tissue were kept on ice for the duration of extraction and processing.
2.5. Flow Cytometry
Single cell suspensions of spleen, blood, and lung tissue (5.0 × 105 cells/sample) were obtained from HCC and SDR-treated animals and then incubated with Fc-receptor block (eBioscience, San Diego, CA; except for panels examining CD16/CD32) for 10 min. Samples were then incubated with 1 μl of fluorescently-labeled monocolonal antibodies or their appropriate isotype control for 45 min at 4 °C in a dark refrigerator, washed, then resuspended in FACS buffer (2% FBS in HBSS and 1 μg/ml sodium azide) for flow analyses. All antibodies were obtained from BD Bioscience (San Diego, CA) or eBioscience (San Diego, CA) including, anti-CD3e, anti-DX5, anti-CD16/CD32, anti-CD69, anti-Ly49a, anti-NGK2a, and anti-CD107a conjugated to the appropriate flurochrome (i.e., FITC, PE, PerCP, or APC). Nonspecific binding was gated on and eliminated by using nonspecific isotype-matched control antibodies. Lymphocyte gates were based on forward versus side scatter, and a total of 10,000–25,000 events were analyzed depending on the NK cell density of the tissue. Data was obtained using a four-color FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) and subsequently analyzed using FlowJo software (Tree Star).
2.6. Natural Killer Cell Isolation and Ex Vivo Cell Culture
Spleens were excised from both HCC and SDR-treated animals, processed, and isolated to obtain single cell suspensions (detailed description in section 2.4.). Purified splenic natural killer cell populations were further isolated for DX5 (CD49b) positivity using MACS magnetic bead separation technology (Miltenyi Biotec, Cambridge, MA) per manufacturer’s instructions.
For ex vivo stimulation experiments examining IFN-γ production from SDR-treated animals following IL-2 and/or IgG stimulation, 96-well flat-bottom tissue culture plates were pre-coated with a suspension of mIgG (100μg/mL in ice cold PBS) overnight at 4 °C. The following day, plates were washed with PBS and then isolated splenic DX5+ cells from HCC and SDR-treated animals were plated at 4.0×105 cells/well in culture media (RPMI-1640 w/o L-glutamine, 100X antibiotic/antimycotic solution, and 10% heat-inactivated FBS) containing either 150 ng/mL of IL-2 or media (served as control). Forty-eight hours post-plating, cell-free supernatants were collected and analyzed for murine IFN-γ production by enzyme-linked immunosorbent assay (ELISA; R&D Systems, Minneapolis, MN).
2.7. 51Cr Cytotoxic Assay
Spleens were harvested and single cell suspensions were obtained from SDR (control and stressed) and propranolol-treated (vehicle and 10 mg/kg) mice (detailed description in section 2.4.). Following isolation, splenocytes were resuspended at 1.0×107 cells/ml and stimulated with 10 ng/ml of human IL-15 (Peprotech; Rocky Hill, NJ) overnight at 37 ºC and 5% CO2. The following day cells were subjected to a 51Cr release assay to determine NK cytotoxicity similar to methods previously described by (Tseng et al., 2005). Briefly, 200 μl (i.e., 2.0×105) of the IL-15 stimulated splenocytes were added to 96-well V-bottom tissue culture plates, then serially diluted by two-fold increments. Radioactively labeled YAC-1 cells (1.0×104 cells/well) were then added to the serially diluted splenocytes in order to obtain effector-to-target (E:T) ratios of 100:1, 50:1, 25:1, and 12.5:1. To obtain maximum cytolytic and spontaneous release values, YAC-1 cells were added to wells containing 10% SDS and RPMI-1640 cell culture media, respectively. Following a 4 hr incubation period at 37 ºC and 5% CO2, 100 μl of cell-free supernatant was collected from each well and 51Cr release was measured using a Wizard2 2470 automatic gamma counter (Perkin Elmer; Waltham, MA). Percent cytolysis was calculated using the following formula:
2.8. Statistical Procedures
Data obtained from splenic weights, cellularity, flow cytometric procedures, cytokine ELISA’s, and individual cytolytic E:T ratios were analyzed using standard one-way ANOVAs, with stress, propranolol treatment, and/or antibody/cytokine stimulation when appropriate as the between-subjects variable. Data collected from the 51Cr release assay were analyzed using a mixed factorial design with propranolol and SDR-treatment as the between-subject variables and E:T ratios as the within-subject variable. Significant interactions among dependent variables were subjected to Fisher’s PLSD post hoc analyses. An alpha level of p≤0.05 was the criterion for rejection of the null hypothesis. All data were analyzed using Statview statistical software (SAS). Results are reported as treatment means ± SEM. Inconsistencies in the number of samples analyzed were due to data being compiled from multiple experiments examining several different hypotheses (i.e., marker and tissue type) and not due to experimenter exclusion.
3. RESULTS
3.1. Splenic Weight and Cellularity Following Repeated Social Disruption
To confirm prior reports that SDR increases both splenic weight and total cell number, spleens were excised from both HCC and SDR-treated animals 14hrs after the 6th cycle of repeated social disruption. As expected, results were consistent with previous findings showing that there was a significant main effect of Stress where SDR-treated animals showed an increased splenic weight compared to control animals (F(1,13)=10.34; p<0.01; see Fig. 1A). Furthermore, there was a significant main effect of Stress on splenic cellularity indicating that repeated social defeat caused an increase in the total number of splenic cells compared to control animals (F(1,21)=8.62; p<0.01; see Fig. 1B).
Fig. 1.

Mean splenic weight (A) and mean total spleen number (B) 14hrs after six consecutive cycles of social disruption (SDR) compared to home cage control (HCC) animals. * Indicates a significant difference from HCC animals (p’s≤0.01). Bars represent group means ± SEM.
3.2. Distribution of Natural Killer Cells in the Spleen, Lung, and Blood 14hrs Following Repeated Social Disruption
Repeated social defeat leads to changes in the proportions of innate immune cells found in both lymphoid and non-lymphoid tissue 14hrs following the last cycle of SDR (Curry et al., 2010; Engler et al., 2004a). Therefore, initial experiments sought to determine if repeated social disruption causes similar alterations in the cellular distribution of natural killer cells in the spleens, lungs, and blood of HCC and SDR-treated animals at the corresponding time-point. Results showed that repeated social disruption significantly decreased the percentage of splenic NK cells, and increased the percentage of NK cells in the lung when compared to control animals. These data were reflected by significant main effects of Stress on the percentage of CD3-/DX5+ (i.e., NK cells) in the spleen (F(1,11)=4.78; p<0.05; see Fig. 2C), and lung (F(1,11)=13.59; p<0.005; see Fig. 2F). Despite altered CD3-/DX5+ NK cell percentages in the spleen and lung resulting from repeated social disruption, there were no significant changes found in the blood at the 14hr assessment time-point (F(1,9)=0.34; ns; data not shown).
Fig. 2.

Representative flow cytometric dot plots for CD3-/DX5+ natural killer cells in the spleen (A and B) and lung (D and E). Average change in the percentage of CD3-/DX5+ natural killer cells in the spleen (C) and lung (F), 14hrs following 6 consecutive cycles of SDR compared to HCC animals. * Indicates a significant difference from HCC animals (p’s≤0.05). Bars represent group means ± SEM.
3.3. Alteration in Activation Receptors in the Spleen, Lung, and Blood 14 hrs Following Repeated Social Disruption
Prior reports have shown repeated social disruption increases activation markers on both peripheral and central innate immune cells 14hrs after the final cycle of SDR (e.g., macrophages/monocytes, microglia, and dendritic cells) (Bailey et al., 2007; Dong-Newsom et al., 2010; Powell et al., 2009; Wohleb et al., 2011). Therefore, to confirm SDR had a similar effect on NK cells in the periphery, blood, lung, and splenic tissue were harvested and analyzed by flow cytometry for the activation markers CD16 and CD69 on CD3-/DX5+ NK cells 14 hrs post-SDR. Results showed SDR-treatment significantly increased both the percentage of the activation markers CD16 and CD69 on splenic CD3-/DX5+ NK cells. These results were reflected in significant main effects of Stress on CD16 (F(1,21)=4.69; p<0.05; see Fig. 3A) and CD69 expression (F(1,21)=14.73; p<0.001; see Fig. 3B). Furthermore, SDR-treatment had a differential effect on activation markers in the lung by significantly increasing CD16 and significantly decreasing CD69 on CD3-/DX5+ NK cells. These findings were revealed by a significant main effect of Stress on CD16 (F(1,11)=11.75; p<0.01; see Fig. 3C) and CD69 expression (F(1,11)=6.08; p<0.05; see Fig. 3D). As for activation marker expression in the blood, SDR-treatment did not significantly alter the expression of either CD16 (F(1,9)=6.08; ns; see Fig. 3E) or CD69 (F(1,9)=0.01; ns; see Fig. 3F) on CD3-/DX5+ NK cells.
Fig. 3.

Mean percentage of CD3-/DX5+ natural killer cells expressing the activation marker CD16 in the spleen (A), lung (C), and blood (E), 14hrs following 6 consecutive cycles of SDR compared to HCC animals. Mean percentage of CD3-/DX5+ natural killer cells expressing the activation marker CD69 in the spleen (B), lung (D), and blood (F), 14hrs following 6 consecutive cycles of SDR compared to HCC animals.* Indicates a significant difference from HCC animals (p’s≤0.05). Bars represent group means ± SEM.
3.4. Alteration in Inhibitory Receptors in the Spleen, Lung, and Blood 14hrs Following Repeated Social Disruption
It is well known that NK inhibitory receptors have a potent regulatory function on the activation of NK cells (Houchins et al., 1997; Karlhofer et al., 1992). To determine possible mechanisms of the reported increase NK cell activation status after SDR, expression of the common inhibitory receptors NKG2a and Ly49a were examined 14hrs following the last cycle of SDR as it was for activation receptors. Once again blood, lung, and spleens were collected from HCC and SDR-treated animals and analyzed for the expression of inhibitory receptors on CD3-/DX5+ NK cells. Results indicated both NKG2a and Ly49a were decreased in the spleens of SDR-treated animals compared to controls. This was reflected in significant main effects of Stress on NKG2a (F(1,19)=5.16; p<0.05; see Fig. 4A) and Ly49a (F(1,19)=4.32; p<0.05; see Fig. 4B) expression. Furthermore, lung Ly49a expression on NK cells was significantly reduced but NKG2a was not, even though there was a trend for a reduction. These results were evident by the main effects of Stress on Ly49a (F(1,11)=7.02; p<0.05; see Fig. 4D) and NKG2a expression (F(1,11)=3.77; p=0.08; see Fig. 4C). Similar to activation receptors, SDR-treatment did not change the expression of the inhibitory receptors NKG2a (F(1,9)=0.08; p=0.79; see Fig. 4E) or Ly49a (F(1,9)=0.07; p=0.80; see Fig. 4F) in the blood.
Fig. 4.

Mean percentage of CD3-/DX5+ natural killer cells expressing the inhibitory receptor NKG2a in the spleen (A), lung (C), and blood (E), 14hrs following 6 consecutive cycles of SDR compared to HCC animals. Mean percentage of CD3-/DX5+ natural killer cells expressing the inhibitory receptor Ly49a in the spleen (B), lung (D), and blood (F), 14hrs following 6 consecutive cycles of SDR compared to HCC animals.* Indicates a significant difference from HCC animals (p’s≤0.05). Bars represent group means ± SEM.
3.5. Splenic Alterations in the Degranulation Marker CD107a 14hrs Following Repeated Social Disruption
Knowing that SDR increases activation and reduces inhibitory markers 14 hr following SDR, we sought to find probable functional consequences in cytolytic potential of these results. Lysosomal-associated membrane protein-1 (LAMP-1), also known as CD107a, is a relativity new marker to assess functionality of activated NK cells that have the capability to degranulate in the absence of cytokine secretion. Furthermore, CD107a has been shown to be highly correlative to cytolytic activity in the 51Cr release assay (Alter et al., 2004). In the current experiments, splenic cells were removed and isolated 14hrs following the last cycle of SDR and stained with CD3, DX5, CD16, and CD107a. Results indicated SDR-treated animals increased the expression of CD107a on CD3-/DX5+ NK cells compared to control animals. These results were reflected in a main effect of Stress on CD3-/DX5+/CD107a+ splenic cells (F(1,11)=8.0; p<0.05; see Fig. 5A).
Fig. 5.

Mean percentage of splenic CD3-/DX5+ cells expressing the degranulation marker CD107a (A), 14hrs following 6 consecutive cycles of SDR compared to HCC animals. Mean percentage of activated splenic CD3-/DX5+/CD16+ cells expressing the degranulation marker CD107a (B), 14hrs following 6 consecutive cycles of SDR compared to HCC animals. * Indicates a significant difference from HCC animals (p’s≤0.05). Bars represent group means ± SEM.
Since SDR-treatment increases both the activation status (i.e., CD16 and CD69) and the overall expression of CD107a on splenic NK cells, we evaluated the potential for degranulation (i.e., CD107a) on activated splenic NK cells (i.e., CD3-/DX5+/CD16+). As expected, SDR increased the cytolytic potential of activated splenic NK cells compared to control animals. These data were reflected in a significant main effect of Stress on splenic CD3-/DX5+/CD16+/CD107a+ cells (F(1,11)=6.79; p<0.05; see Fig. 5B).
3.6. Ex Vivo Analyses of Interferon Gamma Production by Isolated DX5+ Splenic Cells Stimulated with IL-2 and/or Immobile IgG
Natural killer cells are not only capable of exerting their cytolytic function through the release of granules (e.g., perforin and granzyme), but are able to release cytokines such as interferon gamma (IFN-γ) to mount and orchestrate anti-viral defenses. Therefore, we sought to determine if additional NK cellular defense mechanisms, other than the reported increases in the degranulation marker CD107a were altered by social disruption. Results indicated that SDR-treatment did not have a significant effect on the total number of isolated splenic DX5+ cells (F(1,10)=1.01; p=0.34; see Fig. 6A). However, when isolated splenic DX5+ cells were costimulated ex vivo with IL-2 (potent cytokine stimulator of IFN-γ production) and immobilized IgG (ligand for CD16 [FcγRIII]) IFN-γ production was significantly increased. As expected, isolated splenic DX5+ NK cells from SDR-treated animals upon costimulation with IL-2 and immobilized IgG produced the greatest increase in IFN-γ production compared to cells treated with IL-2, IgG, or media alone. These data were reflected by significant main effects for Stress (F(1,20)=6.34; p<0.05; see Fig. 6B), IL-2/IgG stimulation (F(1,20)=8.09; p<0.01; see Fig. 6B), and a significant Stress X IL-2/IgG stimulation interaction (F(1,20)=6.37; p<0.05; see Fig. 6B). Fisher’s PLSD post hoc analyses revealed after ex vivo costimulation of isolated splenic DX5+ NK cells with IL-2 and IgG, both HCC (p<0.01) and SDR-treated (p<0.05) animal NK cells had increased IFN-γ production compared to cells treated with IL-2, IgG, or media alone. Furthermore, when comparing isolated splenic DX5+ NK cells from SDR-treated and control animals that received both IL-2 and immobilized IgG, NK cells from socially defeated animals had a significantly higher IFN-γ production (p<0.05). For all post hoc analyses see Fig. 6B.
Fig. 6.

Mean total isolated DX5+ splenic cell numbers (A), 14hrs following 6 consecutive cycles of SDR compared to HCC animals. Fourteen hours post-SDR, splenic DX5+ cells were isolated, incubated for 48hrs with media, IgG, IL-2, or a combination of IgG/IL-2. Mean IFN-γ concentration was determined by ELISA (B). * Indicates a significant difference from DX5+ NK cells from HCC animals treated with IL-2/IgG and all other control groups (p’s≤0.05). + Indicates a significant difference from DX5+ NK cells from SDR-treated animals treated with IL-2/IgG and all other control groups (p’s≤0.05). Bars represent group means ± SEM.
3.7. Alterations in Activation, Inhibitory, and Degranulation Markers Immediately Following Repeated Social Disruption
Acute stressors are known to alter distribution of NK cells by several different mechanisms. Although the exact mechanism are still under debate, acute stress has been consistently shown to cause a significant increase in the number of circulating NK cells (Engler et al., 2004b; Schedlowski et al., 1993b). Additionally, it has been postulated that acute stress has differential effects on the cytolytic function of NK cells compared to chronic stress and depends on a wide variety of endogenous correlates at the time of assessment (e.g., GCs and catecholamines) (Benschop et al., 1996; Irwin et al., 1990; Sanders et al., 2002; Schedlowski et al., 1993a). Since SDR may be viewed as a stressor that has both acute and chronic stress properties, we wanted to determine if NK cellular distribution, activation and inhibitory status, and degranulation markers were transient in nature or comparable to levels seen at the 14hr post-SDR time-point. In the current set of experiments spleens and blood were collected from HCC and SDR-treated immediately following the last cycle of SDR (i.e., 6th cycle) and assessed for NK cell number and activation status by flow cytometric procedures. Data from flow analyses collected from splenocytes revealed that there were no alterations in the percentage of splenic CD3-/DX5+ NK cells when assessed directly after the last cycle of SDR (F(1,9)=0.083; ns; see Fig. 7A). However, as hypothesized there was a significant increase in the percentage of splenic CD3-/DX5+ NK cells that expressed the activation markers CD16 and CD69 directly after SDR-treatment compared to control animals. These results were evident by a main effect of Stress on splenic CD3-/DX5+/CD16+ (F(1,9)=20.46; p<0.001; see Fig. 7B) and CD3-/DX5+/CD69+ (F(1,9)=40.20; p<0.0001; see Fig. 7C) NK cells. Additionally, SDR-treatment significantly decreased expression of the inhibitory receptors NKG2a and Ly49a on splenic CD3-/DX5+ NK cells. These results were reflected by significant main effects for Stress on CD3-/DX5+/NKG2a+ (F(1,9)=7.17; p<0.05; see Fig. 7D) and CD3-/DX5+/Ly49a+ (F(1,9)=9.25; p<0.01; see Fig. 7E) NK cells. Further examination showed that SDR-treatment significantly increased the percentage of splenic NK cells expressing the degranulation marker CD107a immediately following the last cycle of SDR. These results were evident by a significant main effect of Stress on splenic CD3-/DX5+/CD107a+ NK cells (F(1,9)=5.99; p<0.05; see Fig. 7F).
Fig. 7.

Mean percentage of CD3-/DX5+ natural killer cells (A) expressing the activation receptors CD16 (B) and CD69 (C) in the spleen immediately following the last cycle of SDR. Mean percentage of CD3-/DX5+ natural killer cells expressing the inhibitory receptors NKG2a (D) and Ly49a (E) in the spleen immediately following the last cycle of SDR. Mean percentage of CD3-/DX5+ natural killer cells expressing the degranulation marker CD107a (F) in the spleen immediately following the last cycle of SDR. * Indicates a significant difference from HCC animals (p’s≤0.05). Bars represent group means ± SEM.
When examining flow cytometric data collected from blood samples immediately following SDR, data showed a similar pattern to data collected 14hrs post-SDR. Specifically, examination of activation and inhibitory receptors and degranulation markers showed no significant alterations between SDR-treated animals and controls. However, as hypothesized and consistent with prior reports that show increased number of NK cells following acute stressors, SDR-treatment increased the percentage of blood derived CD3-/DX5+ NK cells (F(1,10)=6.42; p<0.05; data not shown).
3.8. Effects of β-adrenergic Receptor Antagonism on Activation, Inhibitory, and Degranulation Markers 14hrs Following Repeated Social Disruption
SDR causes increased anxiety-like behavior and increased inflammatory phenotypes on immune cells (Bailey et al., 2007; Kinsey et al., 2007; Powell et al., 2009; Wohleb et al., 2011). Our lab has also shown a portion of these SDR-induced alterations are ameliorated by administrating the β-adrenergic receptor antagonist propranolol (Wohleb et al., 2011). Therefore, to examine possible mechanisms for the reported SDR-induced “priming” of NK cells, spleens were harvested from HCC and SDR-treated animals that received either vehicle or 10 mg/kg of propranolol 1 hr prior to 6 consecutive cycles. Flow cytometric data indicated the percentage of splenic CD3-/DX5+ NK cells were unaltered in the SDR-treated animals compared to controls, even though there was a trend for an increase and propranolol treatment ameliorated the effect. These data were reflected by a non-significant main effect of Stress (F(1,26)=2.24; ns; see Fig. 8A) but a significant Stress X Propranolol interaction (F(1,26)=5.33; p<0.05; see Fig. 8A). Fisher’s PLSD post hoc analyses revealed there were no significant differences among treatment groups, however there were trends for an increase in the percentage of CD3-/DX5+ NK cells in SDR-treated animals compared to controls that received vehicle injections (p=0.09) and a decrease in the percentage of CD3-/DX5+ NK cells from propranolol-treated SDR animals compared to SDR-treated animals receiving vehicle injections (p=0.10).
Fig. 8.

Mean percentage of CD3-/DX5+ natural killer cells (A) expressing the activation receptors CD16 (B) and CD69 (C) in the spleen 14hrs following SDR and/or propranolol administration. Mean percentage of CD3-/DX5+ natural killer cells expressing the inhibitory receptors NKG2a (D) and Ly49a (E) in the spleen 14hrs following SDR and/or propranolol administration. Mean percentage of CD3-/DX5+ natural killer cells expressing the degranulation marker CD107a (F) in the spleen 14hrs following SDR and/or propranolol administration. Different letters represents a statistical significance among groups (p’s≤0.05). Bars represent group means ± SEM.
When examining the effects of propranolol pretreatment on splenic NK cell activation receptors, data showed that once again SDR significantly increased CD16 and trended to increase CD69 expression. Furthermore, this increase was able to be blocked with the β-adrenergic receptor antagonist propranolol. These data were revealed by a significant main effect of Stress on the percentage of CD3-/DX5+/CD16+ splenic NK cells (F(1,26)=6.74; p<0.05; see Fig. 8B), in addition to a trending main effect of Stress on the percentage of CD3-/DX5+/CD69+ splenic NK cells (F(1,26)=3.37; p=0.08; see Fig. 8C). Furthermore, data revealed significant Stress X Propranolol treatment interactions for both CD3-/DX5+/CD16+ and CD3-/DX5+/CD69+ splenic NK cells (F(1,26)=7.51; F(1,26)=4.94; p’s<0.05; see Figs. 8B&C, respectively). Fisher’s PLSD post hoc analyses revealed a significant increase in the number of CD3-/DX5+/CD16+ and CD3-/DX5+/CD69+ splenic NK cells in SDR-treated animals compared to controls that received vehicle injections (p<0.005 and p<0.05, respectively). In addition, SDR-treated animals that received propranolol injections had decreased percentages of CD3-/DX5+/CD16+ and CD3-/DX5+/CD69+ splenic NK cells compared to animals receiving the vehicle (p’s<0.05).
Data obtained from the examination of propranolol treatment on inhibitory receptor expression confirmed prior experiments that SDR-treatment significantly decreased NKG2a and Ly49a receptor expression on splenic NK cells. Furthermore, as it was for splenic NK cell activation receptors, this effect was able to be blocked with the administration of propranolol. These data were evident by significant main effects of Stress on NKG2a (F(1,26)=4.84; p<0.05; see Fig. 8D) and Ly49a (F(1,26)=9.30; p<0.01; see Fig. 8E) expression on splenic CD3-/DX5+ NK cells. Additionally, data revealed significant Stress X Propranolol treatment interactions for both CD3-/DX5+/NKG2a+ and CD3-/DX5+/Ly49a+ splenic NK cells (F(1,26)=6.46; F(1,26)=6.98; p’s<0.05; see Figs. 8D&E, respectively). Fisher’s PLSD post hoc analyses revealed significant decreased percentages of CD3-/DX5+/NKG2a+ and CD3-/DX5+/Ly49a+ splenic NK cells in SDR-treated animals compared to controls that received vehicle injections (p’s<0.01). In addition, SDR-treated animals that received propranolol injections had decreased percentages of CD3-/DX5+/NKG2a+ and CD3-/DX5+/Ly49a+ splenic NK cells compared to SDR-treated animals receiving the vehicle (p’s<0.05).
Lastly, flow cytometric analyses confirmed prior results that showed that SDR-treatment significantly increased the degranulation marker CD107a on splenic derived CD3-/DX5+ NK cells. Furthermore, β-adrenergic receptor antagonism by propranolol ameliorated this SDR-induced effect. Statistical analysis of flow cytometric data revealed a significant main effect of Stress (F(1,26)=9.38; p<0.01; see Fig. 8F), as well as a significant Stress X Propranolol treatment interaction (F(1,26)=7.42; p<0.05; see Fig. 8F) for the percentage of CD3-/DX5+/CD107a+ splenic NK cells. Fisher’s PLSD post hoc analyses revealed a significant increased percentage of CD3-/DX5+/CD107a+ splenic NK cells in SDR-treated animals compared to controls that received vehicle injections (p<0.01). Moreover, SDR-treated animals that received propranolol injections had a decreased percentage of CD3-/DX5+/CD107a+ splenic NK cells compared to SDR-treated animals receiving the vehicle (p<0.05).
3.9. Alterations in Splenocyte Cytotoxicity Following Repeated Social Disruption and β-adrenergic Receptor Antagonism Measured by 51Cr Release Assay
Although we have shown SDR increases CD107a (a surrogate marker for cytotoxic activity) and propranolol treatment blocks this SDR-induced increase, a 51Cr release assay was employed to determine additional functional NK cell alterations induced by SDR and β-adrenergic receptor antagonism. Overall, data showed SDR enhanced cytolytic activity against target YAC-1 cells devoid of MHC I. Furthermore, animals that were administered propranolol had further increased cytolytic activity irrespective of repeated social disruption exposure, confirming prior reports that show catecholamines are suppressive in nature to NK cytolytic function. These results were evident by a main effect of Stress (F(1,16)=8.16; p<0.01; see Fig. 9) and Propranolol treatment (F(1,16)=4.47; p<0.05; see Fig. 9) on cytolytic activity. However, there were no interaction effects between repeated social disruption and propranolol treatment. Furthermore, closer examination showed SDR-treated animals had increased cytolytic activity compared to control animals at the 50:1 and 25:1 E:T ratios, irrespective of propranolol treatment (p’s<0.05; see Fig. 9). No other individual groups were statistically significant from one another. Of note, the E:T ratio 100:1 was excluded from analyses due to inconsistent results across multiple experiments.
Fig. 9.
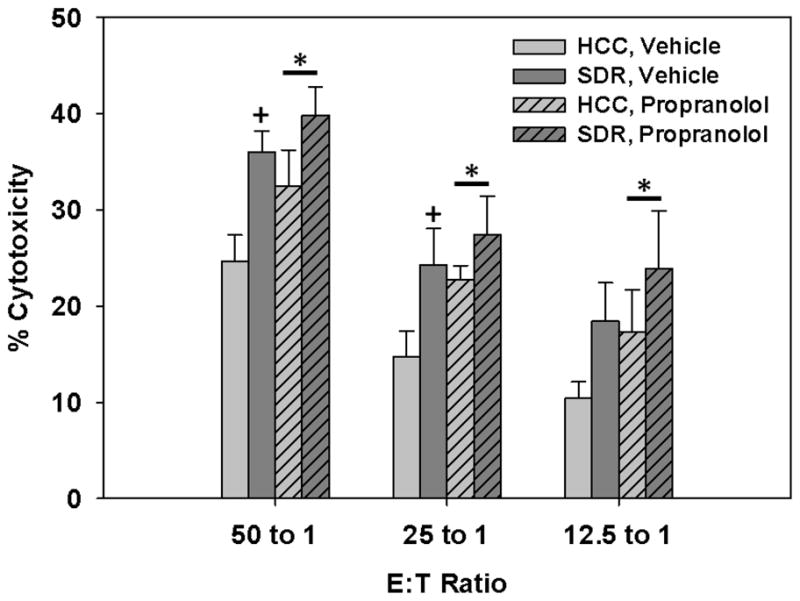
Mean percent cytotoxicity displayed from IL-15 stimulated splenocytes taken from SDR-treated and control animals that received 10mg/kg propranolol or vehicle injections 1hr prior to each cycle of social disruption. * Indicates a main effect of Propranolol treatment regardless of stress condition within each E:T ratio (p’s≤0.05). + Indicates a main effect of Stress within each E:T ratio when comparing HCC to SDR-treated mice receiving vehicle treatment. (p’s≤0.05). Bars represent group means ± SEM.
4. Discussion
As a result of altered local tissue microenvironments through HPA-axis and SNS mediated mechanisms and GC insensitivity, splenic derived CD11b+ monocytes, macrophages, and CD11c+ dendritic cells acquire a “primed” activation state and have enhanced anti-microbial activity (Bailey et al., 2007; Dong-Newsom et al., 2010; Powell et al., 2010). However, as to whether SDR causes alterations in additional innate cell types that are important in resistance to pathogenic challenge has yet to be determined. Additionally, β-AR signaling has been shown to be an important mediator in SDR-induced immune alterations, both in the periphery and the CNS (Hanke, et al., under review; Wohleb et al., 2011). Therefore, the current experiments were designed to determine if SDR causes changes in the activation status and cytolytic/anti-viral potential of natural killer cells, and to determine if these alterations are mediated through β-AR dependent pathways.
CD16 or FcγRIII is a transmembrane protein found on NK cells that facilitates antibody dependent cellular cytotoxicity (ADCC) by binding to the Fc portion of IgG antibody coated target cells (Perussia, 1998). Therefore, an increase in CD16 is associated with an increased activation status and capacity to perform ADCC functions. Studies indicate that CD16/ligand cross-linking also leads to increases in additional activation markers (e.g., CD69) (Marquez et al., 2010). In addition to CD16, CD69 is one of the earliest markers expressed on the cell surface of activated NK cells. Natural killer cells upregulate surface expression of CD69 when stimulated with ligands such as anti-CD16, IL-2, IL12, and IFN-α (Gerosa et al., 1993). CD69 acts as a costimulatory receptor that leads to cytotoxicity, cell proliferation, and/or cytokine secretion (Borrego et al., 1999). The relationship between CD16 and CD69 upregulation is a strong and important one where evidence suggests that both receptors have the capacity to activate NK cytotoxic and anti-viral cytokine mechanisms (Marquez et al., 2010; Moretta et al., 1991). The current experiments indicate that SDR significantly increased both CD16 and CD69 surface expression on splenic CD3-/DX5+ NK cells. Furthermore, lung derived CD3-/DX5+ NK cells had a differential yet significant increase in CD16 and decrease in CD69. Our interpretation for this discrepancy is once circulating CD3-/DX5+ NK cells have “homed” to the lung (a non-lymphoid organ) or spleen (a lymphoid organ) and crossed over to the tissue parenchyma, differences in the types of cells/cytokines present after SDR-treatment alters the CD16 and CD69 expression pattern. Supporting evidence for this hypothesis is that SDR-treatment does not alter levels of either CD16 or CD69 on CD3-/DX5+ NK cells in circulation, but changes become apparent when examining either splenic or lung tissue 14hrs after the last cycle of SDR. Taken together, the current set of experiments show that SDR is capable of increasing/changing the activation pattern of both CD16 and CD69 on spleen and lung derived CD3-/DX5+ NK cells. These results are consistent with prior reports from our laboratory that show repeated social disruption is capable of “priming” immune cells (e.g., macrophages/monocytes, dendritic cells, and memory T-cells) by altering their activation status, rendering them more efficient in expressing anti-bacterial and anti-viral defense mechanisms (Bailey et al., 2007; Dong-Newsom et al., 2010; Mays et al., 2010; Powell et al., 2010).
Natural killer cell inhibitory receptors also have a profound effect on the cytotoxic/cytolytic function of NK cells against virally infected or tumor cells. Inhibitory receptors found on NK cells bind to MHC class I-like molecules on target cells and inhibit cytolytic function through the ITIM pathway, protecting the target cell from lysis (Long, 2008). CD94, which is often dimerized with NKG2a, is a common C-type lectin inhibitory receptor found on NK cells. After cross-linking, the CD94/NKG2a complex is shown to cause a reduction in CD16-dependent killing and CD69-induced cytotoxic function (Borrego et al., 1999; Brooks et al., 1997; Zingoni et al., 2000). Furthermore, other C-type lectin inhibitory receptors such as Ly49a transduce inhibitory signals upon MHC type I-like receptor ligation much like the CD94/NKG2a heterodimer (Held et al., 1996). Results from the current experiments show that SDR significantly decreased splenic levels of NKG2a and Ly49a on CD3-/DX5+ NK cells. Furthermore, SDR significantly decreased Ly49a on lung derived CD3-/DX5+ NK cells, however contrary to our original hypothesis NKG2a was not decreased significantly, though there was a trend for a reduction (p=0.07). Inhibitory markers (i.e., NKG2a and Ly49a) were not altered on CD3-/DX+ NK cells in circulation similar to results obtained for CD16 and CD69. We hypothesize that, as it was for the activation receptors, the requirement of entering an altered tissue microenvironment (i.e., increased cytokines, chemokines, etc.) from circulation is necessary to reduce the inhibitory receptor expression on CD3-/DX5+ NK cells. Taken together, the current set of experiments show that SDR decreased the expression of both NKG2a and Ly49a inhibitory receptor expression on spleen and lung derived CD3-/DX5+ NK cells 14hrs after the last cycle of SDR. Moreover, the decrease in inhibitory receptor expression after exposure to repeated social disruption highlights a potential/probable mechanism for the increase in activation receptors seen after SDR-treatment.
In order to examine possible mechanisms behind SDR-induced increases in activation, inhibitory, and degranulation marker receptor expression, we employed the use of the non-selective β-AR antagonist propranolol. Previous studies from our laboratory has shown propranolol administration attenuates many SDR-induced immunological alterations such as anxiety-like behavior cFos, expression in areas associated with threat appraisal, and inflammatory changes in CNS microglia/macrophages (Wohleb et al., 2011). Additionally, SDR-propranolol-treated mice show reduced IL-6 plasma levels, reduced splenocyte “priming”, and reduced GC insensitivity compared to animals that received SDR treatment alone (Hanke et al., under review). Current data indicate there were no changes in the percentage of splenic CD3-/DX5+ NK cells. However, examination of activation and degranulation status showed consistent findings of SDR-induced increases in CD16 and CD69, and decreased NKG2a and Ly49a expression on splenic CD3-/DX5+ NK cells which was ameliorated with propranolol administration (see Fig. 8B–E). These data provide critical evidence that SDR-induced phenotypic “priming” is mediated through β-AR dependent ligation.
To determine the functional implications of increased activation receptors and decreased inhibitory receptors on CD3-/DX5+ NK cells we chose to examine degranulation markers, cytotoxicity to MHC I devoid YAC-1 cells, and anti-viral cytokine production. NK cells are able to exert their cytolytic/anti-viral effector function by several different mechanisms. One of these mechanisms and possibly the most powerful is the perforin/granzyme exocytosis granule system. Upon recognition of an infected cell, NK cells release both perforins and granzymes into the extracellular space. Current research indicates that the mannose 6-phosphate receptor is able to recognize granzyme B and incorporates a macromolecule which includes perforins, granzymes, and possibly other molecules (Trapani et al., 2002). Once inside the target cell, perforins and granzymes act in synergy to activate endogenous caspases that trigger apoptotic pathways even though cell death is caspase-independent (Trapani et al., 1998). Lysosomal-associated membrane protein 1 (i.e., CD107a) is a granular membrane protein found on cells capable of degranulation such as cytotoxic T-cells and NK cells. Upregulation of CD107a has been found on the surface of cells that are undergoing active degranulation. Although the expression does not measure target cell lysis directly, it does provide evidence of cytolytic potential and is often correlated with IFN-γ secretion (Alter et al., 2004; Betts et al., 2003). The current set of data shows that SDR significantly increased CD107a expression on splenic CD3-/DX5+ cells. Furthermore, of the activated splenic NK cells (i.e., CD3-/DX5+/CD16+), CD107a surface expression was significantly increased in SDR-treated animals compared to controls. These data indicate that social disruption increased degranulation of the overall splenic CD3-/DX5+ population and activation status is a contributing factor. In addition, propranolol was shown to block SDR-induced increases in CD107a expression on CD3-/DX5+ splenocytes, implying β-AR signaling modulates the effects of repeated social disruption on NK cell degranulation. As noted, increased release of apoptotic granules lead to increased killing capability. Therefore, since SDR increases degranulation by NK cells, they are more apt to kill infected target cells. Additionally, we hypothesize that SDR may also decrease the threshold for degranulation making them “primed” and better able to eradicate infected cells upon stimulation, but this has yet to be determined.
As mentioned, another major way that NK cells exert their anti-viral functions is by the release of IFN-γ. Interferon gamma has numerous effector functions such as macrophage, dendritic, and NK cell activation, growth, and differentiation. Additionally, increased B-cell production of IgG has been shown after IFN-γ stimulation. IFN-γ also increases the expression of MHC class I on cells, thereby rendering infected cells more prone to targeting by adaptive CD8+ specific T-cells. Moreover, MHC II is upregulated on B-cells, dendritic cells, and monocytes/macrophages upon IFN-γ stimulation (Boehm et al., 1997; Mach et al., 1996) making them more effective antigen presenting cells to adaptive CD4+ T-cells and better at controlling pathogenic specific responses. Costimulation of CD16 (i.e., Fc receptor for IgG) and IL-2R in most circumstances is necessary for significant IFN-γ production. In fact, research has shown in order to get optimal IFN-γ production a combination of IL-2, IL-12, IL-15, IL-18 and/or CD16 ligation is needed (Carson et al., 1994; Carson et al., 2001; Fehniger et al., 1999a; Fehniger et al., 1999b). Even though IL-2 was originally thought to only act as a proliferative cytokine to NK cells, evidence indicates that IL-2 can be a potent activator of IFN-γ production when acting synergistically with cross-linked CD16 (Carson et al., 2001). To determine if IFN-γ production in NK cells rather than CD8+ T-cells (also known to produce high levels if IFN-γ) is upregulated by social disruption, DX5+ cells from SDR-treated animals and controls were isolated from total splenic cell populations and cultured for 48hrs in the presence of IL-2, immobilized IgG, or a combination of both. Cell free supernatants were then analyzed for IFN-γ production by ELISA. Results indicate that a combination of both IgG and IL-2 stimulation was necessary to induce significant IFN-γ production. Moreover, DX5+ cells from SDR animals produced a dramatically increased level of IFN-γ upon IL-2/IgG stimulation compared to control animals. These data indicate that splenic DX5+ NK cells from SDR-treated animals are more capable of mounting an anti-viral IFN-γ response compared to controls when stimulated with a combination of IL-2 and IgG. We hypothesize that increases in CD16 on splenic NK cells of SDR-treated animals (shown in this report) are a contributing factor to the reported increase in IFN-γ production upon IgG/IL-2 stimulation and potentially mount a greater anti-viral/tumor response than control animals.
As a confirmatory study for above mentioned SDR-induced increased degranulation CD107a marker and increased IFN-γ production after co-stimulation with IgG/IL-2, a 51Cr release assay was performed on whole splenocytes following IL-15 stimulation to determine if SDR causes increased cytotoxicity to MHC I devoid cells. Additionally, animals were given propranolol to determine if β-AR blockade played a role in the hypothesized increase in cytotoxicity exhibited by SDR-treated animals. Current data show that NK cytotoxicity against MHC I devoid cells was increased in animals that underwent repeated social disruption. These data coincide with data collected that showed increased degranulation markers and increased IFN-γ production upon costimulation ex vivo in SDR-treated animals. Interestingly and contrary to our original hypothesis, propranolol administration did not ameliorate this effect, but rather increased cytotoxicity. Evidence to support this finding comes from studies that show that β-AR stimulation reduces NK cytolytic activity both in vivo and in vitro (Dokur et al., 2004; Shakhar et al., 1998; Takamoto et al., 1991). We postulate rather than blocking MHC I-dependent cytotoxicity, the current data provide support for ADCC mechanisms being blocked through β-AR dependent pathways. This hypothesis is corroborated by current data showing propranolol blocking SDR-induced increases in CD16 (IgG receptor) which is a critical receptor in ADCC functioning. Nevertheless, cytotoxicity to MHC I devoid cells was shown to be increased in splenocytes obtained from SDR animals.
Acute versus chronic stressors have a differential impact on the distribution and activation status of NK cells. To determine if our SDR paradigm has any acute stress effects in addition to the alterations already reported on NK cell distribution and activation, these dependent variables were assessed directly following the last cycle of SDR (i.e., 6th cycle). The current data show that immediately following the last cycle of SDR, the percentage of NK cells in the blood were significantly increased. These data coincide with prior reports that show increased numbers of NK cells in circulation following acute stress. Mechanisms for increases in circulating NK cells remain unclear. Some research shows a redistribution of NK cells from tissue parenchyma (e.g., spleen, BM, lymphnodes) after acute stress (Dhabhar et al., 1995; Dhabhar et al., 1996; Dhabhar et al., 1997; Dhabhar, 2000), while others report density, shedding, or changes in affinity in cell adhesion molecules (CAM) (e.g., ICAM, L- and E-selectins) to be the mechanism behind increased circulating NK cells (Benschop et al., 1993; Kimura et al., 2008; Rehman et al., 1997). While both of these explanations are plausible, current data show increased percentage of NK cells in the blood, while no changes are apparent in splenic NK cell percentages immediately after the last cycle of SDR. These data lend support to alterations in aspects of NK cell adhesion to the vascular wall to be the possible mechanism of SDR-induced increases in the percentage of NK cells after SDR. Alternatively, it is possible that egress of NK cells from tissue compartments other than the spleen (i.e., lymphnodes, bone marrow, etc.) to be contributing factors to the effects of SDR on increased circulating NK cell populations. Further experimentation is needed to elucidate these possibilities in our SDR model. Upon closer examination of NK cell phenotypes, SDR-induced alterations in activation, inhibitory, and degranulation markers were similar in the blood and spleen comparing assessment directly after the last cycle of SDR and 14hrs post-stressor. Nevertheless, these data support prior findings that SDR causes both acute and chronic stress-induced immunological alterations and is partially dependent on the time-point of assessment. Furthermore, the current data show that SDR-induced NK cell phenotypic alterations in activation, inhibitory, and degranulation markers are not transient in nature, but remain altered at least 14hrs post-SDR.
Repeated social disruption causes splenomegaly evidenced by increases in splenic mass and cellularity (Avitsur et al., 2003; Avitsur et al., 2002; Bailey et al., 2007). Additionally, prior reports from our laboratory indicate that increases in granulocytes and macrophages/monocytes are contributing factors (Engler et al., 2005; Engler et al., 2004a), presumably due to increased hematopoiesis and egress from the bone marrow and infiltration from circulation. The current data provided in this report confirms that SDR increases both splenic mass and cellularity. Interestingly, and contrary to our original hypothesis, we believe that DX5+ NK cells do not play a significant role in this due to the inability of repeated social disruption to alter the total number of isolated DX5+ cells in the spleen 14 hrs after the last cycle of SDR.
In conclusion, we have demonstrated that repeated social disruption is capable of changing the activation and inhibitory profile of CD3-/DX5+ NK cells in the spleen and lung of non-infected mice potentially rendering them more reactive upon pathogenic challenge. Additionally, we have presented evidence to show that CD3-/DX5+ NK cells from SDR-treated animals increase degranulation and are more apt to eradicate infected target cells. Data show that these SDR-induced alterations in the spleen are at least in part due to β-AR signaling, as the use of propranolol was able to block increases in activation, decreases in inhibition, and increases in degranulation markers. Our current experiments also show that after costimulation with immobilized IgG and IL-2, splenic DX5+ NK cells from SDR animals secrete a significantly greater amount of IFN-γ which may facilitate anti-viral/tumor immune activation. We show that splenic NK cells from SDR-treated mice significantly increase their cytolytic activity to MHC I devoid cells. Lastly, the current data indicate that the SDR-induced phenotypic NK cell alterations are not transient in nature, but can be maintained at least 14hrs post-stressor. Taken together, these data add to the existing literature showing social disruption in the absence of infection “primes” NK cells, increasing anti-microbial potential. These data, as well as prior reports highlight the ability of psychological states (e.g., threat appraisal during SDR) to alter immunological reactivity partially though β-AR mediated mechanisms.
Research Highlight.
Social disruption increases natural killer cell activation, degranulation potential, and anti-viral cytokine production in the absence of infection.
Acknowledgments
This work was supported in full by the NIH/NIDCR grants: RO1-MH046801-18, T32-DE-014320-09, and F32-DE-022230 received by JFS and AJT.
Footnotes
Conflict of Interest Statement: All authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004;294:15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Andoniou CE, van Dommelen SL, Voigt V, Andrews DM, Brizard G, sselin-Paturel C, Delale T, Stacey KJ, Trinchieri G, gli-Esposti MA. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat Immunol. 2005;6:1011–1019. doi: 10.1038/ni1244. [DOI] [PubMed] [Google Scholar]
- Avitsur R, Kavelaars A, Heijnen C, Sheridan JF. Social stress and the regulation of tumor necrosis factor-alpha secretion. Brain Behav Immun. 2005;19:311–317. doi: 10.1016/j.bbi.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Avitsur R, Padgett DA, Dhabhar FS, Stark JL, Kramer KA, Engler H, Sheridan JF. Expression of glucocorticoid resistance following social stress requires a second signal. J Leukoc Biol. 2003;74:507–513. doi: 10.1189/jlb.0303090. [DOI] [PubMed] [Google Scholar]
- Avitsur R, Stark JL, Dhabhar FS, Sheridan JF. Social stress alters splenocyte phenotype and function. J Neuroimmunol. 2002;132:66–71. doi: 10.1016/s0165-5728(02)00310-7. [DOI] [PubMed] [Google Scholar]
- Avitsur R, Stark JL, Sheridan JF. Social stress induces glucocorticoid resistance in subordinate animals. Horm Behav. 2001;39:247–257. doi: 10.1006/hbeh.2001.1653. [DOI] [PubMed] [Google Scholar]
- Bailey MT, Avitsur R, Engler H, Padgett DA, Sheridan JF. Physical defeat reduces the sensitivity of murine splenocytes to the suppressive effects of corticosterone. Brain Behav Immun. 2004;18:416–424. doi: 10.1016/j.bbi.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Bailey MT, Engler H, Powell ND, Padgett DA, Sheridan JF. Repeated social defeat increases the bactericidal activity of splenic macrophages through a Toll-like receptor-dependent pathway. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1180–R1190. doi: 10.1152/ajpregu.00307.2007. [DOI] [PubMed] [Google Scholar]
- Benschop RJ, Jacobs R, Sommer B, Schurmeyer TH, Raab JR, Schmidt RE, Schedlowski M. Modulation of the immunologic response to acute stress in humans by beta-blockade or benzodiazepines. FASEB J. 1996;10:517–524. doi: 10.1096/fasebj.10.4.8647351. [DOI] [PubMed] [Google Scholar]
- Benschop RJ, Oostveen FG, Heijnen CJ, Ballieux RE. Beta 2-adrenergic stimulation causes detachment of natural killer cells from cultured endothelium. Eur J Immunol. 1993;23:3242–3247. doi: 10.1002/eji.1830231230. [DOI] [PubMed] [Google Scholar]
- Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- Borrego F, Robertson MJ, Ritz J, Pena J, Solana R. CD69 is a stimulatory receptor for natural killer cell and its cytotoxic effect is blocked by CD94 inhibitory receptor. Immunology. 1999;97:159–165. doi: 10.1046/j.1365-2567.1999.00738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks AG, Posch PE, Scorzelli CJ, Borrego F, Coligan JE. NKG2A complexed with CD94 defines a novel inhibitory natural killer cell receptor. J Exp Med. 1997;185:795–800. doi: 10.1084/jem.185.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson WE, Giri JG, Lindemann MJ, Linett ML, Ahdieh M, Paxton R, Anderson D, Eisenmann J, Grabstein K, Caligiuri MA. Interleukin (IL) 15 is a novel cytokine that activates human natural killer cells via components of the IL-2 receptor. J Exp Med. 1994;180:1395–1403. doi: 10.1084/jem.180.4.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson WE, Parihar R, Lindemann MJ, Personeni N, Dierksheide J, Meropol NJ, Baselga J, Caligiuri MA. Interleukin-2 enhances the natural killer cell response to Herceptin-coated Her2/neu-positive breast cancer cells. Eur J Immunol. 2001;31:3016–3025. doi: 10.1002/1521-4141(2001010)31:10<3016::aid-immu3016>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Curry JM, Hanke ML, Piper MG, Bailey MT, Bringardner BD, Sheridan JF, Marsh CB. Social disruption induces lung inflammation. Brain Behav Immun. 2010;24:394–402. doi: 10.1016/j.bbi.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhabhar FS. Acute stress enhances while chronic stress suppresses skin immunity. The role of stress hormones and leukocyte trafficking. Ann N Y Acad Sci. 2000;917:876–893. doi: 10.1111/j.1749-6632.2000.tb05454.x. [DOI] [PubMed] [Google Scholar]
- Dhabhar FS, McEwen BS. Acute stress enhances while chronic stress suppresses cell-mediated immunity in vivo: a potential role for leukocyte trafficking. Brain Behav Immun. 1997;11:286–306. doi: 10.1006/brbi.1997.0508. [DOI] [PubMed] [Google Scholar]
- Dhabhar FS, Miller AH, McEwen BS, Spencer RL. Stress-induced changes in blood leukocyte distribution. Role of adrenal steroid hormones. J Immunol. 1996;157:1638–1644. [PubMed] [Google Scholar]
- Dhabhar FS, Miller AH, McEwen BS, Spencer RL. Effects of stress on immune cell distribution. Dynamics and hormonal mechanisms. J Immunol. 1995;154:5511–5527. [PubMed] [Google Scholar]
- Dokur M, Boyadjieva N, Sarkar DK. Catecholaminergic control of NK cell cytolytic activity regulatory factors in the spleen. J Neuroimmunol. 2004;151:148–157. doi: 10.1016/j.jneuroim.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Dong-Newsom P, Powell ND, Bailey MT, Padgett DA, Sheridan JF. Repeated social stress enhances the innate immune response to a primary HSV-1 infection in the cornea and trigeminal ganglia of Balb/c mice. Brain Behav Immun. 2010;24:273–280. doi: 10.1016/j.bbi.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler H, Bailey MT, Engler A, Sheridan JF. Effects of repeated social stress on leukocyte distribution in bone marrow, peripheral blood and spleen. J Neuroimmunol. 2004a;148:106–115. doi: 10.1016/j.jneuroim.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Engler H, Dawils L, Hoves S, Kurth S, Stevenson JR, Schauenstein K, Stefanski V. Effects of social stress on blood leukocyte distribution: the role of alpha- and beta-adrenergic mechanisms. J Neuroimmunol. 2004b;156:153–162. doi: 10.1016/j.jneuroim.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Engler H, Engler A, Bailey MT, Sheridan JF. Tissue-specific alterations in the glucocorticoid sensitivity of immune cells following repeated social defeat in mice. J Neuroimmunol. 2005;163:110–119. doi: 10.1016/j.jneuroim.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Feau S, Facchinetti V, Granucci F, Citterio S, Jarrossay D, Seresini S, Protti MP, Lanzavecchia A, Ricciardi-Castagnoli P. Dendritic cell-derived IL-2 production is regulated by IL-15 in humans and in mice. Blood. 2005;105:697–702. doi: 10.1182/blood-2004-03-1059. [DOI] [PubMed] [Google Scholar]
- Fehniger TA, Carson WE, Caligiuri MA. Costimulation of human natural killer cells is required for interferon gamma production. Transplant Proc. 1999a;31:1476–1478. doi: 10.1016/s0041-1345(99)00011-1. [DOI] [PubMed] [Google Scholar]
- Fehniger TA, Shah MH, Turner MJ, VanDeusen JB, Whitman SP, Cooper MA, Suzuki K, Wechser M, Goodsaid F, Caligiuri MA. Differential cytokine and chemokine gene expression by human NK cells following activation with IL-18 or IL-15 in combination with IL-12: implications for the innate immune response. J Immunol. 1999b;162:4511–4520. [PubMed] [Google Scholar]
- Gerosa F, Tommasi M, Benati C, Gandini G, Libonati M, Tridente G, Carra G, Trinchieri G. Differential effects of tyrosine kinase inhibition in CD69 antigen expression and lytic activity induced by rIL-2, rIL-12, and rIFN-alpha in human NK cells. Cell Immunol. 1993;150:382–390. doi: 10.1006/cimm.1993.1206. [DOI] [PubMed] [Google Scholar]
- Granucci F, Zanoni I, Pavelka N, van Dommelen SL, Andoniou CE, Belardelli F, gli Esposti MA, Ricciardi-Castagnoli P. A contribution of mouse dendritic cell-derived IL-2 for NK cell activation. J Exp Med. 2004;200:287–295. doi: 10.1084/jem.20040370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke ML, Powell ND, Stiner LM, Bailey MT, Sheridan JF. Beta adrenergic blockade decreases the immunomodulatory effects of social disruption stress. Brain Behav Immun. doi: 10.1016/j.bbi.2012.07.011. under review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held W, Cado D, Raulet DH. Transgenic expression of the Ly49A natural killer cell receptor confers class I major histocompatibility complex (MHC)-specific inhibition and prevents bone marrow allograft rejection. J Exp Med. 1996;184:2037–2041. doi: 10.1084/jem.184.5.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henney CS, Kuribayashi K, Kern DE, Gillis S. Interleukin-2 augments natural killer cell activity. Nature. 1981;291:335–338. doi: 10.1038/291335a0. [DOI] [PubMed] [Google Scholar]
- Houchins JP, Lanier LL, Niemi EC, Phillips JH, Ryan JC. Natural killer cell cytolytic activity is inhibited by NKG2-A and activated by NKG2-C. J Immunol. 1997;158:3603–3609. [PubMed] [Google Scholar]
- Irwin M, Patterson T, Smith TL, Caldwell C, Brown SA, Gillin JC, Grant I. Reduction of immune function in life stress and depression. Biol Psychiatry. 1990;27:22–30. doi: 10.1016/0006-3223(90)90016-u. [DOI] [PubMed] [Google Scholar]
- Karlhofer FM, Ribaudo RK, Yokoyama WM. The interaction of Ly-49 with H-2Dd globally inactivates natural killer cell cytolytic activity. Trans Assoc Am Physicians. 1992;105:72–85. [PubMed] [Google Scholar]
- Kimura K, Isowa T, Matsunaga M, Murashima S, Ohira H. The temporal redistribution pattern of NK cells under acute stress based on CD62L adhesion molecule expression. Int J Psychophysiol. 2008;70:63–69. doi: 10.1016/j.ijpsycho.2008.05.580. [DOI] [PubMed] [Google Scholar]
- Kinsey SG, Bailey MT, Sheridan JF, Padgett DA, Avitsur R. Repeated social defeat causes increased anxiety-like behavior and alters splenocyte function in C57BL/6 and CD-1 mice. Brain Behav Immun. 2007;21:458–466. doi: 10.1016/j.bbi.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004;21:107–119. doi: 10.1016/j.immuni.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Liu T, Tang Q, Hendricks RL. Inflammatory infiltration of the trigeminal ganglion after herpes simplex virus type 1 corneal infection. J Virol. 1996;70:264–271. doi: 10.1128/jvi.70.1.264-271.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long EO. Negative signaling by inhibitory receptors: the NK cell paradigm. Immunol Rev. 2008;224:70–84. doi: 10.1111/j.1600-065X.2008.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mach B, Steimle V, Martinez-Soria E, Reith W. Regulation of MHC class II genes: lessons from a disease. Annu Rev Immunol. 1996;14:301–331. doi: 10.1146/annurev.immunol.14.1.301. [DOI] [PubMed] [Google Scholar]
- Marquez ME, Millet C, Stekman H, Conesa A, Deglesne PA, Toro F, Sanctis JD, Blanca I. CD16 cross-linking induces increased expression of CD56 and production of IL-12 in peripheral NK cells. Cell Immunol. 2010;264:86–92. doi: 10.1016/j.cellimm.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Mays JW, Bailey MT, Hunzeker JT, Powell ND, Papenfuss T, Karlsson EA, Padgett DA, Sheridan JF. Influenza virus-specific immunological memory is enhanced by repeated social defeat. J Immunol. 2010;184:2014–2025. doi: 10.4049/jimmunol.0900183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretta A, Poggi A, Pende D, Tripodi G, Orengo AM, Pella N, Augugliaro R, Bottino C, Ciccone E, Moretta L. CD69-mediated pathway of lymphocyte activation: anti-CD69 monoclonal antibodies trigger the cytolytic activity of different lymphoid effector cells with the exception of cytolytic T lymphocytes expressing T cell receptor alpha/beta. J Exp Med. 1991;174:1393–1398. doi: 10.1084/jem.174.6.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orange JS, Biron CA. Characterization of early IL-12, IFN-alphabeta, and TNF effects on antiviral state and NK cell responses during murine cytomegalovirus infection. J Immunol. 1996;156:4746–4756. [PubMed] [Google Scholar]
- Perussia B. Fc receptors on natural killer cells. Curr Top Microbiol Immunol. 1998;230:63–88. doi: 10.1007/978-3-642-46859-9_6. [DOI] [PubMed] [Google Scholar]
- Powell ND, Bailey MT, Mays JW, Stiner-Jones LM, Hanke ML, Padgett DA, Sheridan JF. Repeated social defeat activates dendritic cells and enhances Toll-like receptor dependent cytokine secretion. Brain Behav Immun. 2009;23:225–231. doi: 10.1016/j.bbi.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell ND, Mays JW, Bailey MT, Hanke ML, Sheridan JF. Immunogenic dendritic cells primed by social defeat enhance adaptive immunity to influenza A virus. Brain Behav Immun. 2010 doi: 10.1016/j.bbi.2010.07.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman J, Mills PJ, Carter SM, Chou J, Thomas J, Maisel AS. Dynamic exercise leads to an increase in circulating ICAM-1: further evidence for adrenergic modulation of cell adhesion. Brain Behav Immun. 1997;11:343–351. doi: 10.1006/brbi.1997.0498. [DOI] [PubMed] [Google Scholar]
- Sanders VM, Straub RH. Norepinephrine, the beta-adrenergic receptor, and immunity. Brain Behav Immun. 2002;16:290–332. doi: 10.1006/brbi.2001.0639. [DOI] [PubMed] [Google Scholar]
- Schedlowski M, Jacobs R, Alker J, Prohl F, Stratmann G, Richter S, Hadicke A, Wagner TO, Schmidt RE, Tewes U. Psychophysiological, neuroendocrine and cellular immune reactions under psychological stress. Neuropsychobiology. 1993a;28:87–90. doi: 10.1159/000119006. [DOI] [PubMed] [Google Scholar]
- Schedlowski M, Jacobs R, Stratmann G, Richter S, Hadicke A, Tewes U, Wagner TO, Schmidt RE. Changes of natural killer cells during acute psychological stress. J Clin Immunol. 1993b;13:119–126. doi: 10.1007/BF00919268. [DOI] [PubMed] [Google Scholar]
- Shakhar G, Ben-Eliyahu S. In vivo beta-adrenergic stimulation suppresses natural killer activity and compromises resistance to tumor metastasis in rats. J Immunol. 1998;160:3251–3258. [PubMed] [Google Scholar]
- Stark JL, Avitsur R, Hunzeker J, Padgett DA, Sheridan JF. Interleukin-6 and the development of social disruption-induced glucocorticoid resistance. J Neuroimmunol. 2002;124:9–15. doi: 10.1016/s0165-5728(02)00004-8. [DOI] [PubMed] [Google Scholar]
- Stark JL, Avitsur R, Padgett DA, Campbell KA, Beck FM, Sheridan JF. Social stress induces glucocorticoid resistance in macrophages. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1799–R1805. doi: 10.1152/ajpregu.2001.280.6.R1799. [DOI] [PubMed] [Google Scholar]
- Takamoto T, Hori Y, Koga Y, Toshima H, Hara A, Yokoyama MM. Norepinephrine inhibits human natural killer cell activity in vitro. Int J Neurosci. 1991;58:127–131. doi: 10.3109/00207459108987189. [DOI] [PubMed] [Google Scholar]
- Trapani JA, Jans DA, Jans PJ, Smyth MJ, Browne KA, Sutton VR. Efficient nuclear targeting of granzyme B and the nuclear consequences of apoptosis induced by granzyme B and perforin are caspase-dependent, but cell death is caspase-independent. J Biol Chem. 1998;273:27934–27938. doi: 10.1074/jbc.273.43.27934. [DOI] [PubMed] [Google Scholar]
- Trapani JA, Smyth MJ. Functional significance of the perforin/granzyme cell death pathway. Nat Rev Immunol. 2002;2:735–747. doi: 10.1038/nri911. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. Biology of natural killer cells. Adv Immunol. 1989;47:187–376. doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng RJ, Padgett DA, Dhabhar FS, Engler H, Sheridan JF. Stress-induced modulation of NK activity during influenza viral infection: role of glucocorticoids and opioids. Brain Behav Immun. 2005;19:153–164. doi: 10.1016/j.bbi.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, Nelson RJ, Godbout JP, Sheridan JF. {beta}-Adrenergic Receptor Antagonism Prevents Anxiety-Like Behavior and Microglial Reactivity Induced by Repeated Social Defeat. J Neurosci. 2011;31:6277–6288. doi: 10.1523/JNEUROSCI.0450-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingoni A, Palmieri G, Morrone S, Carretero M, Lopez-Botel M, Piccoli M, Frati L, Santoni A. CD69-triggered ERK activation and functions are negatively regulated by CD94 / NKG2-A inhibitory receptor. Eur J Immunol. 2000;30:644–651. doi: 10.1002/1521-4141(200002)30:2<644::AID-IMMU644>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]