

Abstract
Mitochondrial dysfunction and subsequent energy failure is a contributing factor to degeneration of the substantia nigra pars compacta associated with Parkinson’s disease (PD). In this study, we investigate molecular events trigger by 1-methyl-4-phenylpyridine (MPP+) using whole genome-expression microarray, western blot and HPLC quantification of metabolites. The data show that MPP+ (500μM) evokes obstruction of mitochondrial respiration/oxidative phosphorylation (OXPHOS) in mouse neuroblastoma Neuro-2a cells, juxtapose to accelerated glucose consumption and production of lactic acid. While additional glucose concentrations restored viability at MPP+ (500μM), loss of OXPHOS was sustained suggesting that compensatory anaerobic metabolic systems were fulfilling required energy needs. Under these conditions, MPP+ initiated significant changes to the transcription of 799 genes (596 up-regulated and 203 down-regulated) of which 612 David IDS were applied and 136 functional annotation clusters were identified. Prominent changes were as follows; MPP+ initiated loss of mRNA for mitochondrial encoded NADH dehydrogenase 4, 5 genes, cytochrome b and NADH dehydrogenase (ubiquinone) flavoprotein 3, concomitant to rise in a mitochondrial fission gene; ganglioside-induced differentiation-associated-protein 1 (GDAP1). These negative changes to OXPHOS components were accompanied by a number of protective forces to the mitochondria including elevated ratio of mitochondrial anti/pro-apoptotic processes including a loss of apoptotic Bcl-2/adenovirus E1B 19-kDa-interacting protein (BNIP3) and family with sequence similarity 162, member A (FAM162a) and rise of heat shock protein 1 and Lon peptidase 1. There were no changes indicative of free radical damage (e.g. SOD, GSH-Px), rather MPP+ initiated a large number of significant G protein signaling components (which trigger catabolic processes) and anaerobic metabolic systems involving carboxylic acid/ transamination reactions (e.g. glutamate oxaloacetate transaminase 1 (GOT1), glutamic pyruvate-alanine transaminase 2 (GPT2), cystathionase and redox proteins such as cytochrome b5 reductase 1 and ferredoxin reductase. Counter-intuitively, the data show reduction of mRNA in glycolytic processes [DAVID enrichment score 9.96 p value 1.90E-19], some corroborated by western blot, bringing in to question the sources of lactate observed in the presence of MPP+. Examining this aspect, the data show that diverse carboxylic acids (succinate, oxaloacetate and a-ketoglutarate) are capable of contributing to the lactate pool in addition to phosph(enolpyruvate) or pyruvate in the absence of glucose by this cell line. In conclusion, these findings show that MPP+ negatively affects the transcriptome involved with complex I, but evoked elevation in G protein signaling and anaerobic metabolic systems involved with nitrogen/carboxylic acid metabolism and redox reactions. Anaerobic survival systems appear to be far more complex than previously believed, and future research will be required to elucidate the survival pathways that drive anaerobic substrate level phosphorylation, and define functional ramification to the loss of mitochondrial FAM162a and BNIP3 proteins.
Keywords: MPP+, Neuro 2a, Parkinson’s disease, whole genome, glycolysis, Complex I
1.0 Introduction
Parkinsons disease (PD) is a chronic degenerative disorder of dopaminergic neurons within the substantia nigra pars compacta (SNc). Pathological features of PD involve mitochondrial insufficiencies (Burch and Sheerin, 2005, Lin et al., 2009a, Nishioka et al., 2010), anomalies in autophagolysosome function (Levy et al., 2009, Nagatsu, 2002, Tofaris and Spillantini, 2005), gene aberrations of DJ-1, PTEN-induced kinase 1, leucine-rich repeat kinase 2, park-1/Synuclein, ATP13A2, β-glucocerebrosidase and mitochondrial proteins (park 13 Omi/Htra2, Complex I).(Bertram and Tanzi, 2005, Hyun et al., 2005, Lesage and Brice, 2009, Mortiboys et al., 2010) One of the key pathological features of PD, involve damaged or dysfunctional mitochondria that contribute to bioenergetic crisis and subsequent neurodegeneration.(Winklhofer and Haass, 2010) Mitochondria use O2 to drive cellular respiration in order to generate energy in the form of adenosine triphosphate (ATP), by the process of oxidative phosphorylation (OXPHOS). If damaged, a loss of ATP can precipitate adverse affects on vital systems ranging from cell signaling, neurotransmission, osmotic balance and biosynthetic processes.
In vitro chemical models used to investigate mitochondrial dysfunction include examining the effects of 1-methyl-4-phenylpyridinium (MPP+), paraquat, rotenone or endogenous isoquinolines on diverse cell lines such as murine N2a, rat PC-12 or human SH-SY5Y cells. (Del Zompo et al., 1993, Kotake and Ohta, 2003b, Mazzio and Soliman, 2004b, Nagatsu, 1997) While a variety of cell lines are available, N2a cells are of neuronal phenotype, originally derived from a spontaneous brain tumor and demonstrate greater sensitivity to the lethal effects of MPP+ than human SH-SY5Y cells (LC50~10x lower) and rat PC-12 cells (LC50~2xlower) (Mazzio et al., 2010b). The difference in vulnerability amongst cell lines is unrelated to dopaminergic phenotype, but rather a function of metabolic rate differences that establish velocity of glucose consumption at baseline and are inherent to cell line specie origin. Because MPP+ obstructs OXPHOS, accelerated anaerobic metabolism contributes to rapid depletion of glucose, which then indirectly evokes neuronal death. For this reason, high levels of glucose are known to be protective against MPP+ in vitro, regardless of the type of cell line used. (Akashi et al., 2011, Chalmers-Redman et al., 1999, Di Monte et al., 1988, Maruoka et al., 2007, Mazzio, Soliman, 2010b) N2a cells are particularly vulnerable to mitochondrial toxins, not only to MPP+, but also rotenone (Swarnkar et al., 2012) and isoquinoline derivatives structurally related to MPTP/MPP+, making them a suitable model to evaluate mitochondrial toxins on bioenergetic processes, as in this study. (Storch et al., 2002)
Other factors to consider when studying the effects of MPP+ in vitro, are that immortal cell lines are of malignant origin, and tend to have a higher baseline conversion rate of glucose to lactate relative to normal tissue, even in the presence of O2 (Serganova et al., 2011) known as the Warburg effect. (Minchenko et al., 2002) While glycolytic rates between normal brain tissue and neuroblastoma cell lines may differ, the effects of mitochondrial toxins or loss of O2 are similar; resulting in acceleration of glycolysis (Mazzio and Soliman, 2003b), reduction in OXPHOS, a rise in lactate (Asgari et al., 2011, Chen et al., 2012, Stein et al., 2012) higher lactate/pyruvate ratio and reduction of glucose surrounding hypoxic or damaged tissue relative to baseline. (Karathanou et al., 2011, Omerhodzic et al., 2011, Zweckberger et al., 2011) In rodent models, brain injury, administration of PD model toxins such as MPP+, systemic administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine or even global cerebral ischemia / reperfusion injury can lead to elevation of lactate levels, suggesting bioenergetic crisis. (Koga et al., 2006, Lin et al., 2009b, Rollema et al., 1988) In the human brain tissue, concentrations of lactate remain relatively low unless triggered by hypoxic or traumatic brain injury, in which case the elevation of lactate can exceed 6mM in close proximity to damaged tissue. (Yokobori et al., 2011) These finding stress the importance of bioenergetic systems and the dynamics of anaerobic metabolism in overcoming neurological insult evoked by PD model toxins.
In the current study, we continue our investigation of molecular events altered by MPP+ on the transcriptome of neuroblastoma cells, using whole genomic expression microarrays. In brief, the findings show that MPP+ targets the mitochondria at complex I concomitant to elevated transcription of G protein signal transduction proteins, diverse REDOX systems, carboxylic acid metabolism and transamination processes that could foster recycling of NADH+/NAD in a manner similar to LDH to drive anaerobic energy cycles. These changes were accompanied by a number of survival processes that cultivate anti-apoptotic events. These findings suggest that anaerobic survival systems are more complex than previously thought, and involve a number of pathways that extend beyond glycolysis. While the comprehensive data from whole genomic analysis are presented, we highlight major systems identified through expression profiling.
2.0 Materials and Methods
Dulbecco’s modified Eagle medium (DMEM), L-glutamine, fetal bovine serum (FBS) heat-inactivated, phosphate buffered saline (PBS), Hank’s balanced salt solution (HBSS), penicillin/streptomycin, lactic acid oxidase, and all other materials were purchased from Sigma Chemical (St. Louis, MO, USA). Agilent mouse 4 x 44 k arrays and kits were supplied by Agilent Technologies (Palo Alto, CA) and electrophoresis systems, apparatus and reagents by Biorad Hercules, CA and Promega, Madison, WI.
2.1 Cell culture
CCL-131™ (N-2A) cells are of brain origin and highly vulnerable to the neurotoxic effects of MPP+ (Mazzio et al., 2010c). N2a cells were grown in DMEM containing phenol red, 10% FBS, 4 mM L-glutamine, 20μM sodium pyruvate and penicillin/streptomycin (100 U/0.1 mg/ml). The cells were maintained at 37.5°C in 5% CO2 / atmosphere. Every 2–5 days, the medium was replaced and the cells were sub-cultured. The experimental plating media consisted of DMEM minus phenol red, 1.8% FBS, penicillin/streptomycin (100 U/0.1 mg/ml), / 2 mM sodium pyruvate and 3 mM L-glutamine. For experiments, cells were plated in either 75 cm3 flasks or 96-well plates at a density of approximately 0.5×106 cells/ml.
2.2 Cell Viability
Cell viability was assessed using resazurin oxidoreduction indicator dye. A working solution of resazurin was prepared in PBS minus phenol red (0.5 mg/ml). (Evans et al., 2001) Reduction of the dye by viable cells reduces the amount of oxidized form and increases the amount of its bright red fluorescent intermediate. Quantitative analysis of dye conversion was measured using a microplate fluorometer—Model 7620-version 5.02 (Cambridge Technologies Inc, Watertown, MA, USA) set at 550 / 580 (excitation / emission).
2.3 HPLC Quantification of Lactic Acid and Glucose
Quantification of lactate and glucose was assessed using a Shimadzu HPLC system equipped with an SPD-20A UV detector (set at 210 nm), a RID-10A 120V refractive index detector, a workstation containing EZSTART V7.4 software and an SS420X instrument interface docked to a Waters Autosampler Model 717 Plus (Shimadzu Scientific Instruments, Inc. US; Waters Corp., Milford, MA). The flow rate was isocratic and controlled by a Waters Model 510 pump set at 0.6 ml/min. The mobile phase consisted of 5 mM sulfuric acid, the column; aminex HPX-87H 300 x 7.8 mm, carbohydrate analysis column, 9 μm particle size (Biorad Hercules, CA), run time was 16 min and injection volume 25 μl. Samples were prepared by placing 35 μl cell supernatant into 200ul of 5mM sulfuric acid, immediately stored at −80C. Prior to analysis, samples were thawed and 125 μl was mixed with 275 μl of the 5mM sulfuric acid, vortexed, capped and run by HPLC. Glucose and lactate standard curves were prepared in distilled water and matrix blank controls and spikes were run for every experimental treatment condition tested.
Lactic Acid
For studies evaluating potential energy substrates to produce lactic acid in glucose free media, lactate was determined in 96 well plates using a colorimetric enzymatic assay (Procedure No 735, Sigma Diagnostics, St. Louis, MO). Briefly, lactate was quantified by conversion to pyruvate and H202 by lactate oxidase using a base lactate reagent containing lactate oxidase (400 U/L) and horseradish peroxidase 2400 U/L (Sigma, St. Louis, MO). The reagent was added to a chromogen and samples were incubated for 8 minutes at 37°C. Lactate was quantified at 490 nm on a UV microplate spectrophotometer. The lactic acid standard curve was generated by preparing a dilution of lactic acid (10μM–10mM) in media minus phenol red.
2.4 O2 Consumption—Clark Electrode
A Hanna HI 9142 O2 Meter was used to measure dissolved O2 as an indicator of mitochondrial respiration. The electrode was calibrated with both air saturated de-ionized water and de-ionized water with sodium dithionite. Briefly, 800 μl of cell supernatant compared to blank controls were directly loaded into a small chamber. After rate equilibration, a 30 s reading was taken for each sample. The temperature was maintained at 37°C.
2.5 Whole genome expression profiling
Whole genome expression profiling was carried out on Agilent Mouse or 4 x 44 k arrays (Beckman Coulter Genomics, Morrisville, NC) from total RNA isolated from each sample. Briefly, the quantity of total RNA was determined by spectrophotometry [A260/280 ratio] and the size distribution was assessed by electropherogram using an Agilent Bioanalyzer. 200 ng of total RNA was converted into labeled cRNA with nucleotides coupled to fluorescent Cy3 dye using a Low Input Quick Amp Kit (Agilent Technologies, Palo Alto, CA) following manufacturer’s protocol. Cy3-labeled cRNA (1.65 μg) from each sample was hybridized to an Agilent Mouse Genome 4×44 k array. The hybridized array was then washed and scanned and data was extracted from the scanned image using Feature Extraction version 10.7 software (Agilent Technologies). The data was analyzed by both Gene Sifter, database for annotation, visualization and integrated discovery” (DAVID) and manually. Manual analysis was achieved by normalizing the raw data [gProcessed] signal to the average signal/ average sample hybridization and noise was filtered omitting any gene below a threshold limit of gProcessed >500 to omit false positives from noise/low abundant genes. Ratios for each group were calculated, and p-values determined by t-test, resulting in a final gene list with differential expression profiles at p-Value <0.05 to be 799 genes, 596 up-regulated and 203 down-regulated of which 612 David IDS were applied and 136 functional annotation clusters identified. The data was then analyzed manually by 1) examination of individual genes accounting for potential duplicates in the array using diverse primers for same gene, including observing QC replicates 2) sorting and analysis by greatest difference in intensity (expression dominance) with significance p<0.05 and 3) then fold change. Next, the combined fold-changes, p-values for each set of hybridizations were classified manually by literature review and entering into the DAVID where patterns by enrichment scores averaging less than p<0.01 were focused on in the discussion (Huang da et al., 2009, Huang da et al., 2007).
2.6 Western Blot
Briefly, after treatment, cells were washed, centrifuged and the supernatant discarded using ice cold sterile PBS at 4° C. The pellet was re-suspended and homogenized / sonicated in RIPA lysis buffer containing protease inhibitors. Samples were placed on ice for 30 min, and centrifuged at 10,000 x g for 10 minutes at 4°C. The supernatant was added at 1:1 of Laemmli Sample Buffer (Biorad #161-0737) + fresh β-ME and boiled for 5 minutes. Approximately 50 μg of protein was loaded / lane and separated using 5%-15% SDS-PAGE gels, running buffer, 25 mM Tris, 192 mM glycine, pH 8.3 (Biorad #161-0734) and applying 200 constant V constant ~ 35 min. The proteins were transferred to polyvinylidene fluoride membranes (100V for 30–60 minutes) in ice-cold transfer buffer containing 25mM Tris, 192mM glycine and 20% methanol. The membranes were placed in a blocking buffer consisting of 5% bovine serum albumin Fraction V (BSA) w/v in TBS + 0.05% Tween-20, pH 7.4. The membranes were washed and placed in 1° antibody (1:500–3000) containing 1% BSA in TTBS at RT for 2 Hr. The membranes were washed in TTBS and incubated in 2° anti-mouse/ or rabbit IgG (Fc specific) peroxidase conjugate (1:4000) in 2% non fat dried milk in PBS for 1.5 Hr at RT. After a final wash, peroxidase was detected with Sigma FAST™ DAB (3, 3′-diaminobenzidine tetrahydrochloride) with a metal enhancer cobalt chloride. Images were scanned using an Epson Stylus CX-8400. Intensity analysis was performed using ImageJ software provided from the National Institutes of Health. (Girish and Vijayalakshmi, 2004)
2.7 Data analysis
Statistical analysis was performed using GraphPad Prism (version 3.0; GraphPad Software Inc. San Diego, CA, USA) with significance of difference between the groups assessed using a one-way ANOVA, followed by Tukey post hoc means comparison test, two way ANOVA or Student’s t test.
3.0 Results
A toxicity profile associated with MPP+ ± additional glucose (0.01–10 mM) in baseline low glucose media is shown in Figure 1. Figure 1 shows a dose dependent rise in glucose media concentrations initiated a gradual reduction of MPP+ toxicity. MPP+ mediated obstruction of OXPHOS, where elevated concentrations of glucose worsened the loss of respiration, suggesting that a switch over to anaerobic glucose metabolic systems were completely responsible for sustaining cell viability. Figure 2 is a raw Clark electrode data recording demonstrating that MPP+ mitigates loss to aerobic respiration, where untreated cells have lower levels of remaining dissolved O2 at 24 hours. Treatment for 24 Hrs with MPP+ blocked cell respiration and therefore higher remaining levels of dissolved O2 remained in the media.
Figure 1.

Supplemental glucose protection (.01–10mM) against the toxicity and loss of OXPHOS induced by 500μM MPP+ in N2a cells after 24 Hr cultured in baseline low glucose DMEM (1000mg/L). The data represents cell viability and O2 consumption as % control. Data are presented as the Mean ± S.E.M., n=4. Significance of difference between the control vs. treatment groups were determined by a one–way ANOVA followed by a Tukey post hoc test * p<0.05.
Figure 2.

Oxygen electrode raw data chart recording of dissolved O2 in the media matrix blank vs. N2a cell supernatant ± [MPP+ 500 μM] with variation in additional glucose (0.1 –10 mM) in low glucose DMEM (1000 mg /L). The data represent dissolved O2 (nM / ml at 37.5°C) in one sample set, and all groups were statistically different from cell controls determined by a one–way ANOVA followed by a Tukey post hoc test n=4 * p<0.05.
In the next study, we monitor cell glucose consumption and production of lactic acid quantified by HPLC, while varying concentration of glucose in the media both in a control group (Figure 3a) and upon treatment with MPP+ (500μM) (Figure 3b). Figure 3a shows that relative to the media blank containing 10mM of glucose, with no trace of lactic acid, at 24 hours, N2a cells used approximately half of available glucose and produced 10.65 ± .09 mM of lactate. Interestingly, additional glucose supply in the media above 10mM had no additional effects on production of lactate indicating that the conversion rate of glucose to lactate remains constant. These findings indicate that energy metabolism is conservative and excess energy supplies do not equate to excess energy consumption above what is needed. In the presence of a mitochondrial inhibitor (Figure 3b), there is a marked shift toward anaerobic metabolism amongst cells treated with MPP+ and 10mM Glucose, evidenced by the drop in glucose to 2.7 ± 1.4 mM and rise in lactate to 16.2 ± .28 mM. Again, under these conditions additional concentrations of glucose did not correspond to corresponding rise in lactic acid, suggesting that the rate of glycolysis is likely to be highly controlled, is vastly altered by mitochondrial toxicity, but less influenced by change of pericellular glucose concentrations. In other words, the cells consume glucose at a constant rate as needed.
Figure 3.


Figure 3A. Effects of elevating concentration of glucose on glucose to lactate conversion in N2a cells at 24 Hr. The data represent lactic acid produced (mM) and glucose remaining (mM) [non-utilized glucose] and are presented as the Mean ± S.E.M. n=4. Significance of difference between the cell control vs., treatment groups were determined by a one–way ANOVA followed by a Tukey post hoc test * p<0.05.
Figure 3B. Effects of elevating concentration of glucose on glucose to lactate conversion in N2a cells treated with MPP+ [500 μM] at 24 Hr. The data represent lactic acid produced (mM) and glucose remaining (mM) [non-utilized glucose] and are presented as the Mean ± S.E.M. n=4. Significance of difference between the cell control vs. treatment groups were determined by a one–way ANOVA followed by a Tukey post hoc test * p<0.05.
In order to identify molecular patterns altered by MPP+ independent of toxicity, whole genomic expression microarray analytical studies were conducted on samples collected at 24 Hrs for controls vs. MPP+ [500μM] cultured in high glucose media. Expression profiling was carried out using labeled cRNA coupled to fluorescent Cy3 dye hybridized to an Agilent Mouse Genome 4×44 k array (Beckman Coulter Genomics, Morrisville, NC). Both manual and bio-informatic analysis (Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.7) were used to identify major systems affected. A total of 2740 gene shifts were identified (p-value <0.05) from biological triplicates; of which 1904 were eliminated as noise (low expression volume). Of the remaining 799 genes shifts, 596 were up-regulated and 203 down- regulated. These were further narrowed by eliminating all values less than a 1.5 fold change in expression leaving – a total of 439 genes significantly affected of which 287 DAVID Ids were identified (Table 1). Table 1 displays DAVID enrichment scores for group functional annotation clustering (Huang da, Sherman, 2009, Huang da, Sherman, 2007). Systems largely affected by MPP+ were glycolysis, carboxylic acid metabolism, amino and phosphotransferases, vitamin B6 metabolism, pyruvate and amino acid metabolism, NAD, NADH oxidation-reduction and G protein signaling, most of which suggest changes in energy metabolism.
Table 1. DAVID Functional Annotation Clustering & Enrichment Scores MPP+ (500μM) vs Control in N2a Cells @ 24 Hr.
Functional Annotation Clustering on statistically differentially genes between controls vs. MPP+ treated [500 μM] at 24 Hr.
| Category | Enrichment Score | Term | Average Count | P-Value |
|---|---|---|---|---|
| KP, GPF, SPK, USF | 9.96 | Glycolysis, glucose catabolic processes | 15 | 1.90E-16 |
| GPF, SPK, | 2.88 | Carboxylic Acid, Fatty Acid Metabolic Process | 11 | 9.80E-06 |
| GMF, SPK, UPF | 2.02 | ATP Binding | 31 | 3.50E-03 |
| SPK, USF | 2.02 | Phosphotransferases | 10 | 2.50E-05 |
| GMF, IP, SPK | 1.77 | Vit B6 Binding, Aminotransferase, Transaminase | 7 | 2.10E-03 |
| SMT, IP, GMF, SPK | 1.61 | Calponin-like actin binding, cytoskeleton | 8 | 6.90E-03 |
| GPF | 1.57 | Amino acid, biosynthetic process. serine, glycine, aspartate | 8 | 2.10E-03 |
| GPF | 1.55 | Carbohydrate biosynthesis, Pyruvate metabolic Process | 7 | 1.90E-02 |
| GCF | 1.45 | Melanosome | 5 | 2.20E-02 |
| SPK, USF, IP, GPF, GMF | 1.43 | NAD, NADH oxidation reduction | 16 | 5.30E-03 |
| SPK, CGF | 1.37 | Cytoskeletal, organization, intermediate filament | 7 | 5.20E-03 |
| SPK, GMF, GPF | 1.33 | Amino acid and carboxylic acid transporters | 5 | 2.40E-02 |
| SPK, GMF, SM, IP, USF | 1.32 | GTP Signaling | 12 | 5.70E-03 |
| GPF | 1.26 | Spinal Cord Development | 4 | 1.90E-02 |
| GPF | 1.11 | Cell Response to Unfolded proteins. ER stress | 3 | 1.80E-02 |
| SPK, GBF | 1.03 | Apoptosis | 12 | 2.50E-02 |
| GMF | 1.02 | GTPase activator activity | 8 | 1.00E-02 |
| SPK, GPF, | 0.96 | Cell Cycle | 13 | 7.60E-02 |
| GPF | 0.88 | Programmed Cell Death | 6 | 2.20E-02 |
| GPF, SPK, KP, USF | 0.84 | Mitochondria | 32 | 3.90E-04 |
| SPK, IP, GCF, GPF, USF | 0.67 | Angiogenesis | 5 | 8.30E-02 |
Category Abbreviations: GOTERM_BP_FAT (GPF), SP_PIR_KEYWORDS (SPK), Kegg Pathway (KP), UP_SEQ_FEATURE (USF). GOTERM_MF_FAT (GMF). GOTERM_CC_FAT (GCF), SMART (SM), INTERPRO (IP)
The data represent the database source (category), enrichment score, average # of genes affected in category (average count) and p-values (Fisher Exact/EASE Score).
To gain a clear understanding of the most affected systems, a manual analysis for expression shifts of significance were conducted by viewing individual genes alphabetically (to view replicates or diverse probes for similar genes), by fold change, and fold change by expression dominance. Individual gene expression lists are compiled in Tables 2 (up-regulated by MPP+) and 3 (down-regulated by MPP+); listed by category, gene description, genebank accession #, %, change direction and p-Value. Category classifications were defined both by DAVID, literature and bio-informatics databanks for each gene. While the comprehensive data from whole genomic analysis are presented, we highlight major systems identified through expression profiling also collectively presented in Figure 7.
Table 2. Major Patterns in the Genome: Diferentially Expressed Genes in MPP+ Treated Neuroblastoma Cells after 24 Hours.
Differential whole genome expression in N2a cells treated with 500μM MPP+ vs. controls at 24 Hr.
| R [ Name, Genebank Accession #] | % Change | Direction | p-Value | ||
|---|---|---|---|---|---|
| Metabolism involving Carboxylic Acids, Transamination Reactions, Lipids, non-mitochondrial oxidative reduction and Insulin Signaling | cystathionase (cystathionine gamma-lyase) (Cth), [NM_145953] | MPP ▲ | 384% | <.001 | |
| tribbles homolog 3 (Drosophila) (Trib3), [NM_175093] | MPP ▲ | 339% | 0.004 | ||
| cytochrome b5 reductase 1 (Cyb5r1), [NM_028057] | MPP ▲ | 326% | 0.000 | ||
| 5,10-methylenetetrahydrofolate reductase (Mthfr), tv 2, [NM_010840] | MPP ▲ | 236% | 0.009 | ||
| glutamate oxaloacetate transaminase 1, soluble (Got1), [NM_010324] | MPP ▲ | 214% | 0.002 | ||
| diacylglycerol O-acyltransferase 2 (Dgat2), [NM_026384] | MPP ▲ | 213% | 0.012 | ||
| cysteine conjugate-beta lyase 1 (Ccbl1), [NM_172404] | MPP ▲ | 209% | 0.015 | ||
| pyridoxal (pyridoxine, vitamin B6) kinase (Pdxk), [NM_172134] | MPP ▲ | 206% | 0.039 | ||
| 2 | crystallin, zeta (quinone reductase)-like 1 (Cryzl1), [NM_133679] | MPP ▲ | 198% | 0.001 | |
| acetyl-Coenzyme A acyltransferase 1A (Acaa1a), [NM_130864] | MPP ▲ | 197% | 0.008 | ||
| glutamic pyruvate transaminase (alanine aminotransferase) 2 (Gpt2), [NM_173866] | MPP ▲ | 196% | 0.007 | ||
| ORM1-like 3 (S. cerevisiae) (Ormdl3), [NM_025661] | MPP ▲ | 191% | 0.041 | ||
| starch binding domain 1 (Stbd1), [NM_175096] | MPP ▲ | 190% | 0.034 | ||
| N-acetylglucosamine kinase (Nagk), tv 1, [NM_019542] | MPP ▲ | 189% | 0.017 | ||
| H2-K region expressed gene 6 (H2-Ke6), [NM_013543] | MPP ▲ | 187% | 0.023 | ||
| phosphoserine phosphatase (Psph), [NM_133900] | MPP ▲ | 180% | 0.001 | ||
| ELOVL family member 7, elongation of long chain fatty acids (yeast) (Elovl7), [NM_029001] | MPP ▲ | 175% | 0.028 | ||
| aspartate beta-hydroxylase domain containing 1 (Asphd1), [NM_001039645] | MPP ▲ | 174% | 0.026 | ||
| acyl-Coenzyme A binding domain containing 4 (Acbd4), tv 1, [NM_025988] | MPP ▲ | 172% | 0.003 | ||
| 3-hydroxy-3-methylglutaryl-Coenzyme A lyase (Hmgcl), [NM_008254] | MPP ▲ | 172% | 0.036 | ||
| ceroid-lipofuscinosis, neuronal 8 (Cln8), [NM_012000] | MPP ▲ | 171% | 0.013 | ||
| insulin-like growth factor binding protein 6 (Igfbp6), mRNA [NM_008344] | MPP ▲ | 261% | 0.001 | ||
| similar to 3-phosphoglycerate dehydrogenase (LOC637235), misc RNA [XR_032647] | MPP ▲ | 171% | 0.044 | ||
| ferredoxin reductase (Fdxr), nuclear gene encoding mitochondrial protein, [NM_007997] | MPP ▲ | 170% | 0.014 | ||
|
| |||||
| Angiogenesis | angiopoietin-like 6 (Angptl6), [NM_145154] | MPP ▲ | 291% | <.001 | |
| 2 | pre-B-cell leukemia transcription factor interacting protein 1 (Pbxip1), [NM_146131] | MPP ▲ | 262% | 0.028 | |
| fetuin beta (Fetub), tv 1, [NM_021564] | MPP ▲ | 255% | 0.006 | ||
| erythroid differentiation regulator 1 (Erdr1), [NM_133362] | MPP ▲ | 207% | 0.011 | ||
|
| |||||
| Transporters | solute carrier family 7 (cationic amino acid transporter, y+ system),3(Slc7a3), [NM_007515] | MPP ▲ | 285% | 0.034 | |
| solute carrier family 6 (neurotransmitter transporter, glycine), 9 (Slc6a9), [NM_008135] | MPP ▲ | 270% | 0.014 | ||
| solute carrier family 6 (neurotransmitter transporter, glycine), 9 (Slc6a9), [NM_008135] | MPP ▲ | 255% | 0.011 | ||
| purinergic receptor P2X, ligand-gated ion channel 4 (P2rx4), [NM_011026] | MPP ▲ | 235% | 0.045 | ||
| ATP-binding cassette, sub-family C (CFTR/MRP), member 5 (Abcc5), tv 1, [NM_013790] | MPP ▲ | 217% | 0.039 | ||
| recombination activating gene 1 activating protein 1 (Rag1ap1), [NM_009057] | MPP ▲ | 205% | 0.016 | ||
| solute carrier family 48 (heme transporter), member 1 (Slc48a1), [NM_026353] | MPP ▲ | 196% | 0.020 | ||
| transferrin receptor 2 (Trfr2), [NM_015799] | MPP ▲ | 191% | 0.009 | ||
| solute carrier family 7 (cationic amino acid transporter, y+ system), 5 (Slc7a5),[NM_011404] | MPP ▲ | 180% | 0.010 | ||
| solute carrier family 12, member 4 (Slc12a4), [NM_009195] | MPP ▲ | 177% | 0.013 | ||
|
| |||||
| Mitochondria | solute carrier family 25, member 33 (Slc25a33), [NM_027460] | MPP ▲ | 265% | 0.005 | |
| chloride intracellular channel 4 (mitochondrial) (Clic4), NE protein, [NM_013885] | MPP ▲ | 200% | 0.041 | ||
| lon peptidase 1, mitochondrial (Lonp1), NE protein, [NM_028782] | MPP ▲ | 196% | 0.000 | ||
| heat shock protein 1 (chaperonin 10) (Hspe1), [NM_008303] | MPP ▲ | 178% | 0.043 | ||
| NFU1 iron-sulfur cluster scaffold homolog (S. cerevisiae) NE tv 1, [NM_001170591] | MPP ▲ | 176% | 0.008 | ||
| Coenzyme A synthase (Coasy), NEM, [NM_027896] | MPP ▲ | 175% | 0.010 | ||
| ganglioside-induced differentiation-associated protein 1-like 1 (Gdap1l1) [NM_144891] | MPP ▲ | 140% | 0.002 | ||
|
| |||||
| Response to Stress or Injury | DNA-damage inducible transcript 3 (Ddit3), [NM_007837] | MPP ▲ | 414% | 0.006 | |
| protein phosphatase 1, regulatory (inhibitor) subunit 15A (Ppp1r15a), [NM_008654] | MPP ▲ | 345% | 0.014 | ||
| ChaC, cation transport regulator-like 1 (E. coli) (Chac1), [NM_026929] | MPP ▲ | 266% | 0.001 | ||
| ninjurin 1 (Ninj1), [NM_013610] | MPP ▲ | 247% | 0.033 | ||
| nuclear protein 1 (Nupr1), [NM_019738] | MPP ▲ | 225% | 0.004 | ||
|
| |||||
| G-Protein-Coupled Receptor Signaling | receptor accessory protein 6 (Reep6), [NM_139292] | MPP ▲ | 390% | 0.047 | |
| protein kinase N1 (Pkn1), [NM_177262] | MPP ▲ | 290% | 0.042 | ||
| GTP binding protein 2 (Gtpbp2), tv 1, [NM_019581] | MPP ▲ | 269% | 0.001 | ||
| arginine vasopressin-induced 1 (Avpi1), [NM_027106] | MPP ▲ | 246% | 0.024 | ||
| G protein-coupled rec associated sorting protein 2 (Gprasp2)tv 1[NM_001163015] | MPP ▲ | 228% | 0.020 | ||
| WD repeat domain 67 (Wdr67), tv 1, [NM_001081396] | MPP ▲ | 202% | 0.011 | ||
| rho/rac guanine nucleotide exchange factor (GEF) 2 (Arhgef2), [NM_008487] | MPP ▲ | 202% | 0.001 | ||
| folliculin interacting protein 1 (Fnip1), [NM_173753] | MPP ▲ | 198% | 0.029 | ||
| ras homolog gene family, member U (Rhou), [NM_133955] | MPP ▲ | 197% | 0.031 | ||
| syntaxin 4A (placental) (Stx4a), [NM_009294] | MPP ▲ | 194% | 0.011 | ||
| prostaglandin I receptor (IP) (Ptgir), [NM_008967] | MPP ▲ | 185% | 0.022 | ||
| pleckstrin and Sec7 domain containing (Psd), [NM_028627] | MPP ▲ | 184% | 0.013 | ||
| G protein-coupled receptor 19 (Gpr19), tv 1, [NM_001167693] | MPP ▲ | 182% | 0.024 | ||
| TBC1 domain family, member 9B (Tbc1d9b), [NM_029745] | MPP ▲ | 181% | 0.005 | ||
| folliculin (Flcn), [NM_146018] | MPP ▲ | 180% | 0.025 | ||
| ArfGAP with RhoGAP, ankyrin repeat and PH 3 (Arap3), [NM_139206] | MPP ▲ | 177% | 0.003 | ||
| Mus musculus thyroid hormone receptor interactor 10 (Trip10), [NM_134125] | MPP ▲ | 174% | 0.033 | ||
| membrane associated guanylate kinase, WW and PDZ (Magi1)tv 3[NM_001029850] | MPP ▲ | 174% | 0.041 | ||
| syntaxin 4A (placental) (Stx4a), [NM_009294] | MPP ▲ | 174% | 0.003 | ||
| ral guanine nucleotide dissociation stimulator-like 3 (Rgl3), [NM_023622] | MPP ▲ | 174% | 0.004 | ||
| T-cell lymphoma invasion and metastasis 2 (Tiam2), tv 1, [NM_011878] | MPP ▲ | 173% | 0.015 | ||
| resistance to inhibitors of cholinesterase 8 homolog (C. elegans) [NM_053194] | MPP ▲ | 172% | 0.012 | ||
| Ras-like without CAAX 1 (Rit1), tv 1, [NM_009069] | MPP ▲ | 171% | 0.035 | ||
| Rho family GTPase 1 (Rnd1), [NM_172612] | MPP ▲ | 171% | 0.010 | ||
| Rho, GDP dissociation inhibitor (GDI) beta (Arhgdib), [NM_007486] | MPP ▲ | 170% | 0.010 | ||
|
| |||||
| Signaling | phospholipase C, beta 3 (Plcb3), [NM_008874] | MPP ▲ | 172% | 0.010 | |
| lymphocyte protein tyrosine kinase (Lck), tv 2, [NM_010693] | MPP ▲ | 225% | 0.002 | ||
| protein kinase C, delta (Prkcd), [NM_011103] | MPP ▲ | 217% | 0.029 | ||
| phospholipase C, delta 3 (Plcd3), [NM_152813] | MPP ▲ | 200% | 0.015 | ||
NE = Nuclear gene encoding mitochondrial protein
The data represents genes found to be up-regulated by MPP+ expressed by functional category, gene identification, fold change and significance at p < 0.05. R=gene probe replicates in hybridization array.
Figure 7.


Summary of major MPP+ evoked changes in the transcriptome of N2a cells at 24 hours.
In order to assess protein expression levels, we looked at three major genes of interest including LDH, pyruvate kinase and FAM162a (Figure 4a–c). The data shows gene expression [left] and protein expression [right] with a listing [legend in figure] of various or duplicate probes in the array for the same gene. Western blots for protein quantification are shown in Figure 5a for the three proteins. Figure 5b shows additional gene/protein comparisons for elements of interest. The data in Figure 5 show consistency between microarray genomic expression and protein expression data.
Figure 4.


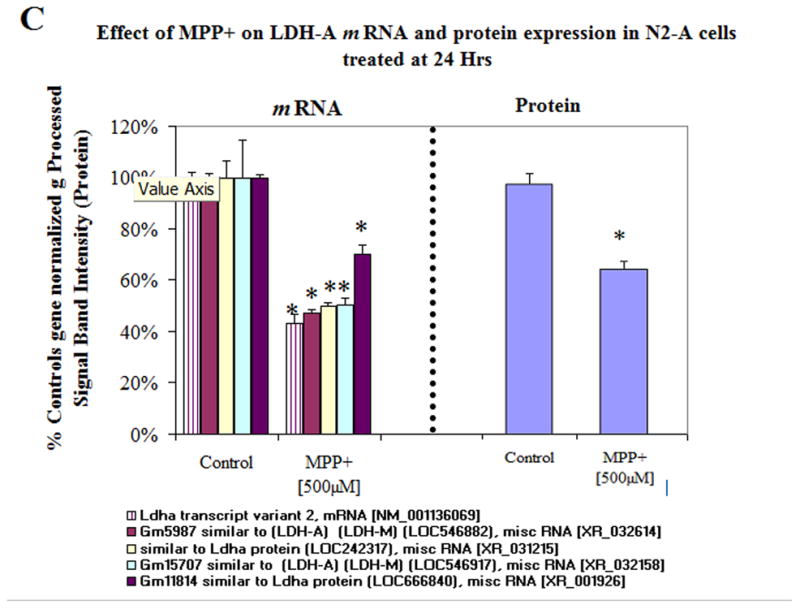
Figure 4A. Variable mRNA and protein expression profiles for family with sequence similarity 162, member A (Fam162a) in N2a cells treated ± 500μM MPP+ at 24 Hr. The data represent mRNA gene (normalized g processed signal) % Ctrl (Left) and protein band intensity determined from Western Blot (Right). The data are presented as the Mean ± S.E.M., n=3. Significance of difference between the control and treatment groups were determined by a students t-test, * p<0.05.
Figure 4B. Variable mRNA and protein expression profiles for pyruvate kinase in N2a cells treated ± 500μM MPP+ at 24 Hr. The data represent mRNA gene normalized g Processed Signal % Ctrl (Left), diverse hybridization probes for similar genes (legend below) and protein band intensity determined from Western Blot (Right). The data are presented as the Mean ± S.E.M., n=3. Significance of difference between the control and treatment groups were determined by a students t-test, * p<0.05.
Figure 4C. Variable mRNA and protein expression profiles for LDH-A in N2a cells treated ± 500μM MPP+ at 24 Hr. The data represent mRNA (normalized g processed signal) % Ctrl (Left), diverse hybridization probes for similar genes (legend below) and protein band intensity determined from Western Blot (Right). The data are presented as the Mean ± S.E.M., n=3. Significance of difference between the control and treatment groups were determined by a students t-test, * p<0.05.
Figure 5.

Figure 5A. Relative protein expression for basic proteins of interest in N2a ± 500μM MPP+ at 12–24 Hours.
Figure 5B. Variable mRNA and expression profiles for proteins of interest in N2a cells treated ± MPP+ [500μM ] at 24 Hr. The data represent mRNA (normalized g processed signal) % Ctrl (Left) presented as the Mean ± S.E.M., n=3. Significance of difference between the control and treatment groups were determined by a students t-test * p<0.05. Corresponding western blots displaying protein expression (Right).
One of the major findings in this paper, revolve around the manner by which N2a cells adapted to the loss of mitochondrial function. The findings in this paper, support greater expression and elevation of molecular components involved with transamination reactions and carboxylic acid metabolism, in particular for pyruvate-alanine transaminase 2 (Gpt2) and glutamate oxaloacetate transaminase 1 (GOT1). These findings corroborate our previous work in which we investigated differential metabolite patterns emerging from treatment of N2a cells with MPP+ (Mazzio et al., 2010a), which were in alignment with bulk of recent literature showing metabolic patterns within tumor cells involve high conversion rates of [1-(13)C]glutamate / pyruvate to alpha-ketoglutarate calatalyzed by alanine transaminase (Gallagher et al., 2011) in addition to high conversion of [1-(13)C]pyruvate to [1-(13)C] alanine (Darpolor et al., 2011, Hu et al., 2011).
Unexpectedly, a rise in these systems paralleled reduction for almost every gene in glycolysis at 24 hour of MPP+ exposure. These data were not anticipated, and bring forth a number of questions as to the source of reducing equivalents and high-energy phosphates that fuel the largest component of anaerobic metabolism. Moreover, there is now question as to the source of lactic acid, which was clearly elevated by MPP+. In the next study, we examined if any class of metabolic substrates or intermediates are capable of producing lactate acid in glucose-free media by N2a cells (Figure 6). The findings suggest that in addition to glucose, metabolites such as oxaloacetic acid, succinic acid and a-ketoglutarate also contribute to the lactate pool. These findings show that anaerobic metabolism involves a number of pathways that extend beyond glycolysis and necessitate further investigation as the to survival pathways by tumor cells responding to lack of functional mitochondria under normoxic conditions.
Figure 6.

Conversion rate of various metabolic substrates (10mM) to lactic acid in N2a cells in the absence of glucose at 24 Hrs. The data represent Lactic Acid Produced (μM/mg protein) and are presented as the Mean ± S.E.M., n=4. N.D. (not detected).
4.0 Discussion
The data from this study provide information on molecular changes incurred by MPP+ related to loss of OXPHOS and compensatory metabolic survival processes through substrate level phosphorylation. While the comprehensive data from whole genomic analysis are presented, we highlight major systems identified through expression profiling.
4.1 Molecular Targets within the Mitochondria
A widely accepted hypothesis describing the toxic effects of MPP+, involve its targeting the mitochondria, initiating a loss of respiration, OXPHOS (Absi et al., 2000) and ATP production (Mizuno et al., 1987, Storch et al., 1999), events which occur through complex I inhibition in a manner that mimics PD pathology. (Ebadi et al., 2001, Kotake and Ohta, 2003a) While many articles preface a main hypothesis with a brief mention regarding MPP+ as a complex I inhibitor, there are very few studies which have explored the molecular patterns with clarity as to the specifics of MPP+ on genetic, proteomic or electron transport chain (ETC) component enzyme kinetic effects. Many of the earlier studies examining the effects of MPP+ involved assays that monitored the oxidation of NADH/NAD+-linked substrates in the TCA cycle on intact mitochondria, demonstrating significant losses to state 3 and 4 respiration; events parallel to the loss of complex I. (Mizuno, 1989, Suzuki et al., 1990) Since then, a number of studies, including our work on intact mitochondria, demonstrate that MPP+ is not only an inhibitor of complex I, but also cytochrome oxidase (complex IV), with the latter being parallel to loss of cell respiration(Mazzio and Soliman, 2004a, Steyn et al., 2005, Sundar Boyalla et al., 2011). If complex I was the only molecular target of MPP+, then fueling energy equivalents through complex II could overcome the loss of OXPHOS, however results from our lab show that not to be the case, suggesting overriding damages occur downstream to complex I. While there are a number of studies investigating impact to the function of intact mitochondria, there are few studies which look at the effects of MPP+ at the transcript level, although some have shown mitigated effects on complex I, III and IV subunits (Zhu et al., 2012) and subunit 4 of complex I. (Conn et al., 2001) In this study, we show that MPP+ adversely affects the transcriptome predominantly at complex I with losses in the NADH dehydrogenase 5 gene, NADH dehydrogenase 4 gene and NADH dehydrogenase (ub) flavoprotein 3. Therefore, the detrimental effect of MPP+ on the mitochondria may involve not only functional loss at complex I and IV, but also losses of nuclear genes that encode for mitochondrial subunits required to sustain OXHPHOS function. These findings suggest that MPP+ adversely impacts the mitochondria through multiple processes, which are likely to be irreversible.
In this study, the loss of complex I transcripts also corresponded to the elevation of ganglioside-induced differentiation-associated protein 1-like 1 (Gdap1) which encodes a protein anchored to the mitochondrial outer membrane (Pedrola et al., 2008), which if present would disrupt mitochondrial networks through fission. (Pedrola et al., 2005) Gdap1 also precipitates functional loss to complex I (Cassereau et al., 2009) and plays a role in axonal / de-myelinating neuropathies (Bouhouche et al., 2007). Proteins such as Gdap1 involved with fission oppose the process of mitochondrial fusion, which would otherwise confer greater collective mitochondrial stability and therefore nutrient accessibility to optimize bioenergetics. (Okamoto and Shaw, 2005) Gdap1 is present in neurons of the spinal cord, olfactory bulb and cortical pyramidal neurons (Pedrola, Espert, 2008) and while there is little to no information of this gene being relevant to PD, it is a correlate to other neurodegenerative diseases involving mitochondrial insufficiency (Cassereau et al., 2011) such as Charcot-Marie-Tooth disease (CMT). (Niemann et al., 2009) It is becoming more apparent that mitochondrial related neurodegenerative disorders such as PD, involve greater mitochondrial fission processes. It is now believed that gene mutations in PINK1, oxidative stress, excitotoxicity or even high levels of nitric oxide associated with inflammation are associated with fission and/or inhibited fusion, which would hamper mitochondrial biogenesis. (Deng et al., 2008, Nakamura and Lipton, 2010)
Damage to the mitochondria can indirectly initiate opening of the mitochondrial permeability transition pore and subsequent triggering of apoptotic cascades. The experimental design of this study allowed for determination of MPP+ molecular targets on OXPHOS, as well as survival processes associated with anaerobic metabolism fueled in high glucose media to prevent cell death. In this aspect, transcriptional losses in OXPHOS genes corresponded to significant rise in protective mitochondrial anti-apoptotic related processes. One of the few notable changes included MPP+ attenuated expression of pro-apoptotic Bnip3 (Bcl-2/adenovirus E1B 19-kDa-interacting protein 3) which at high levels is known to trigger apoptosis through elevating Bax / Bak (Wang et al., 2011a, Wang et al., 2011b), caspase-3 activity and poly (ADP-ribose) polymerase. (Naldini et al., 2011) High glucose concentrations are known to reduce BNIP3, which result in greater cell survival and attenuation of apoptosis / mitochondria autophagy. (Rikka et al., 2011, Thomas et al., 2011, Xu et al., 2011, Zhao et al., 2011a) The data also show that MPP+ evoked elevation of Hsp10 chaperone 10 which is known to induce Bcl-xl, Bcl-2 expression, block caspase-3, poly (ADP-ribose) polymerase, Bax (Shan et al., 2003) and apoptosis. (Ling et al., 2011, Mazzio and Soliman, 2003a) Similarly, MPP+ mediated loss of a gene / protein called FAM162a. Although little is known about this protein, FAM162a is believed to be a HIF-1 alpha-responsive pro-apoptotic molecule also known as human growth and transformation-dependent protein (HGTD-P). FAM162a transmits hypoxic signals to the mitochondria and when over-expressed causes cell death via mitochondrial apoptosis. (Kim et al., 2006, Lee et al., 2004) In neurological models, high levels of FAM162a are associated with activation of caspase-3, translocation of apoptosis inducing factor (AIF) to the nucleus, chromatin condensation and programmed cell death in association with hypoxia ischemia induced brain damage (Qu et al., 2009). This is very interesting, because the changes we see in this particular model using neuroblastoma are actually opposite to hypoxia in many aspects (this being under normal O2 tension) where cells were cultured at 5% CO2 /atm. Other protective factors in the presence of MPP+ include elevation of lon peptidase 1 which would strengthen mitochondria by enhancing degradation of damaged proteins by ROS or other types of stressors in the matrix. (Bayot et al., 2008, Fishovitz et al., 2011, Pinti et al., 2011, Wagatsuma et al., 2011) In summary, the collective general changes in the transcriptome which impacted the mitochondria are associated with either 1) primary loss of OXPHOS genes or 2) rise in systems that attenuate mitochondrial induced apoptosis.
4.2 Energy Metabolism
Changes in metabolic related systems by MPP+ included: elevation in G protein signaling concomitant to a rise in transamination / vitamin B6 and carboxylic acid metabolism. MPP+ triggered a rise in glutamate-pyruvate transaminase (GPT) and glutamate-oxalacetate transaminase (GOT) gene expression, which were expressed in relatively high abundance amongst the entire transcriptome. Elevation of GOT/GPT are typically observed in association with liver / kidney injuries, abnormal glucose metabolism (Ruckert et al., 2011, Song et al., 2011, Zhao et al., 2011b) and ischemic stroke, serving as a means to remove excessive blood/ cerebral glutamate. (Rink et al., 2011) Higher levels of GOT/GPT lead to lower levels of glutamate, and thereby diminished excitotoxicity resulting in reduced infarct size, edema and neurological damage. (Campos et al., 2011a, Campos et al., 2011b, Campos et al., 2011c)
In malignant cells, high levels of GOT / GPT which are believed to play an important role in energy metabolism evidenced by baseline conversion rate of [1-(13)C]pyruvate to [1-(13)C]alanine (Darpolor, Yen, 2011, Hu, Balakrishnan, 2011) and the conversion of [1-(13)C] glutamate and pyruvate to alpha-ketoglutarate calatalyzed by L-alanine transaminase. (Gallagher, Kettunen, 2011) Consistent with these findings, the present study shows that MPP+ triggered an inordinate rise in pyridoxal metabolism (e.g. pyridoxal kinase), which aids in catalytic co-requirement for transamination and other processes that may play a role in anaerobic energy SLP such as cystathionase, which itself is subject to glucagon signaling. (Martin et al., 2007) Cystathionase can catalyze the conversion of (1) cystathionine into cysteine + α-ketobutyrate (2) cysteine / cystine in to pyruvic acid + NH3 and (3) provide a direct route for L-alanine to pyruvic acid conversion. (Ogasawara et al., 2002) Elevated cystathionase expression also serves as a negative feed back loop on glycolysis by inactivation of rate limiting enzymes including phosphofructokinase (PFK) and pyruvate kinase (PK). (Ogasawara et al., 1997) This may possibly contribute to a conservation of glucose. Another contributor to the pyruvate pool could be the pyridoxal 5′-phosphate-requiring enzyme cysteine-S-conjugate beta-lyase; which also converts carboxylic acids into pyruvate + NH3. (Cooper et al., 2011) A rise in many of these similar type systems in the transcriptome by MPP+, suggests there are other metabolic routes to production of pyruvic acid, other than phospho(enol)pyruvate in glycolysis. Previous findings from our work demonstrated metabolic anomalies in neuroblastoma cells exposed to MPP+ (Mazzio, Smith, 2010a) to involve excessive production of L-alanine, likely originating from a bypass route from glycolysis to the pyruvate–alanine transamination cycle. These findings have also been corroborated by a number of other research groups, where it is now believed that cells of tumor phenotype convert a large percentage of hyperpolarized (13)C-labeled pyruvate to both lactate and L-alanine thereby creating a plausible rationale for a three-dimensional (13) C spectroscopic imaging diagnostic platform for tumors. (Albers et al., 2008) These findings are in alignment with studies showing that tumor cells with defective mitochondria survive by reductive carboxylation reactions occurring through TCA cycle driven through transamination reactions (Mullen et al., 2012) also occurring under hypoxia.(Le et al., 2012) However, future research will be required to determine if these systems are attribute to the tumor phenotype of neuroblastoma or similar to what is found in normal respiring brain tissue.
One of the major shifts noted in the transcriptome, was MPP+ induction of G signaling transduction components of which 12 genes were Id’d through DAVID analysis and 25+ via manual analysis (Table 2). The effects of MPP+ on G protein signaling are logical given its major role in triggering switch from anabolic to catabolic cellular metabolic processes under nutrient challenged conditions. Typically, low glucose or nutrient deprivation initiates systemic rise in glucagon or epinephrine, both of which contribute to increased energy through augmenting catabolic breakdown of alternative energy fuels through G signal transduction. Both bind to G protein-coupled receptors which activates adenylyl cyclase prompting a rise in cAMP/ activation of protein kinase A and subsequent phosphorylation of targets leading to inactivation of glycogen synthase, the activation of glycogen phosphorylase (a pyridoxal phosphate dependent enzyme) which breaks down glycogen and elevated phosphorylation of hormone-sensitive lipases (which breaks down fats). G protein signaling can prompt inositol 1,4,5-triphosphate / diacyl glycerol controlled pathways which elevate cytosolic concentrations of calcium, which can also lead to activated glycolygen phosphorylase. These effects could serve in the signaling of catabolic processes, when ATP levels are low from obstruction of OXPHOS.
The findings in this study also show a number of nutrient signaling systems altered by MPP+, including Angptl6 / angiopoietin-related growth factor (Angptl6/AGF) which has capacity to increase insulin receptor substrate 1 phosphorylation and enhance glucose uptake. (Hato et al., 2008) MPP+ also induced a 339% elevation in a largely expressed gene called tribbles homolog 3 (Trib3), a nutrient sensor involved with insulin resistance and hyperglycemia in diabetes. (Liu et al., 2010) Elevated Trib3 is associated with poor prognosis of breast cancer patients, is induced during hypoxia / nutrient deprivation and its silencing results in a dose-dependent increase of hypoxic mediated cell death. (Wennemers et al., 2011) Trib3 enhances the transcriptional activity of SMAD3 and promotes perpetuation of TGF-beta-SMAD3 signaling, partially involved with migration and invasion of tumor cells. (Hua et al., 2011) Again, future research will be required to evaluate if similar glucose sensors or nutrient hormones are involved with overcoming MPP+ mediated mitochondrial damage in normal tissue by optimizing metabolic use of glucose.
4.3 Effects on Tumor Related Processes
MPP+ had substantial effects on a number of genes that accommodate invasive metastasis. Three of the highest expressed genes in terms of dominance from the data of this study (with significant up-regulation) were identified as activating transcription factor 4 (Atf4) mRNA and two genes that are controlled by Atf4 which include 1) nuclear protein 1 (Nupr1) and 2) CHAC1 (cation transport regulator-like protein 1). Nuclear protein 1 is highly up-regulated in response to various stressors (Jin et al., 2009), playing a role in metastasis, progression of cancer and chemo-resistance in various types of malignancies. (Chowdhury et al., 2009) Nupr1 forms a complex with p53 and up-regulates p21, Bcl-x (L), allowing for cells to progress through cell cycle in presence of chemotherapy drugs. (Clark et al., 2008) Similarly, the elevation of CHAC1 results in higher unfolded protein response pathway ATF4-ATF3-CHOP (Mungrue et al., 2009) involved with propagating hormone receptor negative breast and advanced-staged ovarian cancer. (Goebel et al., 2012) Another stress related protein, ninjurin-1 was elevated by MPP+, which is not only associated with tumor progression (Mhawech-Fauceglia et al., 2009) but neurological injury (Kubo et al., 2002) (Araki and Milbrandt, 1996) being present in blood brain barrier epithelial cells and blood monocytes contributing to neuro-inflammatory lesions within the CNS.(Ifergan et al., 2011)
While it is not possible to discuss all aspects of the data presented in Tables 2 and 3, the findings in this paper lead to several general conclusions. MPP+ adversely affects the transcriptome primarily at complex I of the ETC, and in combination to kinetic inactivation of complex I and IV leads to complete loss of mitochondrial respiration. (Mazzio and Soliman, 2004a) In response to ETC damage, several survival processes arise. Survival processes that encompass optimal energy homeostasis in the presence of MPP+ involve elevation of carboxylic acid metabolism GOT1 / GPT2 transamination reactions, cysteine conjugate-beta lyase 1, cystathionase, cytochrome b5r1 and ferridoxin reductase which could contribute to NAD/NADH+ recycling REDOX pathways that may drive production of anaerobic high energy phosphates. MPP+ mediated rise in protective elements within the mitochondrial compartment impart greater resistance to apoptosis including Bnip3, Fam162a, Lon1 and Hspe1. The findings as presented herein are broad but they underlie specific major classes or groups of system affected by MPP+ on the transcriptome in neuroblastoma.
Table 3. Major Patterns in the Genome: Diferentially Repressed Genes in MPP+ Treated Neuroblastoma Cells after 24 Hours.
Differential whole genome expression in N2a cells treated with 500μM MPP+ vs. controls at 24 Hr.
| R [ Name, Genebank Accession #] | Direction | % Change | p-Value | ||
|---|---|---|---|---|---|
| Glycolysis | aldolase C, fructose-bisphosphate (Aldoc), [NM_009657] | MPP ▼ | 552% | 0.036 | |
| phosphofructokinase, liver, B-type (Pfkl), [NM_008826] | MPP ▼ | 263% | <.001 | ||
| phosphoglycerate kinase 1 (Pgk1), [NM_008828] | MPP ▼ | 237% | 0.023 | ||
| sim to phosphoglycerate mutase (LOC432584), [XR_030881] | MPP ▼ | 223% | <.001 | ||
| sim to Phosphoglycerate mutase 1 (brain) (LOC665009), [XR_032298] | MPP ▼ | 219% | <.001 | ||
| lactate dehydrogenase A (Ldha), transcript variant 2, [NM_001136069] | MPP ▼ | 218% | <.001 | ||
| phosphoglycerate mutase 1 (Pgam1), [NM_023418] | MPP ▼ | 217% | 0.009 | ||
| pyruvate kinase, muscle (Pkm2), [NM_011099] | MPP ▼ | 213% | 0.002 | ||
| 2 | triosephosphate isomerase 1 (Tpi1), [NM_009415] | MPP ▼ | 210% | 0.005 | |
| sim to Eno1 protein (LOC667152), [XR_033308] | MPP ▼ | 209% | 0.003 | ||
| sim to fructose-1,6-bisphosphate aldolase A (LOC383862), [XR_035510] | MPP ▼ | 208% | 0.010 | ||
| sim to Aldolase 1, A (LOC665254), [XR_031611] | MPP ▼ | 204% | 0.009 | ||
| 5 | sim to lactate dehydrogenase 1, A chain (LOC666237), [XR_031340] | MPP ▼ | 200% | 0.002 | |
| lactate dehydrogenase A (Ldha), transcript variant 2, [NM_001136069] | MPP ▼ | 199% | 0.002 | ||
| sim to Pyruvate kinase, muscle (LOC241572), [XR_005019] | MPP ▼ | 197% | 0.005 | ||
| sim to Eno1 protein (LOC676933), [XR_004985] | MPP ▼ | 194% | 0.003 | ||
| sim to M2-type pyruvate kinase (LOC100039264), [XR_030770] | MPP ▼ | 192% | 0.004 | ||
| 7 | glyceraldehyde-3-phosphate dehydrogenase (Gapdh), [NM_008084] | MPP ▼ | 191% | 0.010 | |
| sim to Pyruvate kinase, muscle (LOC631301), [XR_031910] | MPP ▼ | 185% | 0.004 | ||
| sim to Ldha protein (LOC242317), [XR_031215] | MPP ▼ | 184% | 0.006 | ||
| sim to aldolase 1, A isoform (LOC667696), [XR_032442] | MPP ▼ | 184% | 0.018 | ||
| 2 | sim to L-lactate dehydrogenase A chain (LDH-A, M)(LOC546917),[XR_032158] | MPP ▼ | 179% | 0.034 | |
| 15 | sim to Glyceraldehyde-3-phosphate dehydrogenase (LOC636859), [XR_032786] | MPP ▼ | 161% | 0.008 | |
| 2 | glucose phosphate isomerase 1 (Gpi1), [NM_008155] | MPP ▼ | 148% | 0.012 | |
| enolase 1, alpha non-neuron (Eno1), [NM_023119] | MPP ▼ | 146% | 0.013 | ||
|
| |||||
| Mitochondria | ME NADH dehydrogenase 5 Gene [*102496] [MUST00000082418] | MPP ▼ | 472% | 0.015 | |
| 3 | BCL2/adenovirus E1B interacting protein 3 (Bnip3), NE, [NM_009760] | MPP ▼ | 443% | 0.010 | |
| ME cytochrome b Gene [*102501] [MUST00000082421] | MPP ▼ | 310% | 0.023 | ||
| cytochrome c oxidase, subunit VIIa 1 (Cox7a1), NE, [NM_009944] | MPP ▼ | 282% | 0.031 | ||
| 10 | family with sequence similarity 162, member A (Fam162a), [NM_027342] | MPP ▼ | 245% | 0.013 | |
| ME NADH dehydrogenase 4 Gene [* 102498] [MUST00000082414] | MPP ▼ | 187% | 0.035 | ||
| NADH dehydrogenase (ub) flavoprotein 3 (Ndufv3), NE, tv1,[NM_030087] | MPP ▼ | 147% | 0.019 | ||
|
| |||||
| Lipids | 3-hydroxybutyratedehydrogenase, type2(Bdh2), tv1, [NM_001172055] | MPP ▼ | 238% | 0.040 | |
| 2 | PTPRF interacting protein, binding protein 2 (liprin beta 2)tv1[NM_008905] | MPP ▼ | 210% | 0.018 | |
| 7-dehydrocholesterol reductase(Dhcr7), [NM_007856] | MPP ▼ | 169% | 0.025 | ||
| fatty acid synthase(Fasn), [NM_007988] | MPP ▼ | 158% | 0.013 | ||
| stearoyl-Coenzyme A desaturase2(Scd2), [NM_009128] | MPP ▼ | 152% | 0.010 | ||
|
| |||||
| Non coding mRNA | 4 | predicted pseudogene 6981(Gm6981), non-codingRNA[NR_023357] | MPP ▼ | 174% | 0.007 |
| 3 | predicted gene 8709(Gm8709), non-codingRNA[NR_033633] | MPP ▼ | 181% | 0.009 | |
| predicted gene 13315(Gm13315), non-codingRNA[NR_028497] | MPP ▼ | 210% | 0.001 | ||
Source:MGI:Symbol:Acc:MGI:
NE = Nuclear gene encoding mitochondrial protein
ME - Mitochondrial encoded
The data represent genes down-regulated by MPP+ and are expressed by functional category, gene identification, fold change and significance at p < 0.05. R=gene probe replicates in hybridization array.
Highlights.
MPP+ administration to N-2A cells affected Complex I transcriptome.
MPP+ caused an elevation of G protein signaling and anaerobic metabolic systems.
MPP+ caused an elevation of carboxylic acid metabolism GOT1 / GPT2 transamination.
MPP+ resulted in increase in cytochrome b5r1 and ferridoxin reductase.
MPP+ caused an increase in protective mitochondrial anti-apoptotic processes.
Acknowledgments
This project was supported by the National Center for Research Resources NIH NCRR RCMI program (G12RR 03020) and the National Institute of Minority Health and Health Disparities, NIH (8G12MD007582-28.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6.0 References
- Absi E, Ayala A, Machado A, Parrado J. Protective effect of melatonin against the 1-methyl-4-phenylpyridinium-induced inhibition of complex I of the mitochondrial respiratory chain. J Pineal Res. 2000;29:40–7. doi: 10.1034/j.1600-079x.2000.290106.x. [DOI] [PubMed] [Google Scholar]
- Akashi S, Kimura T, Takeuchi T, Kuramochi K, Kobayashi S, Sugawara F, et al. Neoechinulin a impedes the progression of rotenone-induced cytotoxicity in PC12 cells. Biol Pharm Bull. 2011;34:243–8. doi: 10.1248/bpb.34.243. [DOI] [PubMed] [Google Scholar]
- Albers MJ, Bok R, Chen AP, Cunningham CH, Zierhut ML, Zhang VY, et al. Hyperpolarized 13C lactate, pyruvate, and alanine: noninvasive biomarkers for prostate cancer detection and grading. Cancer Res. 2008;68:8607–15. doi: 10.1158/0008-5472.CAN-08-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Milbrandt J. Ninjurin, a novel adhesion molecule, is induced by nerve injury and promotes axonal growth. Neuron. 1996;17:353–61. doi: 10.1016/s0896-6273(00)80166-x. [DOI] [PubMed] [Google Scholar]
- Asgari S, Vespa P, Bergsneider M, Hu X. Lack of consistent intracranial pressure pulse morphological changes during episodes of microdialysis lactate/pyruvate ratio increase. Physiol Meas. 2011;32:1639–51. doi: 10.1088/0967-3334/32/10/011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayot A, Basse N, Lee I, Gareil M, Pirotte B, Bulteau AL, et al. Towards the control of intracellular protein turnover: mitochondrial Lon protease inhibitors versus proteasome inhibitors. Biochimie. 2008;90:260–9. doi: 10.1016/j.biochi.2007.10.010. [DOI] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE. The genetic epidemiology of neurodegenerative disease. J Clin Invest. 2005;115:1449–57. doi: 10.1172/JCI24761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhouche A, Birouk N, Benomar A, Ouazzani R, Chkili T, Yahyaoui M. A novel GDAP1 mutation P78L responsible for CMT4A disease in three Moroccan families. Can J Neurol Sci. 2007;34:421–6. doi: 10.1017/s0317167100007290. [DOI] [PubMed] [Google Scholar]
- Burch D, Sheerin F. Parkinson’s disease. Lancet. 2005;365:622–7. doi: 10.1016/S0140-6736(05)17915-X. [DOI] [PubMed] [Google Scholar]
- Campos F, Rodriguez-Yanez M, Castellanos M, Arias S, Perez-Mato M, Sobrino T, et al. Blood levels of glutamate oxaloacetate transaminase are more strongly associated with good outcome in acute ischaemic stroke than glutamate pyruvate transaminase levels. Clin Sci (Lond) 2011a;121:11–7. doi: 10.1042/CS20100427. [DOI] [PubMed] [Google Scholar]
- Campos F, Sobrino T, Ramos-Cabrer P, Argibay B, Agulla J, Perez-Mato M, et al. Neuroprotection by glutamate oxaloacetate transaminase in ischemic stroke: an experimental study. J Cereb Blood Flow Metab. 2011b;31:1378–86. doi: 10.1038/jcbfm.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos F, Sobrino T, Ramos-Cabrer P, Castellanos M, Blanco M, Rodriguez-Yanez M, et al. High blood glutamate oxaloacetate transaminase levels are associated with good functional outcome in acute ischemic stroke. J Cereb Blood Flow Metab. 2011c;31:1387–93. doi: 10.1038/jcbfm.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassereau J, Chevrollier A, Gueguen N, Desquiret V, Verny C, Nicolas G, et al. Mitochondrial dysfunction and pathophysiology of Charcot-Marie-Tooth disease involving GDAP1 mutations. Exp Neurol. 2011;227:31–41. doi: 10.1016/j.expneurol.2010.09.006. [DOI] [PubMed] [Google Scholar]
- Cassereau J, Chevrollier A, Gueguen N, Malinge MC, Letournel F, Nicolas G, et al. Mitochondrial complex I deficiency in GDAP1-related autosomal dominant Charcot-Marie-Tooth disease (CMT2K) Neurogenetics. 2009;10:145–50. doi: 10.1007/s10048-008-0166-9. [DOI] [PubMed] [Google Scholar]
- Chalmers-Redman RM, Fraser AD, Carlile GW, Pong A, Tatton WG. Glucose protection from MPP+-induced apoptosis depends on mitochondrial membrane potential and ATP synthase. Biochem Biophys Res Commun. 1999;257:440–7. doi: 10.1006/bbrc.1999.0487. [DOI] [PubMed] [Google Scholar]
- Chen J, Jin H, Zhang Y, Liang Q, Liao H, Guo Z, et al. MRS and diffusion tensor image in mild traumatic brain injuries. Asian Pacific journal of tropical medicine. 2012;5:67–70. doi: 10.1016/S1995-7645(11)60248-4. [DOI] [PubMed] [Google Scholar]
- Chowdhury UR, Samant RS, Fodstad O, Shevde LA. Emerging role of nuclear protein 1 (NUPR1) in cancer biology. Cancer Metastasis Rev. 2009;28:225–32. doi: 10.1007/s10555-009-9183-x. [DOI] [PubMed] [Google Scholar]
- Clark DW, Mitra A, Fillmore RA, Jiang WG, Samant RS, Fodstad O, et al. NUPR1 interacts with p53, transcriptionally regulates p21 and rescues breast epithelial cells from doxorubicin-induced genotoxic stress. Current cancer drug targets. 2008;8:421–30. doi: 10.2174/156800908785133196. [DOI] [PubMed] [Google Scholar]
- Conn KJ, Ullman MD, Eisenhauer PB, Fine RE, Wells JM. Decreased expression of the NADH:ubiquinone oxidoreductase (complex I) subunit 4 in 1-methyl-4-phenylpyridinium -treated human neuroblastoma SH-SY5Y cells. Neurosci Lett. 2001;306:145–8. doi: 10.1016/s0304-3940(01)01888-2. [DOI] [PubMed] [Google Scholar]
- Cooper AJ, Krasnikov BF, Niatsetskaya ZV, Pinto JT, Callery PS, Villar MT, et al. Cysteine S-conjugate beta-lyases: important roles in the metabolism of naturally occurring sulfur and selenium-containing compounds, xenobiotics and anticancer agents. Amino Acids. 2011;41:7–27. doi: 10.1007/s00726-010-0552-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darpolor MM, Yen YF, Chua MS, Xing L, Clarke-Katzenberg RH, Shi W, et al. In vivo MRSI of hyperpolarized [1-(13)C]pyruvate metabolism in rat hepatocellular carcinoma. NMR Biomed. 2011;24:506–13. doi: 10.1002/nbm.1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Zompo M, Piccardi MP, Ruiu S, Quartu M, Gessa GL, Vaccari A. Selective MPP+ uptake into synaptic dopamine vesicles: possible involvement in MPTP neurotoxicity. Br J Pharmacol. 1993;109:411–4. doi: 10.1111/j.1476-5381.1993.tb13584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Dodson MW, Huang H, Guo M. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A. 2008;105:14503–8. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Monte D, Sandy MS, Blank L, Smith MT. Fructose prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced ATP depletion and toxicity in isolated hepatocytes. Biochem Biophys Res Commun. 1988;153:734–40. doi: 10.1016/s0006-291x(88)81156-2. [DOI] [PubMed] [Google Scholar]
- Ebadi M, Govitrapong P, Sharma S, Muralikrishnan D, Shavali S, Pellett L, et al. Ubiquinone (coenzyme q10) and mitochondria in oxidative stress of parkinson’s disease. Biol Signals Recept. 2001;10:224–53. doi: 10.1159/000046889. [DOI] [PubMed] [Google Scholar]
- Evans SM, Casartelli A, Herreros E, Minnick DT, Day C, George E, et al. Development of a high throughput in vitro toxicity screen predictive of high acute in vivo toxic potential. Toxicology in vitro : an international journal published in association with BIBRA. 2001;15:579–84. doi: 10.1016/s0887-2333(01)00064-9. [DOI] [PubMed] [Google Scholar]
- Fishovitz J, Li M, Frase H, Hudak J, Craig S, Ko K, et al. Active-Site-Directed Chemical Tools for Profiling Mitochondrial Lon Protease. ACS Chem Biol. 2011 doi: 10.1021/cb100408w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher FA, Kettunen MI, Day SE, Hu DE, Karlsson M, Gisselsson A, et al. Detection of tumor glutamate metabolism in vivo using (13)C magnetic resonance spectroscopy and hyperpolarized [1-(13)C]glutamate. Magn Reson Med. 2011;66:18–23. doi: 10.1002/mrm.22851. [DOI] [PubMed] [Google Scholar]
- Girish V, Vijayalakshmi A. Affordable image analysis using NIH Image/ImageJ. Indian J Cancer. 2004;41:47. [PubMed] [Google Scholar]
- Goebel G, Berger R, Strasak AM, Egle D, Muller-Holzner E, Schmidt S, et al. Elevated mRNA expression of CHAC1 splicing variants is associated with poor outcome for breast and ovarian cancer patients. Br J Cancer. 2012;106:189–98. doi: 10.1038/bjc.2011.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hato T, Tabata M, Oike Y. The role of angiopoietin-like proteins in angiogenesis and metabolism. Trends Cardiovasc Med. 2008;18:6–14. doi: 10.1016/j.tcm.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Hu S, Balakrishnan A, Bok RA, Anderton B, Larson PE, Nelson SJ, et al. 13C-pyruvate imaging reveals alterations in glycolysis that precede c-Myc-induced tumor formation and regression. Cell metabolism. 2011;14:131–42. doi: 10.1016/j.cmet.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua F, Mu R, Liu J, Xue J, Wang Z, Lin H, et al. TRB3 interacts with SMAD3 promoting tumor cell migration and invasion. J Cell Sci. 2011;124:3235–46. doi: 10.1242/jcs.082875. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–75. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun DH, Lee M, Halliwell B, Jenner P. Effect of overexpression of wild-type or mutant parkin on the cellular response induced by toxic insults. J Neurosci Res. 2005;82:232–44. doi: 10.1002/jnr.20638. [DOI] [PubMed] [Google Scholar]
- Ifergan I, Kebir H, Terouz S, Alvarez JI, Lecuyer MA, Gendron S, et al. Role of Ninjurin-1 in the migration of myeloid cells to central nervous system inflammatory lesions. Ann Neurol. 2011;70:751–63. doi: 10.1002/ana.22519. [DOI] [PubMed] [Google Scholar]
- Jin HO, Seo SK, Woo SH, Choe TB, Hong SI, Kim JI, et al. Nuclear protein 1 induced by ATF4 in response to various stressors acts as a positive regulator on the transcriptional activation of ATF4. IUBMB life. 2009;61:1153–8. doi: 10.1002/iub.271. [DOI] [PubMed] [Google Scholar]
- Karathanou A, Paterakis K, Pakopoulou M, Tasiou A, Hadjigeorgiou G, Chovas A, et al. Biochemical markers analyzed using microdialysis and traumatic brain injury outcomes. J Neurosurg Sci. 2011;55:173–7. [PubMed] [Google Scholar]
- Kim JY, Kim SM, Ko JH, Yim JH, Park JH, Park JH. Interaction of pro-apoptotic protein HGTD-P with heat shock protein 90 is required for induction of mitochondrial apoptotic cascades. FEBS Lett. 2006;580:3270–5. doi: 10.1016/j.febslet.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Koga K, Mori A, Ohashi S, Kurihara N, Kitagawa H, Ishikawa M, et al. H MRS identifies lactate rise in the striatum of MPTP-treated C57BL/6 mice. Eur J Neurosci. 2006;23:1077–81. doi: 10.1111/j.1460-9568.2006.04610.x. [DOI] [PubMed] [Google Scholar]
- Kotake Y, Ohta S. MPP+ analogs acting on mitochondria and inducing neuro-degeneration. Curr Med Chem. 2003a;10:2507–16. doi: 10.2174/0929867033456558. [DOI] [PubMed] [Google Scholar]
- Kotake Y, Ohta S. MPP+ analogs acting on mitochondria and inducing neuro-degeneration. Curr Med Chem. 2003b;10:2507–16. doi: 10.2174/0929867033456558. [DOI] [PubMed] [Google Scholar]
- Kubo T, Yamashita T, Yamaguchi A, Hosokawa K, Tohyama M. Analysis of genes induced in peripheral nerve after axotomy using cDNA microarrays. J Neurochem. 2002;82:1129–36. doi: 10.1046/j.1471-4159.2002.01060.x. [DOI] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell metabolism. 2012;15:110–21. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Kim JY, Suk K, Park JH. Identification of the hypoxia-inducible factor 1 alpha-responsive HGTD-P gene as a mediator in the mitochondrial apoptotic pathway. Mol Cell Biol. 2004;24:3918–27. doi: 10.1128/MCB.24.9.3918-3927.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage S, Brice A. Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet. 2009;18:R48–59. doi: 10.1093/hmg/ddp012. [DOI] [PubMed] [Google Scholar]
- Levy OA, Malagelada C, Greene LA. Cell death pathways in Parkinson’s disease: proximal triggers, distal effectors, and final steps. Apoptosis. 2009;14:478–500. doi: 10.1007/s10495-008-0309-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TK, Liou CW, Chen SD, Chuang YC, Tiao MM, Wang PW, et al. Mitochondrial dysfunction and biogenesis in the pathogenesis of Parkinson’s disease. Chang Gung medical journal. 2009a;32:589–99. [PubMed] [Google Scholar]
- Lin Y, Zhu N, Yu P, Su L, Mao L. Physiologically relevant online electrochemical method for continuous and simultaneous monitoring of striatum glucose and lactate following global cerebral ischemia/reperfusion. Anal Chem. 2009b;81:2067–74. doi: 10.1021/ac801946s. [DOI] [PubMed] [Google Scholar]
- Ling J, Zhao K, Cui YG, Li Y, Wang X, Li M, et al. Heat shock protein 10 regulated apoptosis of mouse ovarian granulosa cells. Gynecol Endocrinol. 2011;27:63–71. doi: 10.3109/09513590.2010.487594. [DOI] [PubMed] [Google Scholar]
- Liu J, Wu X, Franklin JL, Messina JL, Hill HS, Moellering DR, et al. Mammalian Tribbles homolog 3 impairs insulin action in skeletal muscle: role in glucose-induced insulin resistance. American journal of physiology Endocrinology and metabolism. 2010;298:E565–76. doi: 10.1152/ajpendo.00467.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JA, Pereda J, Martinez-Lopez I, Escrig R, Miralles V, Pallardo FV, et al. Oxidative stress as a signal to up-regulate gamma-cystathionase in the fetal-to-neonatal transition in rats. Cell Mol Biol (Noisy-le-grand) 2007;53(Suppl):OL1010-7. [PubMed] [Google Scholar]
- Maruoka N, Murata T, Omata N, Takashima Y, Fujibayashi Y, Wada Y. Topological and chronological features of the impairment of glucose metabolism induced by 1-methyl-4-phenylpyridinium ion (MPP+) in rat brain slices. J Neural Transm. 2007;114:1155–9. doi: 10.1007/s00702-007-0720-x. [DOI] [PubMed] [Google Scholar]
- Mazzio E, Soliman KF. D-(+)-glucose rescue against 1-methyl-4-phenylpyridinium toxicity through anaerobic glycolysis in neuroblastoma cells. Brain Res. 2003a;962:48–60. doi: 10.1016/s0006-8993(02)03695-8. [DOI] [PubMed] [Google Scholar]
- Mazzio E, Soliman KF. The role of glycolysis and gluconeogenesis in the cytoprotection of neuroblastoma cells against 1-methyl 4-phenylpyridinium ion toxicity. Neurotoxicology. 2003b;24:137–47. doi: 10.1016/s0161-813x(02)00110-9. [DOI] [PubMed] [Google Scholar]
- Mazzio EA, Smith B, Soliman KF. Evaluation of endogenous acidic metabolic products associated with carbohydrate metabolism in tumor cells. Cell Biol Toxicol. 2010a;26:177–88. doi: 10.1007/s10565-009-9138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzio EA, Soliman KF. Effects of enhancing mitochondrial oxidative phosphorylation with reducing equivalents and ubiquinone on 1-methyl-4-phenylpyridinium toxicity and complex I–IV damage in neuroblastoma cells. Biochem Pharmacol. 2004a;67:1167–84. doi: 10.1016/j.bcp.2003.11.016. [DOI] [PubMed] [Google Scholar]
- Mazzio EA, Soliman KF. Effects of enhancing mitochondrial oxidative phosphorylation with reducing equivalents and ubiquinone on 1-methyl-4-phenylpyridinium toxicity and complex I–IV damage in neuroblastoma cells. Biochem Pharmacol. 2004b;67:1167–84. doi: 10.1016/j.bcp.2003.11.016. [DOI] [PubMed] [Google Scholar]
- Mazzio EA, Soliman YI, Soliman KF. Variable toxicological response to the loss of OXPHOS through 1-methyl-4-phenylpyridinium-induced mitochondrial damage and anoxia in diverse neural immortal cell lines. Cell Biol Toxicol. 2010b;26:527–39. doi: 10.1007/s10565-010-9161-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzio EA, Soliman YI, Soliman KF. Variable toxicological response to the loss of OXPHOS through 1-methyl-4-phenylpyridinium-induced mitochondrial damage and anoxia in diverse neural immortal cell lines. Cell Biol Toxicol. 2010c;26:527–39. doi: 10.1007/s10565-010-9161-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhawech-Fauceglia P, Ali L, Cheney RT, Groth J, Herrmann FR. Prognostic significance of neuron-associated protein expression in non-muscle-invasive urothelial bladder cancer. J Clin Pathol. 2009;62:710–4. doi: 10.1136/jcp.2009.066159. [DOI] [PubMed] [Google Scholar]
- Minchenko A, Leshchinsky I, Opentanova I, Sang N, Srinivas V, Armstead V, et al. Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene. Its possible role in the Warburg effect. J Biol Chem. 2002;277:6183–7. doi: 10.1074/jbc.M110978200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y. Contribution of MPTP to studies on the pathogenesis of Parkinson’s disease. Rinsho Shinkeigaku. 1989;29:1494–6. [PubMed] [Google Scholar]
- Mizuno Y, Suzuki K, Sone N, Saitoh T. Inhibition of ATP synthesis by 1-methyl-4-phenylpyridinium ion (MPP+) in isolated mitochondria from mouse brains. Neurosci Lett. 1987;81:204–8. doi: 10.1016/0304-3940(87)90366-1. [DOI] [PubMed] [Google Scholar]
- Mortiboys H, Johansen KK, Aasly JO, Bandmann O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology. 2010;75:2017–20. doi: 10.1212/WNL.0b013e3181ff9685. [DOI] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–8. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungrue IN, Pagnon J, Kohannim O, Gargalovic PS, Lusis AJ. CHAC1/MGC4504 is a novel proapoptotic component of the unfolded protein response, downstream of the ATF4-ATF3-CHOP cascade. J Immunol. 2009;182:466–76. doi: 10.4049/jimmunol.182.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatsu T. Isoquinoline neurotoxins in the brain and Parkinson’s disease. Neurosci Res. 1997;29:99–111. doi: 10.1016/s0168-0102(97)00083-7. [DOI] [PubMed] [Google Scholar]
- Nagatsu T. Parkinson’s disease: changes in apoptosis-related factors suggesting possible gene therapy. J Neural Transm. 2002;109:731–45. doi: 10.1007/s007020200061. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Lipton SA. Redox regulation of mitochondrial fission, protein misfolding, synaptic damage, and neuronal cell death: potential implications for Alzheimer’s and Parkinson’s diseases. Apoptosis : an international journal on programmed cell death. 2010;15:1354–63. doi: 10.1007/s10495-010-0476-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini A, Morena E, Pucci A, Miglietta D, Riboldi E, Sozzani S, et al. Hypoxia affects dendritic cell survival: Role of the hypoxia-inducible factor-1alpha and lipopolysaccharide. J Cell Physiol. 2011 doi: 10.1002/jcp.22761. [DOI] [PubMed] [Google Scholar]
- Niemann A, Wagner KM, Ruegg M, Suter U. GDAP1 mutations differ in their effects on mitochondrial dynamics and apoptosis depending on the mode of inheritance. Neurobiol Dis. 2009;36:509–20. doi: 10.1016/j.nbd.2009.09.011. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Vilarino-Guell C, Cobb SA, Kachergus JM, Ross OA, Hentati E, et al. Genetic variation of the mitochondrial complex I subunit NDUFV2 and Parkinson’s disease. Parkinsonism Relat Disord. 2010;16:686–7. doi: 10.1016/j.parkreldis.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara Y, Ishii K, Tanabe S. Enzymatic assay of gamma-cystathionase activity using pyruvate oxidase-peroxidase sequential reaction. J Biochem Biophys Methods. 2002;51:139–50. doi: 10.1016/s0165-022x(02)00010-6. [DOI] [PubMed] [Google Scholar]
- Ogasawara Y, Suzuki T, Ishii K, Tanabe S. Modification of liver cytosol enzyme activities promoted in vitro by reduced sulfur species generated from cystine with gamma-cystathionase. Biochim Biophys Acta. 1997;1334:33–43. doi: 10.1016/s0304-4165(96)00072-4. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Shaw JM. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet. 2005;39:503–36. doi: 10.1146/annurev.genet.38.072902.093019. [DOI] [PubMed] [Google Scholar]
- Omerhodzic I, Dizdarevic K, Rotim K, Hajdarpasic E, Niksic M, Bejtic-Custovic E, et al. Cerebral microdialysis: perioperative monitoring and treatment of severe neurosurgical patient. Acta clinica Croatica. 2011;50:13–20. [PubMed] [Google Scholar]
- Pedrola L, Espert A, Valdes-Sanchez T, Sanchez-Piris M, Sirkowski EE, Scherer SS, et al. Cell expression of GDAP1 in the nervous system and pathogenesis of Charcot-Marie-Tooth type 4A disease. J Cell Mol Med. 2008;12:679–89. doi: 10.1111/j.1582-4934.2007.00158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrola L, Espert A, Wu X, Claramunt R, Shy ME, Palau F. GDAP1, the protein causing Charcot-Marie-Tooth disease type 4A, is expressed in neurons and is associated with mitochondria. Hum Mol Genet. 2005;14:1087–94. doi: 10.1093/hmg/ddi121. [DOI] [PubMed] [Google Scholar]
- Pinti M, Gibellini L, De Biasi S, Nasi M, Roat E, O’Connor JE, et al. Functional characterization of the promoter of the human Lon protease gene. Mitochondrion. 2011;11:200–6. doi: 10.1016/j.mito.2010.09.010. [DOI] [PubMed] [Google Scholar]
- Qu Y, Mao M, Zhao F, Zhang L, Mu D. Proapoptotic role of human growth and transformation-dependent protein in the developing rat brain after hypoxia-ischemia. Stroke. 2009;40:2843–8. doi: 10.1161/STROKEAHA.109.553644. [DOI] [PubMed] [Google Scholar]
- Rikka S, Quinsay MN, Thomas RL, Kubli DA, Zhang X, Murphy AN, et al. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ. 2011;18:721–31. doi: 10.1038/cdd.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink C, Gnyawali S, Peterson L, Khanna S. Oxygen-inducible glutamate oxaloacetate transaminase as protective switch transforming neurotoxic glutamate to metabolic fuel during acute ischemic stroke. Antioxid Redox Signal. 2011;14:1777–85. doi: 10.1089/ars.2011.3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollema H, Kuhr WG, Kranenborg G, De Vries J, Van den Berg C. MPP+-induced efflux of dopamine and lactate from rat striatum have similar time courses as shown by in vivo brain dialysis. J Pharmacol Exp Ther. 1988;245:858–66. [PubMed] [Google Scholar]
- Ruckert IM, Heier M, Rathmann W, Baumeister SE, Doring A, Meisinger C. Association between markers of fatty liver disease and impaired glucose regulation in men and women from the general population: the KORA-F4-study. PloS one. 2011;6:e22932. doi: 10.1371/journal.pone.0022932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serganova I, Rizwan A, Ni X, Thakur SB, Vider J, Russell J, et al. Metabolic imaging: a link between lactate dehydrogenase A, lactate, and tumor phenotype. Clin Cancer Res. 2011;17:6250–61. doi: 10.1158/1078-0432.CCR-11-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan YX, Liu TJ, Su HF, Samsamshariat A, Mestril R, Wang PH. Hsp10 and Hsp60 modulate Bcl-2 family and mitochondria apoptosis signaling induced by doxorubicin in cardiac muscle cells. J Mol Cell Cardiol. 2003;35:1135–43. doi: 10.1016/s0022-2828(03)00229-3. [DOI] [PubMed] [Google Scholar]
- Song A, Ko HJ, Lai MN, Ng LT. Protective effects of Wu-Ling-Shen (Xylaria nigripes) on carbon tetrachloride-induced hepatotoxicity in mice. Immunopharmacol Immunotoxicol. 2011;33:454–60. doi: 10.3109/08923973.2010.534100. [DOI] [PubMed] [Google Scholar]
- Stein M, Schomacher J, Scharbrodt W, Preuss M, Oertel MF. Cerebrospinal fluid lactate concentration after withdrawal of metabolic suppressive therapy in subarachnoid hemorrhage. Acta neurochirurgica Supplement. 2012;114:333–7. doi: 10.1007/978-3-7091-0956-4_64. [DOI] [PubMed] [Google Scholar]
- Steyn SJ, Pieterse DJ, Mienie LJ, Van der Schyf CJ. Measurement of mitochondrial respiration in permeabilized murine neuroblastoma (N-2alpha) cells, a simple and rapid in situ assay to investigate mitochondrial toxins. J Biochem Biophys Methods. 2005;62:25–40. doi: 10.1016/j.jbbm.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Storch A, Ludolph AC, Schwarz J. HEK-293 cells expressing the human dopamine transporter are susceptible to low concentrations of 1-methyl-4-phenylpyridine (MPP+) via impairment of energy metabolism. Neurochem Int. 1999;35:393–403. doi: 10.1016/s0197-0186(99)00083-2. [DOI] [PubMed] [Google Scholar]
- Storch A, Ott S, Hwang YI, Ortmann R, Hein A, Frenzel S, et al. Selective dopaminergic neurotoxicity of isoquinoline derivatives related to Parkinson’s disease: studies using heterologous expression systems of the dopamine transporter. Biochem Pharmacol. 2002;63:909–20. doi: 10.1016/s0006-2952(01)00922-4. [DOI] [PubMed] [Google Scholar]
- Sundar Boyalla S, Barbara Victor M, Roemgens A, Beyer C, Arnold S. Sex- and brain region-specific role of cytochrome c oxidase in 1-methyl-4-phenylpyridinium-mediated astrocyte vulnerability. J Neurosci Res. 2011;89:2068–82. doi: 10.1002/jnr.22669. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Mizuno Y, Yoshida M. Inhibition of mitochondrial respiration by 1,2,3,4-tetrahydroisoquinoline-like endogenous alkaloids in mouse brain. Neurochem Res. 1990;15:705–10. doi: 10.1007/BF00973651. [DOI] [PubMed] [Google Scholar]
- Swarnkar S, Goswami P, Kamat PK, Gupta S, Patro IK, Singh S, et al. Rotenone-induced apoptosis and role of calcium: a study on Neuro-2a cells. Arch Toxicol. 2012 doi: 10.1007/s00204-012-0853-z. [DOI] [PubMed] [Google Scholar]
- Thomas RL, Kubli DA, Gustafsson AB. Bnip3-mediated defects in oxidative phosphorylation promote mitophagy. Autophagy. 2011;7:775–7. doi: 10.4161/auto.7.7.15536. [DOI] [PubMed] [Google Scholar]
- Tofaris GK, Spillantini MG. Alpha-synuclein dysfunction in Lewy body diseases. Mov Disord. 2005;20 (Suppl 12):S37–44. doi: 10.1002/mds.20538. [DOI] [PubMed] [Google Scholar]
- Wagatsuma A, Kotake N, Yamada S. Muscle regeneration occurs to coincide with mitochondrial biogenesis. Mol Cell Biochem. 2011;349:139–47. doi: 10.1007/s11010-010-0668-2. [DOI] [PubMed] [Google Scholar]
- Wang P, Tang Y, Chen X, Du B. Nuclear translocation of the pro-apoptotic protein BNIP3 in cultured spiral ganglion cells of rat with cisplatin insult. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2011a;46:214–9. [PubMed] [Google Scholar]
- Wang Z, Huang CM, Deng Q, Zeng H, Wang X, Zhang S, et al. Effects of the Proapoptotic Regulator Bcl2/Adenovirus EIB 19 kDa-Interacting Protein 3 on Radiosensitivity of Cervical Cancer. Cancer Biother Radiopharm. 2011b;26:279–86. doi: 10.1089/cbr.2010.0898. [DOI] [PubMed] [Google Scholar]
- Wennemers M, Bussink J, Scheijen B, Nagtegaal ID, van Laarhoven HW, Raleigh JA, et al. Tribbles homolog 3 denotes a poor prognosis in breast cancer and is involved in hypoxia response. Breast cancer research : BCR. 2011;13:R82. doi: 10.1186/bcr2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winklhofer KF, Haass C. Mitochondrial dysfunction in Parkinson’s disease. Biochim Biophys Acta. 2010;1802:29–44. doi: 10.1016/j.bbadis.2009.08.013. [DOI] [PubMed] [Google Scholar]
- Xu P, Das M, Reilly J, Davis RJ. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 2011;25:310–22. doi: 10.1101/gad.1984311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokobori S, Watanabe A, Matsumoto G, Onda H, Masuno T, Fuse A, et al. Lower extracellular glucose level prolonged in elderly patients with severe traumatic brain injury: a microdialysis study. Neurol Med Chir (Tokyo) 2011;51:265–71. doi: 10.2176/nmc.51.265. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Chen G, Zhang W, Xu N, Zhu JY, Jia J, et al. Autophagy regulates hypoxia-induced osteoclastogenesis through the HIF-1alpha/BNIP3 signaling pathway. J Cell Physiol. 2011a doi: 10.1002/jcp.22768. [DOI] [PubMed] [Google Scholar]
- Zhao YL, Zhou GD, Yang HB, Wang JB, Shan LM, Li RS, et al. Rhein protects against acetaminophen-induced hepatic and renal toxicity. Food Chem Toxicol. 2011b;49:1705–10. doi: 10.1016/j.fct.2011.04.011. [DOI] [PubMed] [Google Scholar]
- Zhu JH, Gusdon AM, Cimen H, Van Houten B, Koc E, Chu CT. Impaired mitochondrial biogenesis contributes to depletion of functional mitochondria in chronic MPP(+) toxicity: dual roles for ERK1/2. Cell death & disease. 2012;3:e312. doi: 10.1038/cddis.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweckberger K, Hackenberg K, Jung CS, Hertle DN, Kiening KL, Unterberg AW, et al. Cerebral metabolism after early decompression craniotomy following controlled cortical impact injury in rats. Neurol Res. 2011;33:875–80. doi: 10.1179/1743132811Y.0000000017. [DOI] [PubMed] [Google Scholar]